Impact Factor ISSN: 1449-1907

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2026; 23(7):2290-2301. doi:10.7150/ijms.124444 This issue Cite
Research Paper
Plumbagin sensitizes leukemia cells to cisplatin by promoting oxidative stress, apoptosis, and DNA damage
Yan-Ning Chen1#, Jai-Wei Lee2#, Tsu-Ming Chien3,4,5, Chia-Hung Yen1, Bing-Hung Chen6 ![]() , Hsueh-Wei Chang1,7,8,9,10
, Hsueh-Wei Chang1,7,8,9,10 ![]()
1. Graduate Institute of Natural Products, Kaohsiung Medical University, Kaohsiung 80708, Taiwan.
2. Department of Tropical Agriculture and International Cooperation, National Pingtung University of Science and Technology, Pingtung 912301, Taiwan.
3. School of Post-Baccalaureate Medicine, Kaohsiung Medical University, Kaohsiung 80708, Taiwan.
4. Department of Urology, Kaohsiung Medical University Hospital, Kaohsiung Medical University, Kaohsiung 80708, Taiwan.
5. Department of Urology, Kaohsiung Gangshan Hospital, Kaohsiung Medical University, Kaohsiung 820111, Taiwan.
6. Department of Biotechnology, College of Life Science, Kaohsiung Medical University, Kaohsiung 80708, Taiwan.
7. Department of Biomedical Science and Environmental Biology, College of Life Science, Kaohsiung Medical University, Kaohsiung 80708, Taiwan.
8. Department of Medical Research, Kaohsiung Medical University Hospital, Kaohsiung 80708, Taiwan.
9. Center for Cancer Research and Research Center for Molecular Medicine, Kaohsiung Medical University, Kaohsiung 80708, Taiwan.
10. Drug Development and Value Creation Research Center, Kaohsiung Medical University, Kaohsiung 80708, Taiwan.
# Equal contribution.
Received 2025-8-30; Accepted 2026-5-19; Published 2026-5-29
Abstract

This study focused on validating the synergistic antiproliferative activity and mechanism of cisplatin/plumbagin (PLB) in leukemic cells versus normal macrophages. Leukemic cells (MOLT-4) treated with cisplatin/PLB (3 μM/0.4 μM) for 24 h showed markedly reduced viability (45.51%) compared to cisplatin or PLB (81.07% or 76.36%), while normal macrophages (NR8383) retained high viability in cisplatin/PLB (~93.15%). The enhanced cytotoxicity in leukemia cells was driven by oxidative stress: the general antioxidant N-acetylcysteine (NAC) and the mitochondrial ROS scavenger MitoTEMPO substantially reversed the combination effect. Cisplatin/PLB increased total ROS and mitochondrial superoxide in MOLT-4 cells more than in macrophages, and provoked loss of mitochondrial membrane potential and depletion of glutathione; these alterations were mitigated by NAC and MitoTEMPO. Oxidative stress led to higher apoptosis in leukemia cells than normal macrophages, shown by increased subG1 fraction (validated via NAC), higher annexin V positivity (validated via NAC and MitoTEMPO), elevated cleaved PARP and caspases-9/-3, Bax upregulation, and a reduced p-Bcl-2 (anti-apoptotic Ser70)/Bcl-2 ratio (validated via NAC), whereas caspase-8 changed only modestly. DNA damage markers (γH2AX and 8-OHdG) were also increased in MOLT-4 cells and attenuated by NAC and MitoTEMPO. Overall, cisplatin/PLB triggers selective and oxidative stress-dependent in leukemia cells while sparing normal macrophages, supporting the combination as a promising antileukemic approach with limited toxicity.
Keywords: plumbagin, combined treatment, cisplatin, leukemia, oxidative stress
Introduction
Leukemia is a type of hematological malignancy with the mass production characteristics of abnormal immature blood cells, blocking the generation of normal blood cells. There were 66,890 estimated new cases and 23,540 estimated deaths worldwide caused by leukemia in “Cancer statistics, 2025” [1]. Males show a mildly higher number of cases of fatalities from leukemia than females. Generally, four common types of leukemia have been classified, i.e., acute myeloid leukemia (AML) [2], acute lymphoblastic leukemia (ALL) [3], chronic lymphocytic leukemia (CLL) [4], and chronic myeloid leukemia (CML) [5]. ALL is the most common malignancy in children and teenagers [6, 7]. Therefore, the present study focused on improving the inhibition of ALL cell proliferation.
Combined treatment is an improved anticancer strategy that combines two or more therapeutic agents [8]. The ideal combined treatment is effective cancer therapy with fewer toxic side effects because it uses a low dose but generates synergistic effects. Leukemia therapy favors combined treatments, including chemotherapy, radiotherapy, targeted therapy, and bone marrow transplantation [9, 10]. However, patients occasionally experience adverse effects from receiving the leukemia treatments [11]. Investigating additional potential anti-leukemia drugs for combined treatment with clinical medications is necessary to alleviate side effects.
Cisplatin is the first-line anticancer drug for several cancers, such as oral, breast, bladder, testicular, ovarian, prostate, cervical, lung, leukemia, and others [12, 13]. Although cisplatin generally has practical chemotherapeutic functions, its potential side effects limit cancer treatment [14]. A combination of natural products with cisplatin has been reported in several anticancer studies [15, 16], thereby enhancing cisplatin sensitivity in cancer cells and reducing cytotoxicity in normal cells.
Natural products have made a significant contribution to anticancer drug discovery [17]. Several natural products exhibit high antiproliferative efficacy against cancer cells with low cytotoxicity to normal cells [18, 19]. For example, Nepenthes plants, such as N. mirabilis, N. alata, N. khasiana, and N. rajah, are commonly used as traditional herbs in Southeast Asia [20]. In addition to the antibacterial, antifungal [21], and anti-inflammatory [22] effects, several Nepenthes extracts exhibit antiproliferative functions against cancer cells [23]. For example, the ethyl acetate extract of N. thorelii x (ventricosa x maxima) (EANT) shows antiproliferative activity in leukemia cells [24]. Plumbagin (PLB), the main bioactive compound (naphthoquinone) of EANT, showed antiproliferative effects on leukemia cells [24], cervical [25], and tongue [26] cancer cells. Furthermore, PLB induces apoptosis of CML (K562) [27]. The safety of PLB was validated in normal peripheral blood mononuclear cells [28] and in animal studies [28].
Prooxidant drugs can suppress leukemia; for example, arsenic trioxide generates ROS that induce mitotic arrest and apoptosis in AML [29], while all-trans retinoic acid derivative elevates ROS to promote differentiation and inhibit leukemic cell growth [30]. Because both PLB [24] and cisplatin [31] induce ROS in leukemia cells, their combination may further elevate ROS levels and enhance antileukemic activity. While cisplatin/PLB combined treatment has been described for cervical [25] and tongue [26] cancer cell models, it has not yet been reported in leukemia cells. Consequently, the potential antiproliferative effects of cisplatin/PLB on leukemia cells warrant a detailed assessment.
The present study aims to evaluate the antiproliferative effects and assess the anticancer mechanism of cisplatin/PLB on leukemia cells, particularly for ALL.
Materials and Methods
Cell cultures, viability, synergy index, and reagents
Two kinds of ATCC human T-cell ALL cell lines (Manassas) derived from leukemia were chosen, including MOLT-4 [32] and J45.01. NR8383 [33], the normal rat lung macrophage from ATCC, is regarded as the normal control cell. MOLT-4 is a primary cortical T-cell ALL line with intact CD45 expression and functional T-cell receptor signaling [34], while J45.01 is a CD45-deficient Jurkat mutant lacking this signaling [35]. MOLT-4 and J45.01 cells were kept in RPMI medium (Gibco) and supplemented with 10% FBS and antibiotics. NR8383 cells were kept in F12 medium containing 15% FBS and antibiotics. ATP-lite reagents (PerkinElmer Life Sciences) were used to assess cell viability, according to the user instructions. Briefly, after removing the medium, 100 μL of serum-free medium and 50 μL of lysis buffer were added per well and shaken at 100 rpm for 5 min. Then 100 μL of lysate was transferred to a white 96-well plate, 12.5 μL of D-luciferin/luciferase substrate was added, and the plate was shaken at 100 rpm for 5 min. Finally, luminescence was measured with a luminometer [36]. The synergy index (α value) for the combined cisplatin/PLB was calculated as previously described [37]: α = (viability fraction of cisplatin × viability fraction of PLB) / viability fraction of cisplatin/PLB. Values of α = 1, > 1, and < 1 indicate additive, synergistic and antagonistic interactions, respectively.
PLB (Selleckchem) was dissolved in DMSO. The DMSO concentration in all experiments was the same (0.1%). N-acetylcysteine (NAC; 5 mM, 1 h) (Sigma-Aldrich) [38] and MitoTEMPO (20 μM, 1 h) (Cayman Chemical) [39, 40] were used as inhibitors for reactive oxygen species (ROS) and mitochondrial superoxide (MitoSOX). These inhibitors were pretreated to validate the role of ROS and MitoSOX in PLB treatments.
ROS, MitoSOX, mitochondrial membrane potential (MMP), and glutathione (GSH) assay
According to the user's manual, flow cytometry reagents were applied to detect several oxidative stress-related indicators, i.e., dihydroethidium (DHE) (Sigma-Aldrich) [39] (5 μM for 30 min) for ROS, MitoSOX™ Red (Molecular Probes, Invitrogen, Eugene, OR, USA) [41] (50 nM for 30 min) for MitoSOX, JC-1 (Sigma-Aldrich) [42] (0.2 μM for 30 min) for MMP, and 5-chloromethylfluorescein diacetate (CMF-DA) (Thermo Fisher Scientific) [41] (0.1 μM, 20 min) for GSH detections. Finally, these oxidative stress-related responses were detected using flow cytometry analysis. Notably, JC-1 green appears in damaged MMP, while JC-1 red appears in healthy MMP. A high ratio (JC-1 green/JC-1 red) and GSH (-) proportion indicate the MMP and GSH depletion.
Cell cycle assay
Cells were fixed with 75% ethanol overnight and stained with 7-aminoactinomycin D (7AAD) (Biotium) [19] (1 μg/mL, 30 min, 37 °C). After washing, cells were conducted with a flow cytometer (Guava easyCyte, Luminex, TX, USA).
Annexin V-apoptosis assay
Annexin V can bind to phosphatidylserine in apoptotic cells. Reagents containing annexin V/7AAD kit (Strong Biotech) [19] were used for this double staining. After washing, cells were conducted with a flow cytometer.
Western blotting for apoptotic signaling
Apoptotic signaling was detected using antibody sampler kits (#9915/#9941/#9942, Cell Signaling Technology), recognizing proapoptotic (Poly (ADP-ribose) polymerase (PARP), Bcl-2 associated X (Bax), and cleaved caspases 3/8/9) and anti-apoptotic proteins (B-cell lymphoma 2 (Bcl-2) and phosphorylated Bcl-2 (Ser70) (5H2) (anti-apoptotic p-Bcl-2 [43], #2827)).
γH2AX and 8-hydroxy-2′-deoxyguanosine (8-OHdG) assays
γH2AX primary antibody (Santa Cruz Biotechnology) was diluted to 500X. After washing, the fixed cells were incubated with the Alexa Fluor 488-secondary antibody (1:10000) (Jackson Laboratory). Without the secondary antibody step, the 8-OHdG-FITC antibody was diluted to 10000X (Santa Cruz Biotechnology) for mixing with cell suspensions. Both γH2AX and 8-OHdG were detected using flow cytometry analysis [41].
Statistics
The ANOVA assay and Tukey post hoc test were conducted using JMP 12 software (SAS Institute) for most experiments except for western blotting (Student's t-test). In multi-comparison, JMP gave low-case letters for different treatments. The statistical difference between treatments was judged by non-overlapped lower-case letters (p < 0.05). Data were derived from triplicated experiments and are represented as the means ± SDs.
Results
Synergistic antiproliferative effects of cisplatin/PLB
Cisplatin/PLB, the combined treatment, showed cell viability of 45.51% and 49.94% in MOLT-4 and J45.01 cells, respectively, lower than cisplatin (81.07% and 81.79%) or PLB (76.36% and 77.40%) (Figure 1A). In comparison, the normal cell line (NR8383) showed limited cytotoxicity. The synergy index (α value) of cisplatin/PLB for the MOLT-4 and J45.01 cells was 1.36 and 1.27. This result revealed that cisplatin/PLB synergistically inhibited the proliferation of ALL cells. Since the MOLT-4 cells had a higher synergy index (α value) than J45.01 cells showed similar antiproliferative effects for cisplatin/PLB (1.36 and 1.27), MOLT-4 cells were chosen for the following experiments.
Cell viability for cisplatin and/or PLB treatments. Except for NAC or MitoTEMPO pretreatment or not, the cells were treated with the control (0.1% DMSO), cisplatin (3 μM), PLB (0.4 μM), and cisplatin/PLB (3 μM/0.4 μM) for 24 h, and their viabilities were analyzed by ATP assay. The data are marked at the top with lower-case letters, and nonoverlapping notes indicate statistical differences in multi-comparisons (p < 0.05).
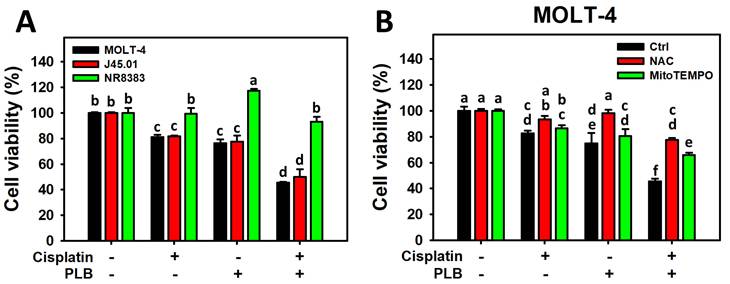
The ROS and MitoSOX inhibitors (NAC and MitoTEMPO) pretreatments were conducted before cisplatin or/and PLB treatments on MOLT-4 cells to assess the participation of cellular and mitochondrial oxidative stress. NAC and MitoTEMPO rescued the cisplatin/PLB-inhibiting proliferation (Figure 1B). Consequently, the results suggest ROS and MitoSOX contribute to the synergistic antiproliferative effect of cisplatin/PLB on MOLT-4 cells, although pharmacological inhibitor studies alone do not establish direct causality.
Enhanced ROS and MitoSOX generation of cisplatin/PLB
As mentioned above, the role of cellular and mitochondrial oxidative stress (ROS and MitoSOX) on the synergistic antiproliferation of MOLT-4 cells treated with cisplatin/PLB was validated by their inhibitors such as NAC and MitoTEMPO, respectively (Figure 1B). However, oxidative stress, such as ROS and MitoSOX, was not assessed.
The ROS and MitoSOX response of cisplatin/PLB of MOLT-4 cells was determined via flow cytometry in parallel with normal cells (NR8383). Cisplatin/PLB caused a more significant ROS and MitoSOX proportion (+) (%) in MOLT-4 cells than in cisplatin, PLB, or the control (Figures 2A, 2C). In comparison, normal cells (NR8383) showed limited ROS and MitoSOX response in the cisplatin and/or PLB treatments (Figures 2B,2D).
ROS and MitoSOX assays for cisplatin and/or PLB treatments. (A,C) ROS and MitoSOX levels of MOLT-4 cells. Except for NAC or MitoTEMPO pretreatment or not, MOLT-4 cells were treated with the control (0.1% DMSO), cisplatin (3 μM), PLB (0.4 μM), and cisplatin/PLB (3 μM/0.4 μM) for 24 h. (B,D) ROS and MitoSOX levels of normal cells (NR8383). Cells were treated in the same condition as (A,C), but no inhibitor was used. The ROS and MitoSOX-positive proportions (%) are labeled with (+) (%). The data are marked at the top with lower-case letters, and different letters indicate statistical differences in multi-comparisons (p < 0.05). Gating for ROS and MitoSOX produced similar proportions of positive events in the control, NAC, and MitoTEMPO groups under PLB(-)/cisplatin(-) conditions; the same gate positions were applied to all other treatments.
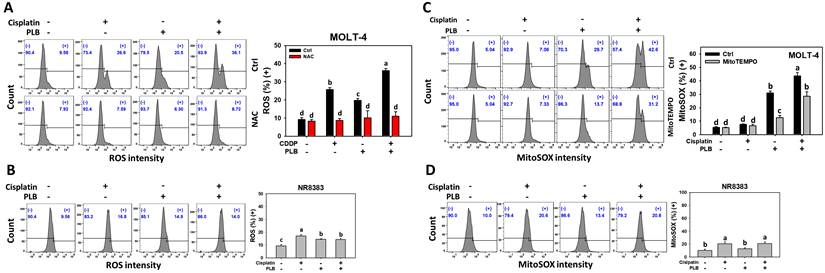
NAC and MitoTEMPO pretreatments were conducted before cisplatin or/and PLB treatments on MOLT-4 cells to further confirm the cellular and mitochondrial oxidative stress status. NAC and MitoTEMPO rescued the cisplatin or/and PLB-induced ROS and MitoSOX increment (Figures 2A,2C). Consequently, cisplatin/PLB demonstrated the enhanced burst effects of ROS and MitoSOX on MOLT-4 cells.
Enhanced MMP and GSH depletion of cisplatin/PLB
In addition to ROS and MitoSOX burst, MMP depletion is another sign of oxidative stress [44]. The MMP response of cisplatin/PLB was determined via flow cytometry. Cisplatin/PLB exhibited a higher ratio (JC-1 green/red) in MOLT-4 cells than in cisplatin, PLB, or the control (Figure 3A), indicating MMP depletion. In comparison, normal cells (NR8383) showed limited MMP response in cisplatin and/or PLB treatments (Figure 3B).
MMP and GSH assays for cisplatin and/or PLB treatments. (A,C) MMP and GSH levels of MOLT-4 cells. Except for NAC or MitoTEMPO pretreatment or not, MOLT-4 cells were treated with the control (0.1% DMSO), cisplatin (3 μM), PLB (0.4 μM), and cisplatin/PLB (3 μM/0.4 μM) for 24 h. (B,D) MMP and GSH levels of normal cells (NR8383). Cells were treated in the same condition as (A,C), but no inhibitor was used. The MMP status was evaluated by the JC-1 green/red ratio, where a high ratio indicated MMP depletion. The GSH-negative proportion (%) is labeled with (-) (%), where a high (-) (%) indicates GSH depletion. The data are marked at the top with lower-case letters, and different letters indicate statistical differences in multi-comparisons (p < 0.05). Gating for MMP and GSH produced similar proportions of positive events in the control, NAC, and MitoTEMPO groups under PLB(-)/cisplatin(-) conditions; the same gate positions were applied to all other treatments.
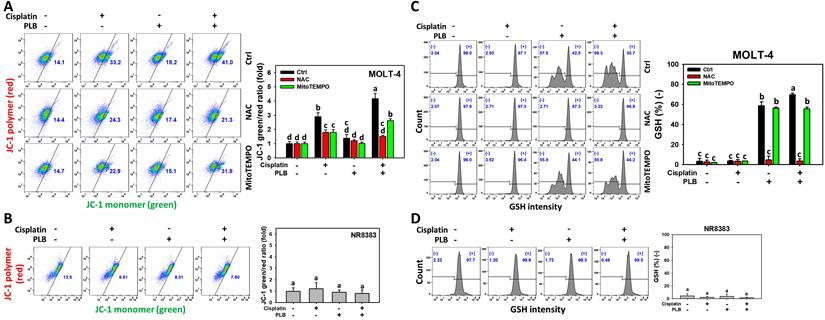
NAC and MitoTEMPO pretreatments were conducted before cisplatin or/and PLB treatments on MOLT-4 cells to further confirm the oxidative stress status. NAC and MitoTEMPO rescued the cisplatin or/and PLB-induced MMP depletion, as evidenced by decreasing the ratio (JC-1 green/red) (Figure 3A). Consequently, the results suggest ROS and MitoSOX contribute to the enhanced MMP depletion of cisplatin/PLB on MOLT-4 cells, although pharmacological inhibitor studies alone do not establish direct causality.
GSH can counteract oxidative stress and the GSH level is associated with the modulation of oxidative stress [45]. The GSH response of cisplatin/PLB was determined via flow cytometry. Cisplatin/PLB caused a more significant GSH proportion (-) (%) in MOLT-4 cells than in cisplatin, PLB, or the control (Figure 3C). In comparison, normal cells (NR8383) showed limited GSH response in cisplatin and/or PLB treatments (Figure 3D).
To further confirm the GSH status, NAC and MitoTEMPO pretreatments were conducted before cisplatin or/and PLB treatments on MOLT-4 cells. NAC and MitoTEMPO rescued the cisplatin or/and PLB-induced GSH depletion (Figure 3C). Consequently, the results suggest ROS and MitoSOX contribute to the enhanced GSH depletion of cisplatin/PLB on MOLT-4 cells, although pharmacological inhibitor studies alone do not establish direct causality.
Enhanced subG1 increment of cisplatin/PLB
The participation of cell cycle interference in the synergistic antiproliferative effects of cisplatin/PLB was assessed (Figure 4A). Cisplatin/PLB increased the subG1 proportion (%) of MOLT-4 cells more than cisplatin, PLB, or the control. In comparison, normal cells (NR8383) showed limited subG1 response to the cisplatin and/or PLB treatments (Figure 4B). This subG1 phenomenon is an apoptosis-like indicator and warrants careful investigation.
Cell cycle assay for cisplatin and/or PLB treatments. (A) SubG1 proportion of MOLT-4 cells. Except for NAC pretreatment or not, MOLT-4 cells were treated with the control (0.1% DMSO), cisplatin (3 μM), PLB (0.4 μM), and cisplatin/PLB (3 μM/0.4 μM) for 24 h. (B) SubG1 proportion of normal cells (NR8383). Cells were treated in the same condition as (A), but no inhibitor was used. The data are marked at the top with lower-case letters, and different letters indicate statistical differences in multi-comparisons (p < 0.05). Gating for G1 and G2/M was based on DNA content (2n and 4n, respectively); S phase was defined as the region between G1 and G2/M, and sub-G1 as events with DNA content below the G1 peak.
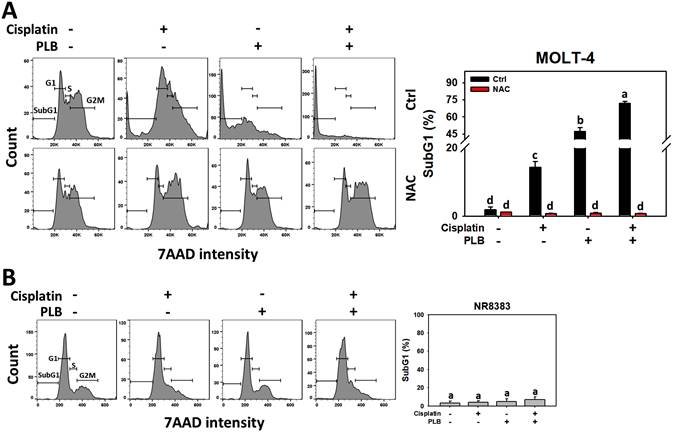
Global cellular ROS has a more critical and direct role in broader cell cycle progression [46] than mitochondrial ROS alone [47]. To assess the participation of global oxidative stress in cell cycle progression, NAC but not MitoTEMPO was introduced for pretreatment in the treatment of cisplatin or/and PLB on MOLT-4 cells. NAC rescued the cisplatin or/and PLB-induced subG1 increment (Figure 4A). Consequently, the results suggest ROS contributes to the enhanced subG1 accumulative effects of cisplatin/PLB on MOLT-4 cells, although pharmacological inhibitor studies alone do not establish direct causality.
Enhanced apoptosis (annexin V) increment of cisplatin/PLB
The potential for apoptosis of MOLT-4 cells in the treatments of cisplatin or/and PLB was evaluated using the annexin V/7AAD method. Cisplatin/PLB caused more significant apoptosis (%) in MOLT-4 cells than in cisplatin, PLB, or the control (Figure 5A). Notably, its quadrant Q2 (late apoptosis) was higher than Q3 (early apoptosis). In comparison, normal cells (NR8383) showed limited apoptosis response to the cisplatin and/or PLB treatments (Figure 5B), which dominantly showed the early apoptosis (Q3). This suggests that cisplatin/PLB exerts more severe apoptosis in leukemia cells than normal cells.
Annexin V/7AAD assay for cisplatin and/or PLB treatments. (A) Annexin V levels of MOLT-4 cells. Except for NAC or MitoTEMPO pretreatment or not, MOLT-4 cells were treated with the control (0.1% DMSO), cisplatin (3 μM), PLB (0.4 μM), and cisplatin/PLB (3 μM/0.4 μM) for 24 h. (B) Annexin V level of normal cells (NR8383). Cells were treated in the same condition as (A), but no inhibitor was used. The annexin V-positive proportion (%) is counted as apoptosis (%). The data are marked at the top with lower-case letters, and different letters indicate statistical differences in multi-comparisons (p < 0.05). Gating for annexin V produced similar proportions of positive events (Q2+Q3) in the control, NAC, and MitoTEMPO groups under PLB(-)/cisplatin(-) conditions; the same gate positions were applied to all other treatments.
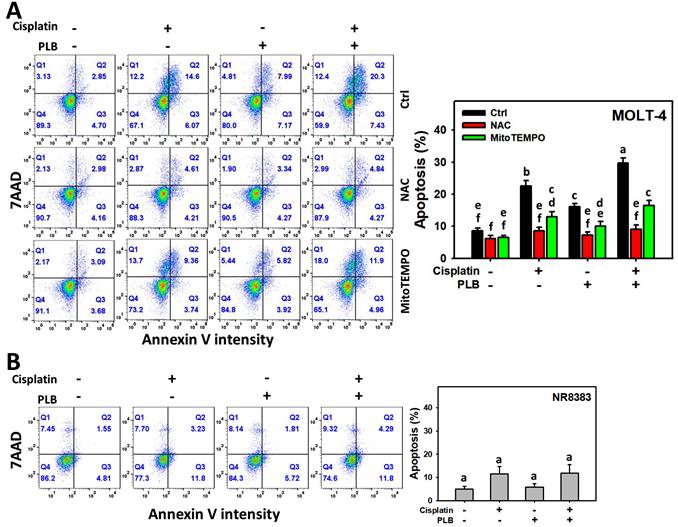
To assess the participation of oxidative stress in apoptosis, NAC and MitoTEMPO pretreatments were conducted before cisplatin or/and PLB treatments on MOLT-4 cells. NAC and MitoTEMPO rescued the cisplatin or/and PLB-induced annexin V increment, i.e., apoptosis (Figure 5A). Consequently, the results suggest ROS and MitoSOX contribute to the enhanced apoptotic effects of cisplatin/PLB on MOLT-4 cells, although pharmacological inhibitor studies alone do not establish direct causality.
Enhanced activation for intrinsic apoptotic signaling of cisplatin/PLB
PARP, the DNA repair enzyme, is cleaved by activated caspase 3 and is regarded as a hallmark of apoptosis [48]. To assess the involvement of extrinsic and intrinsic caspases, the levels of cleaved caspases 8 and 9 were examined. Cisplatin/PLB caused higher expressions of cleaved caspases 3 and 9 in MOLT-4 cells than in cisplatin, PLB, or the control (Figure 6). However, the extrinsic caspase 8 levels were non-significantly different between cisplatin/PLB and cisplatin alone, although they were higher than the control and PLB alone.
Apoptotic signaling assays. Except for NAC pretreatment or not, MOLT-4 cells were treated with the control (0.1% DMSO), cisplatin (3 μM), PLB (0.4 μM), and cisplatin/PLB (3 μM/0.4 μM) for 24 h, and their apoptotic signaling status was analyzed via Western blotting. The data are marked at the top with lower-case letters, and different letters indicate statistical differences in multi-comparisons (p < 0.05). p-Bcl-2 (Ser70) is used as anti-apoptotic marker.
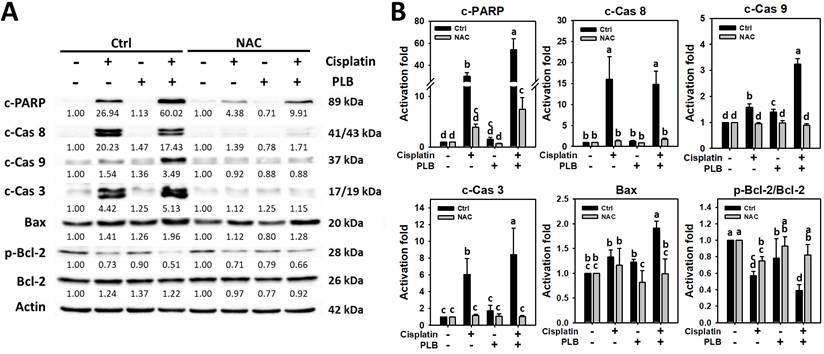
To further assess the role of intrinsic caspases, the levels of caspase 9's upstream proapoptotic and anti-apoptotic proteins, such as Bax and Bcl-2, were examined [49]. When the p-Bcl-2/Bcl-2 ratio is larger than 1, it indicates that cells were triggered to intrinsic apoptosis [50]. Consistently, cisplatin/PLB upregulated higher Bax in MOLT-4 cells than in other treatments. In contrast, cisplatin/PLB downregulated the p-Bcl-2 (anti-apoptotic Ser70)/Bcl-2 ratio more than the single treatments (Figure 6).
Since Figure 5A showed that NAC is more effective in suppressing apoptosis (as indicated by annexin V), we therefore take advantage of this to use NAC to investigate its apoptotic signaling without MitoTEMPO. To assess the participation of cellular oxidative stress in PARP cleavage and caspases 8/9/3 activation, NAC pretreatment was conducted before cisplatin or/and PLB treatments on MOLT-4 cells. NAC rescued the cisplatin or/and PLB-induced apoptotic signaling (Figure 6). Consequently, the results suggest ROS contributes to the enhanced intrinsic apoptotic signaling of cisplatin/PLB on MOLT-4 cells, although pharmacological inhibitor studies alone do not establish direct causality.
Enhanced γH2AX and 8-OHdG increment of cisplatin/PLB
The γH2AX and 8-OHdG response of cisplatin/PLB was determined via flow cytometry. Cisplatin/PLB caused a more significant γH2AX and 8-OHdG proportion (+) (%) in MOLT-4 cells than in cisplatin, PLB, or the control (Figures 7A,7C). In comparison, normal cells (NR8383) showed limited γH2AX and 8-OHdG response to the cisplatin and/or PLB treatments (Figures 7B,7D).
γH2AX and 8-OHdG assays for cisplatin and/or PLB treatments. (A,C) γH2AX and 8-OHdG levels of MOLT-4 cells. Except for NAC or MitoTEMPO pretreatment or not, MOLT-4 cells were treated with the control (0.1% DMSO), cisplatin (3 μM), PLB (0.4 μM), and cisplatin/PLB (3 μM/0.4 μM) for 24 h. (B,D) γH2AX and 8-OHdG levels of normal cells (NR8383). Cells were treated in the same condition as (A,C), but no inhibitor was used. The γH2AX and 8-OHdG-positive proportion (%) is labeled with (+) (%). The data are marked at the top with lower-case letters, and different letters indicate statistical differences in multi-comparisons (p < 0.05). Gating for γH2AX and 8-OHdG produced similar proportions of positive events in the control, NAC, and MitoTEMPO groups under PLB(-)/cisplatin(-) conditions; the same gate positions were applied to all other treatments.
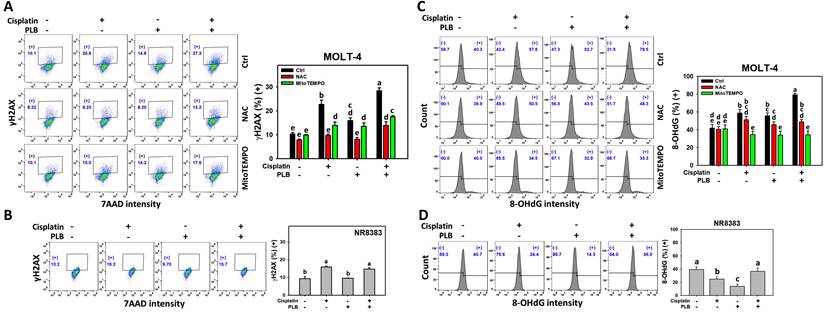
To further confirm the DNA damage status, NAC and MitoTEMPO pretreatments were conducted before cisplatin or/and PLB treatment on MOLT-4 cells. NAC and MitoTEMPO rescued the cisplatin or/and PLB-induced γH2AX and 8-OHdG increment (Figures 7A,7C). Consequently, the results suggest ROS and MitoSOX contribute to the enhanced γH2AX and 8-OHdG DNA damage of cisplatin/PLB on MOLT-4 cells, although pharmacological inhibitor studies alone do not establish direct causality.
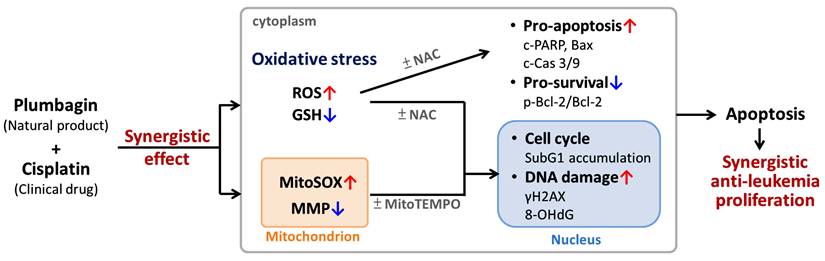
Overview of the effects and mechanisms of cisplatin/PLB synergistic anti-leukemia proliferation. As shown in Figures 1-7, all cisplatin/PLB-induced synergistic effects in Figure 8 were higher in leukemia cells than in normal macrophages (not shown in this figure). NAC and MitoTEMPO are inhibitors of cellular and mitochondrial oxidative stress. Mechanistically, the synergistic antiproliferative effect of cisplatin/PLB on MOLT-4 cells was attenuated by NAC and MitoTEMPO. The enhanced oxidative stresses were confirmed, because the induction of ROS/MitoSOX and the depletion of MMP/GSH of cisplatin/PLB were more highly regulated in leukemia cells than in normal macrophages. These oxidative stress responses were also attenuated by NAC and/or MitoTEMPO, suggesting a potential role for enhanced oxidative stress in the synergistic antiproliferative effect of cisplatin/PLB treatment in leukemia cells. Moreover, these enhanced oxidative stresses caused by cisplatin/PLB were validated as triggering apoptosis (subG1 and annexin V results) and intrinsic apoptotic signaling, as evidenced by NAC, as well as triggering DNA damage (γH2AX and 8-OHdG), as evidenced by NAC and MitoTEMPO. Notably, these cisplatin/PLB mechanisms exhibited higher performance in leukemia cells than in normal macrophages.
Discussion
In this investigation, we assessed the proliferation-regulating effects of the combined treatment of cisplatin and PLB in leukemia cells. Mechanisms underlying cell cycle, oxidative stress, apoptosis, and DNA damage induced by cisplatin/PLB were explored and compared with those induced by cisplatin or PLB alone.
Natural products have been applied to sensitize the chemotherapy responses for cancer cells [51, 52]. For example, ethyl acetate Nepenthes extract (EANV)/cisplatin combined treatment shows the synergistic antiproliferation of oral cancer cells [51]. Artocarpus heterophyllus-derived artocarpanone and cycloartocarpin combined with cisplatin synergistically suppress lung and breast cancer cell proliferation [52].
Several literature reports have explored the potential reasons for the cisplatin-sensitizing effects of natural products. Both cisplatin and several natural products exhibit ROS-modulating potential that may converge to promote this, potentially leading to the antiproliferation of cancer cells. For example, low-dose cisplatin upregulates oxidative stress-responsive enzyme activity (superoxide dismutase and catalase) and lipid peroxidation [53] of leukemia (HL-60) cells. Similarly, Lithospermum erythrorhizon-derived shikonin [54] enhance cisplatin-induced ROS generation in colon cancer cells. Therefore, the synergistic antiproliferation effects of combined treatment (cisplatin/natural products) are partly attributed to their enhanced generation of oxidative stress.
Similarly, the examples of cisplatin/PLB combined treatment show the synergistic antiproliferative effects of cisplatin/PLB on several types of cancer cells, such as cervical [25] and tongue [26]. However, cisplatin/PLB for leukemia cells has not yet been reported. The enhanced ROS generation of cisplatin/PLB has been reported in tongue cancer cells [26], improving the antiproliferative effect. Although PLB exhibits anti-leukemia effects [27, 55], its combined effects with cisplatin have been rarely investigated. In the present investigation, the antiproliferative effects of cisplatin/PLB in leukemia cells (MOLT-4 and J45.01) were validated synergistically (synergy index = 1.36 and 1.27) (Figure 1A). Because the cervical study [25] used a 48 h MTT assay and the tongue study [26] used a 24 h CCK-8 assay, their cisplatin/plumbagin results are not directly comparable with our leukemia data obtained by a 24 h ATP assay. However, these findings suggest that the cisplatin/PLB combination could be a promising anticancer therapy. PLB shows good drug safety in normal peripheral blood mononuclear cells [55] and animal studies [28]. Similarly, cisplatin/PLB exhibits non-cytotoxicity in normal cells (Figure 1A). Therefore, cisplatin/PLB offers anti-leukemia functions without significant side effects to normal cells. Given that the cervical [25], tongue [26], and our leukemia experiments were all in vitro, conclusions about therapeutic promise and minimal toxicity remain tentative; in vivo validation is necessary to advance the cisplatin/PLB combination toward clinical translation.
These synergistic antiproliferative effects of cisplatin/PLB in leukemia cell treatment were attenuated by NAC and MitoTEMPO (Figure 1B). This raises the possibility that cellular and mitochondrial oxidative stress play a vital role in the anti-leukemia effects. It warrants a deep assessment of cellular and mitochondrial oxidative stress in cisplatin/PLB. Notably, cisplatin/PLB induces greater cellular and mitochondrial oxidative stress via ROS and a MitoSOX burst than either cisplatin or PLB alone (Figure 2), as confirmed by their respective inhibitors. Consequently, the impacts of cellular and mitochondrial oxidative stress were confirmed in the present study. Notably, cisplatin primarily increases cellular ROS, whereas PLB induces both cellular ROS and mitochondrial MitoSOX in leukemia cells (Figure 2). NAC pretreatment reduces cisplatin-induced cytotoxicity in tongue cancer cells [56], implying that ROS contributes to cisplatin efficacy. Therefore, PLB-driven ROS—including cellular and mitochondrial ROS—may amplify cisplatin's activity and underlie the synergistic response observed in MOLT-4 cells. Moreover, the antioxidant Nrf2/HO-1 pathway is frequently upregulated in malignancies (such as cisplatin-resistant A549 lung cancer cells [57]) and contributes to apoptosis evasion. Therefore, assessing how the cisplatin/PLB combination affects Nrf2 and HO-1 expression in MOLT-4 cells should be investigated in future studies.
This enhanced oxidative stress of leukemia cells was also validated by downregulating MMP (Figure 3A), an oxidative stress indicator [44]. Since oxidative stress is counteracted by cellular antioxidants such as GSH [58], the downregulation of GSH appropriately causes oxidative stress generation. Consistently, cisplatin/PLB shows higher GSH (-) proportions in leukemia cells than cisplatin or PLB (Figure 3C), indicating that GSH depletion is highly induced by cisplatin/PLB. Hence, the enhanced GSH depletion may contribute to the enhanced oxidative stress exerted by cisplatin/PLB. Moreover, both cisplatin/PLB-induced MMP and GSH depletion in leukemia cells were attenuated by NAC and MitoTEMPO, suggesting that cisplatin/PLB enhances cellular and mitochondrial oxidative stress in leukemia cells.
Oxidative stress may stimulate apoptosis [59] and DNA damage [60] in cancer cells. These oxidative stress responses are prone to trigger high levels of apoptosis and DNA damage. Cisplatin upregulates oxidative stress, DNA damage [53], and apoptosis [61] in leukemia (HL-60) cells. Several examples of combined treatment for cisplatin and anticancer drugs are provided as follows. For instance, the nitrated [6,6,6] tricycle derivative (SK2)/cisplatin [62] induce high apoptosis and DNA damage due to increased oxidative stress in oral cancer cells. Shikonin synergistically induces oxidative stress to promote cisplatin-induced apoptosis in colon cancer cells [54]. Thymoquinone promotes cisplatin-triggered apoptosis and oxidative DNA damage (γH2AX and 8-OHdG) of osteosarcoma cells, accompanied by synergistically inducing ROS [62]. Similarly, cisplatin/PLB triggers more subG1 accumulation (Figure 4), annexin V-detected apoptosis (Figure 5), and DNA damage (γH2AX and 8-OHdG) (Figure 7) in leukemia cells than single treatments. Furthermore, both cisplatin/PLB-induced γH2AX and 8-OHdG expression in leukemia cells were attenuated by NAC and MitoTEMPO, suggesting that cisplatin/PLB enhances DNA damage in leukemia cells via oxidative stress.
Moreover, higher intrinsic caspase 9 and the apoptosis executioner caspase 3 were activated by cisplatin/PLB than by single treatments (Figure 6), accompanied by enhanced oxidative stress in leukemia cells. This caspase 9 activation in cisplatin/PLB was also supported by the upregulated Bax and downregulated Bcl-2 activation (p-Bcl-2 (Ser70, anti-apoptotic [43])/Bcl-2 ratio). As shown in Figure 6, cisplatin/PLB reduced Bcl-2 phosphorylation at Ser70, an anti-apoptotic marker, and this decrease was reversed by NAC, indicating that Ser70 dephosphorylation of p-Bcl-2 is driven by oxidative stress from the cisplatin/PLB combination. Because AKT (protein kinase B) upregulates Bcl-2 expression [63], AKT may be downregulated by cisplatin/PLB, causing apoptosis. Moreover, NAC attenuated the activation of caspase 3 and 9 in cisplatin/PLB. These results suggest cellular oxidative stress triggers intrinsic caspase signaling and apoptosis. Oxidative stress can inhibit AKT signaling in Chinese herb rhubarb-derived rhein-triggered apoptosis of pancreatic cancer cells [64]. Since oxidative stress was upregulated in cisplatin/PLB, it warrants a deep investigation of the role of AKT in the synergistic regulation of antiproliferation in cisplatin/PLB.
PLB has been investigated as a chemotherapy sensitizer. In addition to leukemia cells, in a phase I trial, PLB improved the efficacy of androgen deprivation therapy in prostate cancer [65]. PLB also enhances cisplatin sensitivity in gastric cancer cells [66] and potentiates tamoxifen-induced apoptosis in breast cancer cells [67]. Together, these findings support PLB as a broad chemosensitizer that can increase the effectiveness of diverse anticancer therapies.
There are some limitations in the current study. Although NAC and MitoTEMPO are widely used to implicate oxidative stress, they have pleiotropic effects beyond ROS and MitoSOX scavenging; therefore, complementary analyses of antioxidant substrates and enzymes are needed to avoid over-attributing causality to ROS and MitoSOX in NAC and MitoTEMPO rescue experiments. Because NR8383 rat lung macrophages were the only normal control, the observed selectivity may be model-specific; using human normal hematopoietic cells or peripheral blood mononuclear cells, as well as animal models, in follow-up experiments would substantially bolster translational relevance. Moreover, another limitation of the present study is that it investigated optimal conditions for combined PLB/cisplatin treatment but measured only cell viability at fixed doses rather than determining IC50 values. The Chou-Talalay method for drug combination synergy analysis relies on the median effect equation, in which the median effect dose Dm corresponds to the drug's IC50 derived from a dose-response curve. Because IC50 values were not available, the Chou-Talalay method could not be applied and was therefore not used in this study. Overall, this limitation restricts the study by assessing interactions at specific fixed doses, potentially overlooking effects that change with doses.
Conclusions
The cisplatin/PLB combined treatment has not been reported previously in leukemia cells. This study validates the synergistic and selective antiproliferative activity and mechanisms of cisplatin/PLB against leukemia cells in vitro, with limited cytotoxicity to the normal NR8383 macrophage model. Mechanistic experiments implicate combined cellular and mitochondrial oxidative stress driving apoptosis and DNA damage, and antioxidant probes (NAC and MitoTEMPO) modulate these effects. These results support the therapeutic potential of cisplatin/PLB but remain preliminary because they are based on in vitro assays and a single normal cell comparator.
Acknowledgements
Funding
This work was partly supported by funds from the National Science and Technology Council (NSTC 114-2320-B-037-015 and NSTC 114-2314-B-037-032), NPUST-KMU joint research project (NPUST-KMU-112-P008), Kaohsiung Medical University (KMU-TB114009-2), and the Kaohsiung Medical University Research Center (KMU-TC114A04 and KMU-TC114A203).
Data availability
Data will be made available on request.
Author contributions
Conceptualization, Y.-N.C., J.-W.L., B.-H.C. and H.-W.C; data curation, Y.-N.C.; formal analysis, Y.-N.C.; methodology, Y.-N.C. and C.-H.Y.; supervision, B.-H.C. and H.-W.C.; writing—original draft, Y.-N.C., J.-W.L., T.-M.C., and H.-W.C.; writing—review and editing, B.-H.C. and H.-W.C. All the authors have read and agreed to this version of the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Siegel RL, Kratzer TB, Giaquinto AN, Sung H, Jemal A. Cancer statistics, 2025. CA Cancer J Clin. 2025;75:10-45
2. Lowenberg B, Downing JR, Burnett A. Acute myeloid leukemia. N Engl J Med. 1999;341:1051-62
3. Pui C-H, Relling MV, Downing JR. Acute lymphoblastic leukemia. N Engl J Med. 2004;350:1535-48
4. Chiorazzi N, Rai KR, Ferrarini M. Chronic lymphocytic leukemia. N Engl J Med. 2005;352:804-15
5. Sawyers CL. Chronic myeloid leukemia. N Engl J Med. 1999;340:1330-40
6. Inaba H, Mullighan CG. Pediatric acute lymphoblastic leukemia. Haematologica. 2020;105:2524-39
7. Hunger SP, Mullighan CG. Acute lymphoblastic leukemia in children. N Engl J Med. 2015;373:1541-52
8. Bayat Mokhtari R, Homayouni TS, Baluch N, Morgatskaya E, Kumar S, Das B. et al. Combination therapy in combating cancer. Oncotarget. 2017;8:38022-43
9. Short NJ, Konopleva M, Kadia TM, Borthakur G, Ravandi F, DiNardo CD. et al. Advances in the treatment of acute myeloid leukemia: New drugs and new challenges. Cancer Discov. 2020;10:506-25
10. Karr M, Roeker L. A history of targeted therapy development and progress in novel-novel combinations for chronic lymphocytic leukemia (CLL). Cancers (Basel). 2023;15:1018
11. Crossnohere NL, Richardson DR, Reinhart C, O'Donoghue B, Love SM, Smith BD. et al. Side effects from acute myeloid leukemia treatment: results from a national survey. Curr Med Res Opin. 2019;35:1965-70
12. Ghosh S. Cisplatin: The first metal based anticancer drug. Bioorg Chem. 2019;88:102925
13. Seiter K, Katragadda S, Ponce D, Rasul M, Ahmed N. Temozolomide and cisplatin in relapsed/refractory acute leukemia. J Hematol Oncol. 2009;2:21
14. Florea AM, Busselberg D. Cisplatin as an anti-tumor drug: cellular mechanisms of activity, drug resistance and induced side effects. Cancers (Basel). 2011;3:1351-71
15. de Oliveira Junior RG, Christiane Adrielly AF, da Silva Almeida JRG, Grougnet R, Thiery V, Picot L. Sensitization of tumor cells to chemotherapy by natural products: A systematic review of preclinical data and molecular mechanisms. Fitoterapia. 2018;129:383-400
16. Rajendran G, Taylor JA 3rd, Woolbright BL. Natural products as a means of overcoming cisplatin chemoresistance in bladder cancer. Cancer Drug Resist. 2021;4:69-84
17. Atanasov AG, Zotchev SB, Dirsch VM, International Natural Product Sciences T, Supuran CT. Natural products in drug discovery: advances and opportunities. Nat Rev Drug Discov. 2021;20:200-16
18. Liang X, Wang P, Yang C, Huang F, Wu H, Shi H. et al. Galangin inhibits gastric cancer growth through enhancing STAT3 mediated ROS production. Front Pharmacol. 2021;12:646628
19. Shiau JP, Chuang YT, Yang KH, Chang FR, Sheu JH, Hou MF. et al. Brown algae-derived fucoidan exerts oxidative stress-dependent antiproliferation on oral cancer cells. Antioxidants (Basel). 2022;11:841
20. Sanusi SB, Bakar MFA, Mohamed M, Sabran SF, Mainasara MM. Ethnobotanical, phytochemical, and pharmacological properties of Nepenthes species: A review. Asian J Pharm Clin Res. 2017;10:16-9
21. Ismail NA, Kamariah AS, Lim LBL, Ahmad N. Phytochemical and pharmacological evaluation of methanolic extracts of the leaves of Nepenthes bicalcarata. Int J Pharma Phyto Res. 2015;7:1127-38
22. Thao NP, Luyen BT, Koo JE, Kim S, Koh YS, Thanh NV. et al. In vitro anti-inflammatory components isolated from the carnivorous plant Nepenthes mirabilis (Lour.) Rafarin. Pharm Biol. 2016;54:588-94
23. De U, Son JY, Jeon Y, Ha SY, Park YJ, Yoon S. et al. Plumbagin from a tropical pitcher plant (Nepenthes alata Blanco) induces apoptotic cell death via a p53-dependent pathway in MCF-7 human breast cancer cells. Food Chem Toxicol. 2019;123:492-500
24. Liu W, Lin LC, Wang PJ, Chen YN, Wang SC, Chuang YT. et al. Nepenthes ethyl acetate extract provides oxidative stress-dependent anti-leukemia effects. Antioxidants (Basel). 2021;10:1410
25. Sidhu H, Capalash N. Plumbagin downregulates UHRF1, p-Akt, MMP-2 and suppresses survival, growth and migration of cervical cancer CaSki cells. Toxicol In Vitro. 2023;86:105512
26. Xue D, Pan ST, Zhou X, Ye F, Zhou Q, Shi F. et al. Plumbagin enhances the anticancer efficacy of cisplatin by increasing intracellular ROS in human tongue squamous cell carcinoma. Oxid Med Cell Longev. 2020;2020:5649174
27. Sun J, McKallip RJ. Plumbagin treatment leads to apoptosis in human K562 leukemia cells through increased ROS and elevated TRAIL receptor expression. Leuk Res. 2011;35:1402-8
28. Sumsakul W, Plengsuriyakarn T, Chaijaroenkul W, Viyanant V, Karbwang J, Na-Bangchang K. Antimalarial activity of plumbagin in vitro and in animal models. BMC Complement Altern Med. 2014;14:15
29. Cai X, Yu Y, Huang Y, Zhang L, Jia PM, Zhao Q. et al. Arsenic trioxide-induced mitotic arrest and apoptosis in acute promyelocytic leukemia cells. Leukemia. 2003;17:1333-7
30. Feng Y, Hua X, Niu R, Du Y, Shi C, Zhou R. et al. ROS play an important role in ATPR inducing differentiation and inhibiting proliferation of leukemia cells by regulating the PTEN/PI3K/AKT signaling pathway. Biol Res. 2019;52:26
31. Kumar S, Tchounwou PB. Molecular mechanisms of cisplatin cytotoxicity in acute promyelocytic leukemia cells. Oncotarget. 2015;6:40734-46
32. Wang KC, Liu YC, El-Shazly M, Shih SP, Du YC, Hsu YM. et al. The antioxidant from ethanolic extract of Rosa cymosa fruits activates phosphatase and tensin homolog in vitro and in vivo: A new insight on its antileukemic effect. Int J Mol Sci. 2019;20:1935
33. Shih SP, Lu MC, El-Shazly M, Lin YH, Chen CL, Yu SS. et al. The antileukemic and anti-prostatic effect of aeroplysinin-1 is mediated through ROS-induced apoptosis via NOx activation and inhibition of HIF-1a activity. Life (Basel). 2022;12:687
34. Alabed HBR, Pellegrino RM, Buratta S, Lema Fernandez AG, La Starza R, Urbanelli L. et al. Metabolic profiling as an approach to differentiate t-cell acute lymphoblastic leukemia cell lines belonging to the same genetic subgroup. Int J Mol Sci. 2024;25:3921
35. Koretzky GA, Picus J, Schultz T, Weiss A. Tyrosine phosphatase CD45 is required for T-cell antigen receptor and CD2-mediated activation of a protein tyrosine kinase and interleukin 2 production. Proc Natl Acad Sci U S A. 1991;88:2037-41
36. Chen CY, Yen CY, Wang HR, Yang HP, Tang JY, Huang HW. et al. Tenuifolide B from Cinnamomum tenuifolium stem selectively inhibits proliferation of oral cancer cells via apoptosis, ROS generation, mitochondrial depolarization, and DNA damage. Toxins (Basel). 2016;8:319
37. Chuang YT, Shiau JP, Yen CY, Hou MF, Jeng JH, Tang JY. et al. Fucoidan/UVC combined treatment exerts preferential antiproliferation in oral cancer cells but not normal cells. Antioxidants (Basel). 2022;11:1797
38. Chang CC, Kuan CP, Lin JY, Lai JS, Ho TF. Tanshinone IIA facilitates TRAIL sensitization by up-regulating DR5 through the ROS-JNK-CHOP signaling axis in human ovarian carcinoma cell lines. Chem Res Toxicol. 2015;28:1574-83
39. Wang HR, Chen PH, Tang JY, Yen CY, Su YC, Huang MY. et al. Manoalide shows mutual interaction between cellular and mitochondrial reactive species with apoptosis in oral cancer cells. Oxid Med Cell Longev. 2021;2021:6667355
40. Wang TS, Lin CP, Chen YP, Chao MR, Li CC, Liu KL. CYP450-mediated mitochondrial ROS production involved in arecoline N-oxide-induced oxidative damage in liver cell lines. Environ Toxicol. 2018;33:1029-38
41. Chen YN, Chan CK, Yen CY, Shiau JP, Chang MY, Wang CC. et al. Antioral cancer effects by the nitrated [6,6,6]tricycles compound (SK1) in vitro. Antioxidants (Basel). 2022;11:2072
42. Shahali A, Ghanadian M, Jafari SM, Aghaei M. Mitochondrial and caspase pathways are involved in the induction of apoptosis by nardosinen in MCF-7 breast cancer cell line. Res Pharm Sci. 2018;13:12-21
43. Ke D, Yu Y, Li C, Han J, Xu J. Phosphorylation of BCL2 at the Ser70 site mediates RANKL-induced osteoclast precursor autophagy and osteoclastogenesis. Mol Med. 2022;28:22
44. Satoh T, Enokido Y, Aoshima H, Uchiyama Y, Hatanaka H. Changes in mitochondrial membrane potential during oxidative stress-induced apoptosis in PC12 cells. J Neurosci Res. 1997;50:413-20
45. Zitka O, Skalickova S, Gumulec J, Masarik M, Adam V, Hubalek J. et al. Redox status expressed as GSH:GSSG ratio as a marker for oxidative stress in paediatric tumour patients. Oncol Lett. 2012;4:1247-53
46. Burhans WC, Heintz NH. The cell cycle is a redox cycle: linking phase-specific targets to cell fate. Free Radic Biol Med. 2009;47:1282-93
47. Kirova DG, Judasova K, Vorhauser J, Zerjatke T, Leung JK, Glauche I. et al. A ROS-dependent mechanism promotes CDK2 phosphorylation to drive progression through S phase. Dev Cell. 2022;57:1712-27 e9
48. Boulares AH, Yakovlev AG, Ivanova V, Stoica BA, Wang G, Iyer S. et al. Role of poly(ADP-ribose) polymerase (PARP) cleavage in apoptosis. Caspase 3-resistant PARP mutant increases rates of apoptosis in transfected cells. J Biol Chem. 1999;274:22932-40
49. Russo A, Cardile V, Graziano ACE, Avola R, Bruno M, Rigano D. Involvement of Bax and Bcl-2 in induction of apoptosis by essential oils of three lebanese salvia species in human prostate cancer cells. Int J Mol Sci. 2018;19:292
50. Dai Y, Jin S, Li X, Wang D. The involvement of Bcl-2 family proteins in AKT-regulated cell survival in cisplatin resistant epithelial ovarian cancer. Oncotarget. 2017;8:1354-68
51. Tang JY, Li LJ, Ou-Yang F, Wang CL, Shu CW, Wu KH. et al. Ethyl acetate extract of Nepenthes ventricosa x maxima exerts preferential killing to oral cancer cells. DNA Cell Biol. 2019;38:763-72
52. Daud NNNNM, Septama AW, Simbak N, Bakar NHA, Rahmi EP. Synergistic effect of flavonoids from Artocarpus heterophyllus heartwoods on anticancer activity of cisplatin against H460 and MCF-7 cell lines. Nat Prod Sci. 2019;25:311-6
53. Dasari SR, Velma V, Yedjou CG, Tchounwou PB. Preclinical assessment of low doses of cisplatin in the management of acute promyelocytic leukemia. Int J Cancer Res Mol Mech. 2015;1:10.16966 /2381-3318.113
54. He G, He G, Zhou R, Pi Z, Zhu T, Jiang L. et al. Enhancement of cisplatin-induced colon cancer cells apoptosis by shikonin, a natural inducer of ROS in vitro and in vivo. Biochem Biophys Res Commun. 2016;469:1075-82
55. Bae KJ, Lee Y, Kim SA, Kim J. Plumbagin exerts an immunosuppressive effect on human T-cell acute lymphoblastic leukemia MOLT-4 cells. Biochem Biophys Res Commun. 2016;473:272-7
56. Xue DF, Pan ST, Huang G, Qiu JX. ROS enhances the cytotoxicity of cisplatin by inducing apoptosis and autophagy in tongue squamous cell carcinoma cells. Int J Biochem Cell Biol. 2020;122:105732
57. Silva MM, Rocha CRR, Kinker GS, Pelegrini AL, Menck CFM. The balance between NRF2/GSH antioxidant mediated pathway and DNA repair modulates cisplatin resistance in lung cancer cells. Sci Rep. 2019;9:17639
58. Kwon DH, Cha HJ, Lee H, Hong SH, Park C, Park SH. et al. Protective effect of glutathione against oxidative stress-induced cytotoxicity in RAW 264.7 macrophages through activating the nuclear factor erythroid 2-related factor-2/heme oxygenase-1 pathway. Antioxidants (Basel). 2019;8:82
59. Huang CH, Huang ZW, Ho FM, Chan WH. Berberine impairs embryonic development in vitro and in vivo through oxidative stress-mediated apoptotic processes. Environ Toxicol. 2018;33:280-94
60. Wu CF, Lee MG, El-Shazly M, Lai KH, Ke SC, Su CW. et al. Isoaaptamine induces T-47D cells apoptosis and autophagy via oxidative stress. Mar Drugs. 2018;16:18
61. Velma V, Dasari SR, Tchounwou PB. Low doses of cisplatin induce gene alterations, cell cycle arrest, and apoptosis in human promyelocytic leukemia cells. Biomark Insights. 2016;11:113-21
62. Ahmadzadeh H, Ahmadi M, Golchin A, Malakoti F, Maleki M, Alemi F. et al. Investigating the role of thymoquinone in increasing the rate of cisplatin-induced apoptosis through oxidative DNA damage in Saso-2 cancer cells. Drug Res. 2022;72:171-6
63. Pugazhenthi S, Nesterova A, Sable C, Heidenreich KA, Boxer LM, Heasley LE. et al. Akt/protein kinase B up-regulates Bcl-2 expression through cAMP-response element-binding protein. J Biol Chem. 2000;275:10761-6
64. Liu Y, Shi C, He Z, Zhu F, Wang M, He R. et al. Inhibition of PI3K/AKT signaling via ROS regulation is involved in Rhein-induced apoptosis and enhancement of oxaliplatin sensitivity in pancreatic cancer cells. Int J Biol Sci. 2021;17:589-602
65. Chrastina A, Baron VT, Abedinpour P, Rondeau G, Welsh J, Borgstrom P. Plumbagin-loaded nanoemulsion drug delivery formulation and evaluation of antiproliferative effect on prostate cancer cells. Biomed Res Int. 2018;2018:9035452
66. Li J, Shen L, Lu FR, Qin Y, Chen R, Li J. et al. Plumbagin inhibits cell growth and potentiates apoptosis in human gastric cancer cells in vitro through the NF-kappaB signaling pathway. Acta Pharmacol Sin. 2012;33:242-9
67. Kawiak A, Domachowska A, Jaworska A, Lojkowska E. Plumbagin sensitizes breast cancer cells to tamoxifen-induced cell death through GRP78 inhibition and Bik upregulation. Sci Rep. 2017;7:43781
Author contact
![]() Corresponding authors: bhchenedu.tw (B.-H.C.); changhwedu.tw (H.-W.C.); Tel.: +886-7-312-1101 (ext. 2676) (B.-H.C.); +886-7-312-1101 (ext. 2691) (H.-W.C.).
Corresponding authors: bhchenedu.tw (B.-H.C.); changhwedu.tw (H.-W.C.); Tel.: +886-7-312-1101 (ext. 2676) (B.-H.C.); +886-7-312-1101 (ext. 2691) (H.-W.C.).