Impact Factor ISSN: 1449-1907
- Issue 9; 2026
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Volume 23; 2026
- Past Issues
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Clinical management
The differences in cellular and...
Potential treatment targets for...
Conclusion and perspectives
Abbreviations
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2025; 22(16):4451-4468. doi:10.7150/ijms.124332 This issue Cite
Review
New Exploration of Therapeutic Targets for Radiation Pneumonitis: Comparative Analysis of Molecular Pathways in Radiation-Induced and LPS-Induced Pneumonitis
Siyi Niu1*, Zhen Li2*, Nan Wang1, Guoliang Xue1, Huiwen Xue1, Qian Hong1, Wei Xu3 ![]() , Zhigang Wei1,4
, Zhigang Wei1,4 ![]() , Xin Ye1
, Xin Ye1 ![]() , Qi Xie1,5
, Qi Xie1,5 ![]()
1. Department of Oncology, Lung Cancer Center, The First Affiliated Hospital of Shandong First Medical University & Shandong Provincial Qianfoshan Hospital, Shandong Lung Cancer Institute, Jinan, China, 250014.
2. School of Preventive Medicine Science, Shandong First Medical University & Shandong Academy of Medical Sciences, Jinan, China, 250117
3. Department of Clinical Pharmacy, The First Affiliated Hospital of Shandong First Medical University & Shandong Provincial Qianfoshan Hospital, Jinan, China, 250014.
4. Cheeloo College of Medicine, Shandong University, Jinan, China, 250033.
5. Shandong Provincial Lab for Clinical Immunology Translational Medicine in Universities, Jinan, China, 250014.
* They equally contributed to this article.
Received 2025-8-27; Accepted 2025-10-2; Published 2025-10-24
Abstract

Radiation pneumonitis (RP) is a common complication of radiotherapy that significantly limits the tolerable radiation dose, thereby compromising treatment outcomes. In severe cases, it can become life-threatening. Currently, the management of RP relies primarily on glucocorticoids. However, this approach is associated with several drawbacks, including markedly increased risks of immunosuppression and infection, metabolic disturbances, musculoskeletal injury, gastrointestinal adverse effects, and most importantly- a potential compromise of antitumor efficacy. Moreover, current treatments for RP and radiation induced pulmonary fibrosis remain unsatisfactory, underscoring the need for mechanistic studies and the exploration of novel therapeutic targets and strategies. RP and lipopolysaccharide (LPS) -induced pneumonitis, originating from Gram-negative bacteria, represent two distinct forms of pulmonary inflammation. We compare their molecular mechanisms—with both shared pathways and key distinctions despite clinical similarities—and explore diverse therapies, including anti-inflammatory responses, antioxidant defenses, gut microbiota regulation, cell death modulation, mitophagy enhancement, among others. In particular, we highlight therapies and targets that have shown efficacy in LPS-induced pneumonitis but have not yet been investigated in the context of RP. These insights may offer valuable guidance for both clinical management and fundamental research on RP.
Keywords: radiation pneumonitis (RP), radiation induced lung injury (RILI), lipopolysaccharide (LPS)-induced pneumonitis, therapeutic targets, molecular pathways
Introduction
Radiation pneumonitis (RP) is a common side effect of radiotherapy, frequently arising after the irradiation of malignant tumors in the chest and breast, particularly in the treatment of lung cancer. Radiation exposure inflicts damage on cells in lung, resulting in DNA damage and an excessive accumulation of reactive oxygen species (ROS) [1]. This cascade of events subsequently triggers an inflammatory response, ultimately causing damage to alveolar epithelial cells and impairment of adjacent endothelial tissue [2]. The diagnosis of RP typically relies on the patient's treatment history and the manifestation of a spectrum of clinical symptoms [3]. Common symptoms often include dyspnea (shortness of breath), a dry cough devoid of sputum, low-grade fever, chest pain, and overall feelings of malaise [4]. RP predominantly manifests within the irradiated region and seldom affects distant lung tissues [1]. This condition significantly constrains the permissible radiation dose, thereby impacting treatment efficacy, and in severe instances, can pose a life-threatening risk to the patient [5].
Hospital-acquired pneumonia is predominantly caused by Gram-negative bacilli, with common culprits including Klebsiella pneumoniae, Escherichia coli, Proteus, Haemophilus influenzae, Pseudomonas aeruginosa, etc. [6]. Lipopolysaccharide (LPS), a major constituent of the Gram-negative bacterial cell wall, plays a pivotal role in mediating the pathological progression of pneumonitis and is classified as an endotoxin. The structure of LPS comprises hydrophilic polysaccharide chains referred to as O-antigens, an oligosaccharide core, and lipid A, which is highly toxic [7]. Lipid A, serving as the biologically active core and primary toxic component of LPS, acts as a potent trigger for innate immune responses. LPS has the potential to trigger a spectrum of acute pulmonary conditions, such as pneumonitis and acute lung injury (ALI), and it may also contribute to the development of various chronic diseases [8, 9]. ALI is marked by the aggregation of white blood cells, epithelial cell damage, pulmonary edema, heightened alveolar permeability, and extensive diffuse alveolar damage [10].
Currently, the management of RP relies primarily on glucocorticoids. However, this approach is associated with several drawbacks, including markedly increased risks of immunosuppression and infection, metabolic disturbances, musculoskeletal injury, gastrointestinal adverse effects, and most importantly- a potential compromise of antitumor efficacy. Identifying new therapeutic targets is particularly crucial. This review examines the similarities and differences in the pathogenic mechanisms between RP and LPS-induced pneumonitis, summarizing potential therapeutic strategies and shared molecular targets for both conditions. The goal is to identify new effective drugs for RP.
Clinical management
Clinical management for RP
There exists an incubation period for the clinical manifestations of RP, typically spanning from several weeks to months following radiation exposure. Although overt symptoms of pneumonitis may not be evident at the conclusion of radiotherapy, subtle pathological alterations can be identified within the lung tissue. Upon confirmation of a RP diagnosis, the most immediate and critical intervention is the cessation of radiotherapy. To ensure standardized treatment protocols, patients are evaluated and categorized based on the severity of their pneumonitis. The Radiation Therapy Oncology Group (RTOG) and the Common Terminology Criteria for Adverse Events (CTCAE) are two widely adopted grading systems utilized in clinical practice to assess radiation toxicity [11].
Glucocorticoids serve as the primary treatment for RP, renowned for their robust anti-inflammatory and antifibrotic effects. For patients exhibiting mild symptoms, it is advisable to contemplate initiating treatment with non-steroidal anti-inflammatory drugs (NSAIDs) or inhaled corticosteroids as a viable therapeutic option [4]. For patients who exhibit intolerance to steroids, other immunosuppressive agents such as azathioprine and cyclosporine may be considered as potential alternatives. However, such applications have been documented only in case reports, and there currently exists a dearth of robust clinical trial data to substantiate their efficacy in the management of RP [12].
Theoretically, RP is considered a sterile inflammatory condition; however, patients are at heightened risk of secondary lung infections due to the immunosuppressive effects that may arise from glucocorticoids therapy. Upon the earliest indication of infection, empirical anti-infective therapy should be initiated without delay. Additionally, the antibiotic regimen should be promptly adjusted based on the results of sputum culture and drug sensitivity testing [13]. Particular vigilance is warranted for potential infections with Pneumocystis and other pulmonary fungal pathogens [14]. Furthermore, several medications have demonstrated the efficacy in inhibiting fibrosis, thereby decelerating the progression of RP [15]. Moreover, the treatment plan should be meticulously tailored to accommodate the diverse clinical presentations of each patient. The prevalent symptoms of RP encompass dyspnea [16], which can range from mild to severe, and a persistent dry cough devoid of sputum. Symptomatic interventions, such as cough suppression, expectorant therapy, and oxygen administration, may be employed as needed to alleviate these symptoms.
Clinical management for LPS-induced pneumonitis
LPS constitutes a crucial structural element of the cell wall in Gram-negative bacteria, functioning as a potent endotoxin and playing a pivotal role in the pathophysiological mechanisms underlying bacterial infections. The prevalent gram-negative bacteria responsible for triggering pneumonitis primarily encompass Haemophilus influenzae, Klebsiella pneumoniae, Escherichia coli, Pseudomonas aeruginosa, and Acinetobacter baumannii, among others [6]. The current primary treatment strategies are centered around anti-inflammatory and anti-infective interventions. Among these, anti-infective therapy is the core, necessitating the selection of appropriate antibiotics based on the specific pathogen and its antibiotic sensitivity characteristics [17, 18]. Glucocorticoids are the preferred anti-inflammatory agents, owing to their demonstrated efficacy in mitigating lung inflammation and fibrosis [19]. Beyond these primary treatments, supportive measures like oxygen therapy will be provided. Should it be required, immunomodulators can be administered to adjust the immune system, effectively diminishing the exaggerated inflammatory response. Moreover, antioxidants may be employed to mitigate oxidative stress and preserve the structural integrity of lung tissue [20]. In addition, embracing prophylactic vaccination serves as a foresighted preventive strategy [21-23]. Endotoxin is a key instigator of cytokine storms in sepsis. Recent studies have elucidated that polymyxin B binds to endotoxin and effectively inactivates it [24]. For those battling severe acute respiratory distress syndrome (ARDS) alongside multiple organ dysfunction syndrome (MODS), hemoperfusion with polymyxin B-embedded fibers could potentially yield considerable therapeutic benefits [25]. Nevertheless, the efficacy of this therapeutic approach necessitates further validation through a substantial number of trials to definitively elucidate its clinical utility.
Similarities and distinctions
Both RP and LPS-induced pneumonitis typically necessitate anti-inflammatory treatment with glucocorticoids. LPS-induced pneumonitis generally requires anti-infective therapy, whereas RP may also warrant antibiotic treatment if complicated by an infection. Additionally, antioxidant therapy can be employed as needed to mitigate oxidative stress [26]. RP may further benefit from antifibrotic therapies designed to counteract and diminish the fibrotic processes impacting lung tissue. In the management of RP, anti-fibrotic agents represent a pivotal therapeutic strategy for counteracting progressive pulmonary damage (Table 1). These pharmacologic interventions specifically target radiation-induced fibrogenesis. By inhibiting molecular pathways driving fibroblast activation and collagen dysregulation, anti-fibrotic therapies such as pirfenidone and nintedanib demonstrate clinical potential to attenuate radiation-triggered tissue stiffening while preserving alveolar-capillary gas exchange capacity [27, 28]. Current clinical guidelines increasingly highlight the integration of antifibrotic drugs into multimodal therapies, acknowledging their role in preventing the insidious progression from inflammatory pneumonitis to irreversible pulmonary fibrosis. In conclusion, while LPS-induced pneumonitis treatment primarily emphasizes bacterial eradication via antimicrobial therapy, RP management focuses on inflammatory suppression, symptom alleviation, and prevention of progressive lung damage.
Clinical management for RP and LPS-induced pneumonitis.
| Category | Radiation Pneumonitis | LPS-induced pneumonitis |
|---|---|---|
| Diagnostic Criteria | - Mandatory: History of thoracic radiotherapy - Imaging: Ground-glass opacities confined to radiation field (CT) - Clinical presentation | - Mandatory: Bacteriological evidence (e.g.,Bacterial culture/smear) - Imaging: Diffuse alveolar damage (HRCT) |
| Conventional Treatment | - High-dose Corticosteroids - Supportive oxygen therapy - Immunosuppressants: Azathioprine, mycophenolate - Antifibrotic agents: Pirfenidone/Nintedanib (for chronic progression) | - Combination or monotherapy of pathogen-sensitive antibiotics - Low-dose corticosteroids - Supportive oxygen therapy - Conditional use of antifibrotic agents |
| Immunity Modulation | - Immunosuppressed patients: Avoid prolonged steroids; prioritize antifibrotics | - Neutropenic patients: Augment with granulocyte transfusions |
| Preventive Strategies | - Reducing radiation-induced toxicity: IMRT and VMAT - Prophylaxis: Amifostine (limited evidence) | - LPS exposure control: Environmental decontamination - Prophylaxis: Probiotics |
| Emerging Therapies | - Stem cell therapy: Mesenchymal stromal cells (MSCs) for fibrosis reversal (limited evidence) | - Anti-LPS vaccines - Microbiome modulation: Fecal microbiota transplantation |
The clinical management of RP also presents emerging challenges. The use of glucocorticoids, while beneficial, may precipitate a spectrum of side effects, including hyperglycemia, hypertension, osteoporosis, and weight gain [19]. Notably, glucocorticoids may compromise immune system homeostasis, consequently attenuating immunotherapy efficacy in tumor treatment. RP, when faced with the daunting challenge of extensive pulmonary fibrosis, encounters a paucity of effective treatments [29]. Although glucocorticoid interventions are employed, they fail to reverse fibrotic changes and merely decelerate disease progression. In conclusion, current treatments for RP and pulmonary fibrosis present limitations, necessitating further mechanistic elucidation and exploration of novel therapeutic targets and strategies.
The differences in cellular and molecular mechanism between RP and LPS-induced pneumonitis
Cellular and molecular mechanisms for RP
In the pathogenesis of RP, ionizing radiation induces lung tissue damage via two primary mechanisms: direct DNA injury and the indirect production of ROS [30] (Figure 1). ROS can exacerbate damage to the DNA, proteins, and lipid membranes of target cells. Subsequently, the activation of two distinct mechanisms triggers intracellular signaling, leading to the secretion of a diverse array of molecules and cytokines. This cascade ultimately promotes both inflammatory and immune responses. Ionizing radiation can directly impair deoxyribonucleic acid (DNA), such as by inducing base deletions, DNA single-strand breaks (SSBs), and DNA double-strand breaks (DSBs) [31]. According to the current study, the main target cells of radiation-induced lung injury (RILI) are vascular endothelial cells and alveolar epithelial cells. The primary constituent of alveolar epithelial cells, Type I alveolar epithelial cells, do not possess the capacity for proliferation and hence exhibit relative radio-resistance [12]. Nevertheless, they can still undergo necrosis or apoptosis upon exposure to certain doses of radiation [32]. Radiation impairs both type II alveolar epithelial cells and vascular endothelial cells. Type II alveolar epithelial cells serve as crucial precursor cells to type I alveolar cells, and upon radiation exposure, they can initiate the excessive proliferation of fibroblasts [12]. Furthermore, abnormal proliferation diminishes the secretion of alveolar surfactant substances, resulting in a reduction of alveolar surface tension. This decrement subsequently induces lung tissue edema and pulmonary atelectasis. Conversely, vascular endothelial cells exhibit heightened vascular permeability and inflammatory exudation [33].
The mechanism of RP. Ionizing radiation induces DNA damage through direct and indirect pathways, with the direct pathway causing DNA damage (e.g., SSB and DSB) that leads to vascular endothelial cell injury and alveolar epithelial cell injury, apoptosis, increased cytokine secretion, and EMT/ EndMT, ultimately resulting in increased vascular permeability, pulmonary edema, pulmonary fibrosis, epithelial barrier damage, and alveolar dysfunction, while the indirect pathway involves ionization of water molecules in irradiated cells to generate reactive oxygen species (ROS) that damage DNA, activate the NF-κB pathway to promote cytokine secretion and participate in the development of RP, and disrupt junction proteins to further increase vascular permeability, with these radiation-induced changes collectively driving the pathogenesis of RP and RIPF.
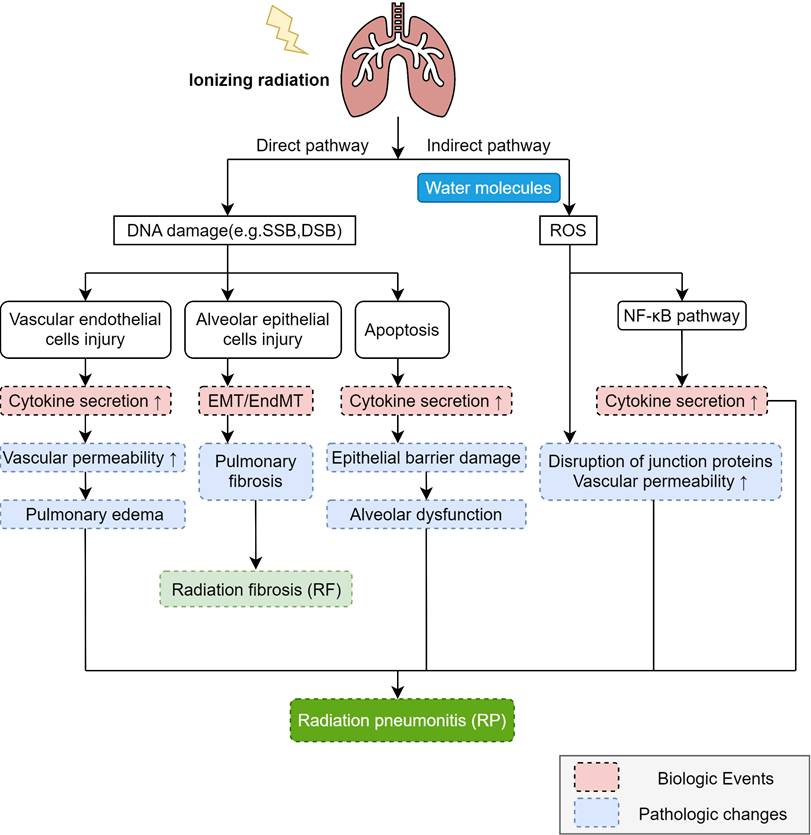
Epithelial-mesenchymal transition (EMT) is pivotal in the pathogenesis of RILI. This process entails the gradual loss of epithelial traits and the acquisition of mesenchymal fibroblast-like characteristics by epithelial cells. Endothelial cells, as specialized epithelioid cells, undergo endothelial-mesenchymal transition (EndMT), triggering collagen deposition with subsequent declines in lung compliance and diffusion capacity. Radiation induces EMT and/or EndMT, culminating in pulmonary fibrosis [33]. These cells may undergo post-radiation mitosis [34]. At this stage, if the damaged DNA is not fully repaired or if the DNA damage is excessively severe, it may result in apoptosis. This ultimately compromises the alveolar barrier function and initiates an inflammatory response. Damaged cells release inflammatory cytokines that recruit inflammatory cells into the alveoli and interstitium, thereby precipitating pneumonitis [11]. The severity of acute-phase RP appears dose-dependent, with higher doses correlating with more severe lung injury [35].
Ionizing radiation generates ROS through water ionization, creating an indirect pathway for DNA damage (Figure 1). ROS damage not only nuclear DNA but also mitochondrial DNA (mtDNA) [1]. Research indicates that ROS can activate the nuclear factor κB (NF-κB) signaling pathway- a key transcription factor regulating immune and inflammatory responses. NF-κB activation and nuclear translocation stimulate inflammation, triggering chemokine and cytokine production. ROS also disrupt intercellular junction proteins (e.g., vascular endothelial cadherin, VE-cadherin) in vascular endothelial cells, impairing the endothelial barrier. This increased vascular permeability promotes massive leukocyte migration across the endothelium and protein leakage into the alveolar lumen, ultimately causing pulmonary oedema.
Damage-associated Molecular Patterns (DAMPs) are a class of molecules that are released into the extracellular environment during cell damage or death [36]. Under homeostatic conditions, DAMPs reside intracellularly. Cellular disruption from physical, chemical, or biological trauma triggers their release, enabling recognition by immune pattern recognition receptors (PRRs). In the immediate aftermath of radiotherapy, DAMPs mediate the chemotaxis of neutrophils and macrophages from circulation to sites of lung tissue damage [11]. These activated cells subsequently secrete pro-inflammatory mediators including IL-6, TNF-α, and TGF-β. These cytokines exhibit dual functionality: while inducing inflammatory responses that may lead to pulmonary tissue injury, they simultaneously facilitate tissue remodeling processes [37].
Cellular and molecular mechanism for LPS-induced pneumonitis
The LPS molecule comprises an O-specific side chain, a core region, and a lipid A component, predominantly located in the outer membrane of Gram-negative bacteria [38]. LPS-induced pneumonitis mainly acts on lung epithelial cells, neutrophils, macrophages and endothelial cells through Toll-like receptor signaling pathway, cytokines and chemokines.
LPS engages with the LPS binding protein (LBP) situated external to the cell membrane, thereby forming a complex known as LPS-LBP [39]. CD14 is a protein present on the cell membrane that recognizes and binds LPS [40]. CD14 does not bind to Toll-like receptor (TLR) 4 directly, but rather delivers LPS to the complex formed by TLR4 and MD2. MD2 is an accessory protein that binds LPS and helps TLR4 recognize LPS [41]. When LPS binds to MD2, it will lead to a conformational change of MD2, which in turn activates the intracellular signaling domain of TLR4 and initiates a series of signal transduction (Figure 2). In the TLR4/MD2 complex, MD2 serves as a bridge, connecting LPS to the host's immune response. Without MD2, TLR4 is unable to effectively recognize LPS, making MD2 an important component of the innate immune system's defense against Gram-negative bacterial infections [42]. CD11b is an integrin that plays an auxiliary role in the binding of LPS to TLR4. It is able to interact with the TLR4-MD2 complex and enhance TLR4 sensitivity to LPS.
The mechanism of LPS-induced pneumonitis. In myeloid cells, membrane-bound CD14 (mCD14) is expressed, whereas in non-myeloid cells, CD14 exists as the soluble plasma form (sCD14). LPS forms a complex with lipopolysaccharide-binding protein (LBP) and interacts with CD14, which facilitates the delivery of LPS to TLR4. The LPS-TLR4-MD2 complex activates MyD88-dependent signaling, driving the production of pro-inflammatory cytokines via the NF-κB pathway. Simultaneously, it engages the TRIF-dependent pathway to internalize the complex, thereby inducing IFN-β generation. During LPS-induced lung injury, TLR5 may also participate. TLR5 is expressed in certain non-myeloid cells (e.g., epithelial and endothelial cells) and myeloid cells (e.g., alveolar macrophages and neutrophils). The presence of TLR5 promotes a bias toward the MyD88-dependent pathway in TLR4 signaling.
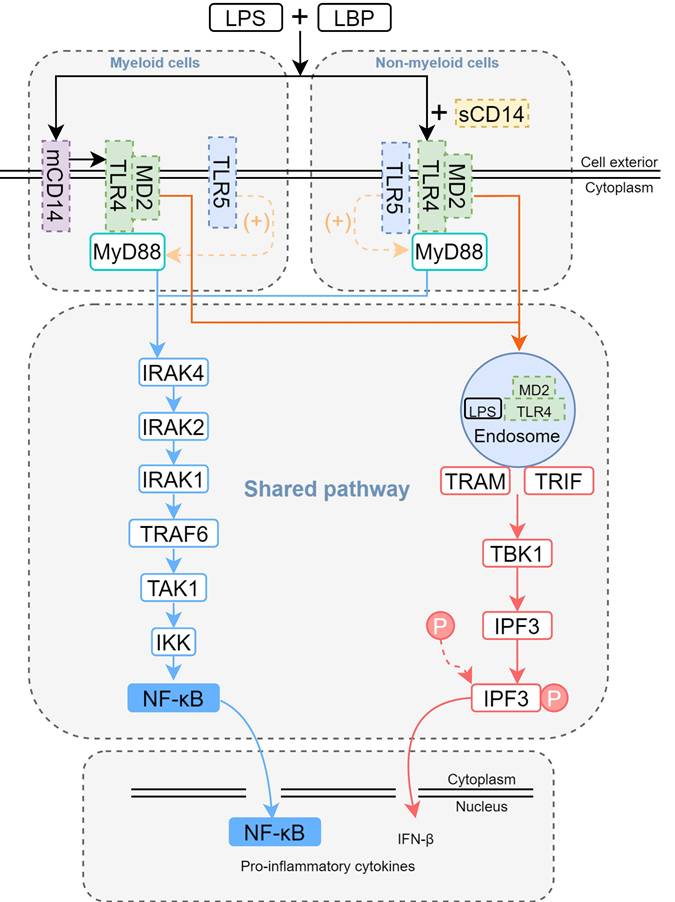
Upon binding to LPS, TLR4 activates both the myeloid differentiation primary response 88 (MyD88)-and toll/interleukin-1 receptor (TIR)-domain-containing adapter inducing interferon-β(TRIF)-dependent signaling cascades [43]. MyD88-dependent signaling rapidly initiates downstream activation of the mitogen-activated protein kinase (MAPK) family and NF-κB, driving the upregulation of pro-inflammatory cytokines such as TNF-α, IL-1β, and IL-6 (Figure 2). Recent studies demonstrate a key role for TLR5 in LPS-mediated lung injury. Specifically, mechanistic findings reveal that TLR5 promotes assembly of the MyD88 complex, which amplifies the TLR4/MyD88-dependent signaling cascade [44] (Figure 2). The functional interaction was evidenced by significantly reduced production of pro-inflammatory cytokines (30-50% decrease) in TLR5-deficient mouse models. Notably, TLR5 imparts signaling bias within the TLR4 complex, preferentially engaging MyD88-dependent over TRIF-mediated pathways for downstream signal transduction. Subsequently, the TLR4/MD-2/LPS complex is internalized via endocytosis, thereby activating the TRIF-dependent signaling pathway. Within intracellular compartments, TLR4 recruits TRIF through the adaptor protein TRAM. TRIF subsequently interacts with TRAF3 while simultaneously recruiting TBK1 and IKKε, initiating the phosphorylation of IRF3. Phosphorylated IRF3 then translocates to the nucleus, thereby promoting the transcription of IFN-β. Additionally, TRIF engages TRAF6 to activate TAK1, thereby triggering a delayed NF-κB-mediated inflammatory response (Figure 2).
Distinctions and similarities on cellular and molecular mechanism
Inflammatory response
Both LPS-induced pneumonitis and RP activate key inflammatory signaling pathways - notably NF-κB and p38 MAPK - which drive the recruitment and activation of inflammatory cells (primarily neutrophils and macrophages) in pulmonary tissue. These cells coordinate inflammatory responses by phagocytosing pathogens, secreting mediators, and generating ROS. Upon activation, these inflammatory cells secrete key mediators - including TNF-α, IL-1β, and IL-6 -which both amplify inflammation and exert chemotactic effects, recruiting more immune cells to pulmonary sites. This mediator release elevates vascular permeability, facilitating protein extravasation and cellular infiltration into lung tissue, ultimately triggering pulmonary edema. Mounting evidence highlights the pivotal role of NLRP3 inflammasome activation in the pathogenesis of both LPS-induced pneumonitis and RP. As a canonical PRR, NLRP3 constitutes the structural and functional core of the inflammasome complex [45]. It is responsible for sensing intracellular and extracellular danger signals and initiates immune defense by activating inflammatory responses, which plays a dual role in defense against infection and disease development. NLRP3 inflammasome activation requires both a priming signal and an activating signal. In both pneumonitis models, NF-κB upregulation provides the priming signal, while ROS serve as the activating signal. These dual stimuli trigger assembly of the NLRP3-ASC-caspase-1 complex, leading to caspase-1 activation. Active caspase-1 processes and releases IL-1β/IL-18, which recruit neutrophils -culminating in alveolar damage and edema. NLRP3 activation dynamics differ between the two pneumonitis models. LPS rapidly induces NLRP3 expression via NF-κB activation through the TLR4-MyD88/TRIF pathway [46, 47]. In contrast, RP indirectly activates NF-κB through ROS [48]. Furthermore, ionizing radiation triggers mitochondrial damage, releasing mtROS and mtDNA that directly activate NLRP3 (Table 2).
Similarities and distinctions in cellular and molecular mechanisms between RP and LPS-induced pneumonitis.
| Category | Features | Radiation Pneumonitis | LPS-induced Pneumonitis | References |
|---|---|---|---|---|
| Inflammatory Response | Inflammasome | NLRP3 inflammasome | [48, 128] | |
| Key cytokines/chemokines | TNF-α, TGF-β, IL-1β, IL-6, IL-8, IL-10, PDGF | TNF-α, IL-1β, IL-6, IL-8, IL-18 | [129, 130] | |
| Oxidative Stress | Antioxidant defense | Nrf2 activation | [52, 131] | |
| Major ROS sources | Direct ionizing radiation-induced water radiolysis; mitochondrial electron transport chain disruption | TLR4/NF-κB-driven NOX activation | [20, 50] | |
| Triggers | Mechanistic cascade | Radiation → DNA damage/DAMPs → Sterile inflammation | LPS →TLR4 → Anti-pathogen immunity | [132, 133] |
| Signaling Pathways | Dominant pathways | NF-κB signaling pathway MAPK signaling pathway | [134, 135] | |
| ATM-Chk2-p53 signaling pathway TGF-β/Smad signaling pathway | USP7/MAPK14 axis Rho A/ROCK1/2 signaling pathway | [74, 136-138] | ||
| Death of cells | Primary modes | Pyroptosis, poptosis, ferroptosis, NETosis | [86, 88] | |
| Senescence, autophagy | Necroptosis | [80] | ||
Oxidative stress
ROS production plays a critical role in innate immune defense against invading pathogens, primarily through its potent antimicrobial activity [49]. However, oxygen free radicals induce the peroxidation of membrane lipids in cellular and subcellular organelles, and the excessive generation of ROS can lead to severe damage to cellular structure and function. Both LPS-induced pneumonitis and RP can harm lung tissues through oxidative stress. LPS activates the NF-κB signaling pathway through direct binding to TLR4 [20, 49]. Activation of NF-κB induces the expression of NADPH oxidase (NOX), which catalyzes the conversion of oxygen into superoxide anion (O₂⁻), leading to the generation of hydrogen peroxide (H₂O₂) and hydroxyl radicals (·OH). This process initiates oxidative stress through the production of ROS. In RP, ionizing radiation directly hydrolyzes intracellular water molecules, resulting in the substantial production of ROS [50]. On one hand, ROS can activate the NF-κB signaling pathway, thereby perpetuating the generation of additional ROS. On the other hand, ROS can disrupt the mitochondrial electron transport chain, resulting in the release of even more ROS, thereby establishing a deleterious feedback loop. Persistent ROS stimulation prompts the release of TGF-β, which activates fibroblasts, fostering collagen deposition and ultimately leading to pulmonary fibrosis. The accumulation of lipid peroxides triggers iron-dependent cell death, further accelerating the fibrotic process [51]. Nuclear factor erythroid 2-related factor 2 (Nrf2) is a pivotal transcription factor that plays a crucial role in the regulation of redox homeostasis. The upregulation of Nrf2 can mitigate the pro-inflammatory response orchestrated by the NF-κB transcription factor [52]. Meanwhile, Nrf2 negatively regulates the activation of the NLRP3 inflammasome during the regulation of ROS. Considering the strong correlation among Nrf2, NF-κB, and NLRP3, the Nrf2/NF-κB/NLRP3 axis is regarded as an effective therapeutic approach to alleviate severe pulmonary inflammation [53] (Table 2). The Nrf2/ARE signaling pathway also makes an important contribution to maintaining cellular homeostasis under oxidative stress [54, 55].
Death of cells
Pyroptosis, a programmed cell death mechanism, plays a critical role in the pathogenesis of both RP and LPS-induced pneumonitis. As radiation exposure and LPS stimulation activate inflammasomes (including NLRP3 and AIM2)—key molecular complexes that trigger pyroptosis—this process directly contributes to disease development in both pneumonitis types [56-60]. Additionally, both LPS and radiation activate death receptors (e.g., Fas/FasL) on cell membranes, initiating caspase-8-mediated cleavage of downstream caspase-3 to execute apoptosis [61-64]. Critically, ROS are also fundamentally implicated in both pathways. ROS directly activate pro-apoptotic proteins Bax/Bak, inducing mitochondrial damage and subsequent apoptotic cell death [65, 66].
The pathogenesis of both RP and LPS-induced ALI is mechanistically linked to ferroptosis (Table 2). Gu et al. demonstrated that LPS stimulation activates ferroptosis, as evidenced by elevated levels of multiple ferroptosis markers 24 hours post-exposure [67]. Radiation induces ferroptosis through a triad of mechanisms: accelerated iron metabolism, increased generation of ROS, and functional suppression of the system Xc⁻/glutathione peroxidase 4 (GPX4) axis [68, 69]. It upregulates acyl-CoA synthetase long-chain family member 4 (ACSL4), which catalyzes the conversion of polyunsaturated fatty acids (PUFAs) to PUFA-CoA esters [70]. This PUFA-CoA undergo enzymatic oxidation by arachidonate lipoxygenases (ALOXs) to generate lipid hydroperoxides (LOOHs) [71, 72]. Simultaneously, radiation suppresses the function of the Xc⁻ system, reduces glutathione (GSH) synthesis, and impairs GPX4's ability to detoxify lipid peroxides [69]. Accumulated lipid peroxides react with intracellular free iron (Fe²⁺) via the Fenton reaction, generating substantial ROS [70]. This ultimately leads to rupture of the plasma membrane and organelle membranes, triggering cell death [73].
Although RP and LPS-induced pneumonitis exhibit substantial pathophysiological similarities, distinct regulatory mechanisms govern their cell death modalities. In RP, the principal modes of programmed cell death comprise pyroptosis, apoptosis, ferroptosis, and cellular senescence [55] (Table 2). These four modes interact to form a vicious cycle through oxidative stress, inflammation, and fibrosis signaling. Unlike LPS-induced pneumonitis, RP involves direct DNA damage caused by ionizing radiation, resulting in DSBs. These DSBs are then initiated and amplified through the coordinated activation of multiple DNA damage response (DDR) pathways, mediated by DDR kinases such as ATM, DNA-PK, and CHK2, along with their downstream effector molecules (e.g., p53 and NF-κB) [74, 75]. Furthermore, recent studies have revealed that DNA damage triggers the overproduction of poly ADP-ribose (PAR) by PARP1 [76]. Consequently, DNA damage signals are amplified, initiating the apoptotic signaling cascade. Following radiation exposure, unrepaired double-strand breaks (DSBs) activate the p53/p21 pathway [77, 78], resulting in cell cycle arrest. As a result, senescence-associated secretory phenotype (SASP) factors activate neighboring fibroblasts, thereby promoting fibrosis [79]. Research demonstrates that radiation dose variations lead to distinct cell death modalities: low-dose exposure predominantly induces apoptosis, while high-dose irradiation favors autophagy [80]. Autophagy functions as a double-edged sword, with its outcomes dependent on the magnitude and duration of its activation. Under specific conditions, autophagy suppression can induce pulmonary inflammation [81].
LPS-induced lung injury exhibits a pathogenic nexus with necroptosis - an inflammation-dependent regulated cell death pathway. Unlike apoptosis, necroptosis serves dual roles as an executor of cellular demise and an amplifier of inflammatory signaling. This necrosis-associated process triggers substantial release of DAMPs, ultimately driving robust inflammatory cascades. Following LPS stimulation, RIPK1 and RIPK3 assemble into a kinase complex that mediates phosphorylation-dependent activation of MLKL [82]. Phosphorylated MLKL, a key facilitator of necroptosis, triggers cell membrane disruption, resulting in cell demise. Recent studies have revealed that the TBK1/ IKKε signaling pathway exerts a suppressive effect on the inflammatory process. The LPS-TLR4-TBK1 axis functions as a negative regulator of necroptosis in polymorphonuclear neutrophils (PMNs), suggesting its potential as a therapeutic target for controlling PMN mortality and inflammation [83]. NETosis constitutes a specialized cell-death program exclusive to neutrophils, releasing neutrophil extracellular traps (NETs) to capture and eliminate invading microorganisms [84] (Table 2). Notwithstanding this antimicrobial defense, exaggerated NET may instigate tissue injury and propagate inflammatory pathology [85]. In LPS-induced ALI, activated platelets drive NET formation, thereby amplifying tissue damage and inflammatory cascades [84]. Significantly, DNase I-dependent NET degradation promoted removal of NET-bound proteins and conferred protection against ALI in murine models [86]. Although RP is typically regarded as a sterile inflammation, evidence indicates that low-dose ionizing radiation elicits NET formation. This radiation ionizes oxygen molecules to produce ROS, which then activate neutrophil NADPH oxidase. Subsequent amplification of ROS triggers degranulation and IL-8 release. Through autocrine signaling via CXCR1/2 receptors, IL-8 further potentiates NETosis, creating a self-reinforcing loop that drives NET production [87]. Signaling through DAMPs and ROS potently drives NETosis, a process that fuels chronic inflammation and fibrotic progression [86, 88].
Signaling pathways
While overlap exists between RP and LPS-induced pneumonitis in signaling pathways, their principal inflammatory cascades are not entirely identical (Table 2). Radiation directly inflicts DNA lesions, initiating the DDR pathway as the primary early reaction. This cascade activates the ATM-Chk2-p53 axis to execute apoptotic cell death while concurrently imposing cell cycle arrest [58]. Such arrest provides a temporal window for damage repair. When DNA damage proves irreparable, p53 can additionally drive cellular senescence and apoptosis. The TGF-β/Smad signaling pathway serves as a core regulator in RILI pathogenesis [59]. Specifically, TGF-β1 transcriptionally activates SERPINE1 (PAI-1)-a potent pro-fibrotic mediator in stromal cells [60]. Cells compromised by ionizing radiation secrete TGF-β1. This cytokine induces phosphorylation of downstream SMAD2/3 and, through p53-dependent mechanisms, upregulates PAI-1- culminating in fibrotic tissue remodeling [60]. Separately, alveolar macrophages from thoracic radiotherapy patients constitutively secrete PDGF-BB [61]. Concurrently, irradiated lung fibroblasts exhibit heightened chemotactic potency toward PDGF-BB, correlating with PDGFRB overexpression and potentially driving RP and radiation induced pulmonary fibrosis (RIPF) pathogenesis.
During LPS-induced pneumonitis, LPS primarily engages TLR4 receptors, initiating the TLR4/MyD88/NF-κB cascade. This signaling prompts the release of pro-inflammatory mediators (TNF-α, IL-1β, IL-6) and the production of ROS. NLRP3 inflammasome activation by ROS culminates in pyroptosis accompanied by IL-1β maturation. Evidence further reveals involvement of the USP7/MAPK14 axis in LPS-driven pulmonary injury. Functionally, USP7 catalytically stabilizes MAPK14 via its deubiquitinase activity and potentiates it signaling capacity, thereby amplifying inflammatory tissue destruction [62]. Preclinical models demonstrate that USP7 inhibition alleviates LPS-induced pulmonary damage in mice. Separately, LPS-triggered endothelial barrier impairment correlates with RhoA/ROCK1/2 pathway activation. RhoA drives cellular proliferation, motility, and invasiveness through ROCK1/2-mediated protein modulation - a key mechanism underlying contraction-mediated endothelial hyperpermeability [63]. LPS exposure significantly elevates RhoA and ROCK1/2 expression levels.
Potential treatment targets for RP
RP and LPS-induced pneumonitis exhibit overlapping mechanisms while maintaining their own specific targets. Exploiting these shared mechanisms, it is possible to identify potential therapeutic agents for both conditions.
The NF-κB pathway and NLRP3 inflammasome contribute to both diseases. Amentoflavone (AF), previously shown to exert anti-inflammatory and antioxidant effects, was recently reported by Sun et al. to alleviate LPS-induced ALI by inhibiting the NLRP3/ASC/Caspase-1 axis and reducing pyroptosis [89]. While the role of AF in RILI has not yet been investigated, its efficacy in mitigating LPS-induced ALI suggests potential therapeutic value for RP.
Targeting the modulation of cell death pathways, including apoptosis, pyroptosis, and ferroptosis, emerges as a promising therapeutic approach for addressing RP. Dapagliflozin (DPG) ameliorates LPS-induced lung injury by upregulating SIRT-1-mediated deacetylation to activate PGC-1α activity, suppressing the NF-κB inflammatory signaling pathway, enhancing mitochondrial antioxidant capacity, restoring the Bcl-2/Bax balance, and reducing caspase-3-dependent apoptosis [90]. Currently, there is limited research exploring whether the SIRT-1/PGC-1α pathway can ameliorate radiation-induced damage. As a selective DRP1 inhibitor, the cell-permeable quinazolinone compound Mdivi-1 attenuates ALI by suppressing mitochondrial ROS (mtROS)/NLRP3 signaling, which consequently blocks M1 alveolar macrophage polarization and pyroptosis [91]. These mechanistic insights support DRP1-dependent mitochondrial fission as a promising therapeutic target for RP requiring further validation. Obacunone (OB), a natural limonoid compound with demonstrated anti-inflammatory and antioxidant activities, confers protection against LPS-induced ALI by enhancing pulmonary antioxidant capacity, suppressing ferroptosis, and stabilizing Nrf2 through inhibition of ubiquitin-proteasomal degradation [92]. Given the critical involvement of Nrf2 dysfunction and oxidative stress in RILI, OB's mechanisms position it as a promising therapeutic candidate for RP.
Regulating gut flora homeostasis might improve RP. A recent study revealed that alterations in gut microbiota can impact intestinal digestion and immune function. It was found that the lung microbiomes of mouse sepsis and human acute respiratory distress syndrome patients were enriched with bacteria of intestinal origin [93]. Meanwhile, septic ALI can be accompanied by changes in the composition and function of gut microbiota, which might in turn affect distal organs through the gut-lung axis. Wang et al. discovered that Cordyceps militaris solid medium extract (CMME) decreased the levels of inflammatory factors and oxidative stress, reduced macrophage activation and neutrophil recruitment, and ultimately exerted a modulating effect on LPS-induced lung inflammation [94]. This might be accomplished by regulating the gut flora and correcting metabolic disturbances. Radiation can modify the composition of the intestinal flora, resulting in a reduction in intestinal flora diversity [95]. It has been shown that faecal microbiota transplantation (FMT) can maintain intestinal flora homeostasis and significantly enhance the survival rate of radiation-exposed mice, and this alteration might be achieved through the regulation of immune function [96]. Whether this treatment by regulating gut flora homeostasis can improve RP requires experimental confirmation.
New methods for removing ROS may play a significant role in the treatment of RP. Manganese superoxide dismutase (MnSOD), a crucial member of the superoxide dismutase (SOD) family, functions as a ROS scavenger. Recent studies have demonstrated that plasmid-mediated MnSOD gene delivery (pMnSOD) presents a promising therapeutic strategy for radiation-induced dermal injury [97]. Furthermore, systemic administration of MnSOD-engineered mesenchymal stem cells (MnSOD-MSCs) has shown significant efficacy in mitigating pulmonary inflammation [98]. However, aerosolized delivery remains a technical challenge for such genetic therapies. Although preliminary evidence suggests the biosafety of nebulized MSC-derived exosomes and their potential to enhance pulmonary lesion resolution, the absence of double-blind clinical trials renders current therapeutic efficacy assessments inconclusive. The clinical translation of aerosolized MnSOD-MSCs inhalation delivery for pulmonary antioxidant therapy remains a persistent challenge [99].
Nanoparticles may also become effective drugs for the treatment of RP as ALI treatment. The design of nanoparticles for different applications can achieve objectives such as targeted drug delivery, improved bioavailability, extended sustained release, and minimized drug toxicity. Zhang et al. proposed a non-invasive inhalation of NPs for the safe and effective treatment of ALI. This formulation demonstrates excellent biocompatibility and exhibits a sustained-release pharmacological profile. It not only inhibits the release of cytokines but also blocks the triggering pathway of ALI, thereby reducing lung injury [100]. Sun et al. discovered that curcumin-loaded ROS-responsive hollow mesoporous silica nanoparticles (Cur@HMSN-BSA) achieve specific drug release in high-ROS pneumonitis microenvironments [101]. These nanoparticles effectively scavenge excessive intracellular ROS. Further studies confirmed its capability to promote macrophage polarization from the pro-inflammatory M1 phenotype to the anti-inflammatory M2 phenotype. Such ROS-responsive nanoparticles effectively avoided non-specific release of the drug and improved targeting. A novel X-ray-responsive nanocomposite, Au@mSiO2@Mn(CO)5Br (ASMB), was designed to mitigate RILI through localized release of CO and Mn²⁺ upon irradiation. CO suppresses NLRP3 inflammasome activation, reduces pyroptosis, scavenges ROS, and enhances DNA repair, while Mn²⁺ alleviates hypoxia via H₂O₂-to-O₂ conversion and downregulates fibrotic markers (e.g., TGF-β1, α-SMA). In RILI models, ASMB NPs reduced pulmonary edema, inflammation, and fibrosis while enhancing treatment outcomes, demonstrating dual therapeutic-radiosensitizing potential [102]. An analysis of the transcriptomic profile of RILI in non-human primates revealed that the expression of SERPINA3, ATP12A, GJB2, CLDN10, TOX3, and LPA was significantly up-regulated, suggesting their potential as biomarkers and therapeutic targets for RILI [103].
Excessive production of NETs has been shown to promote ROS generation, which contributes to tissue damage. Limiting NETs overproduction therefore emerges as a promising therapeutic approach. In vitro experiments have demonstrated that knocking out CXCL2 significantly reduces NET formation and ROS production. Epigallocatechin-3-gallate (EGCG), a natural compound derived from green tea, exhibits anti-cancer, anti-inflammatory, and antioxidant properties. Experimental data reveal that EGCG suppresses NET formation by downregulating CXCL2, thereby reducing pulmonary inflammation [104]. EGCG has demonstrated significant therapeutic benefits in LPS-induced ALI. Given the mechanistic similarities in the pathogenesis, it is plausible that EGCG could also be a promising treatment for RILI. Nevertheless, the efficacy of EGCG in treating RILI needs to be further investigated through additional experimental and clinical research.
Studies have shown that hypoxia-inducible factor-1α (HIF-1α) upregulates phosphofructokinase/fructose-2,6-bisphosphatase 2 (PFKFB2), which increases glycolysis, accelerates dendritic cell (DC) maturation, and amplifies immune activation. These processes exacerbate inflammatory responses and contribute to ALI progression. In a mouse model of LPS-induced ALI, both DC-specific PFKFB2 knockout and DC-targeted delivery of HIF-1α inhibitor-loaded nanoparticles effectively suppressed DC maturation and alleviated ALI severity [105]. Calcitonin gene-related peptide (CGRP) has been shown to inhibit the HIF-1α pathway and modulate macrophage polarization balance [106]. Additionally, ophiopogonin D (OP-D) improves pulmonary microvascular endothelial barrier dysfunction by targeting the HIF-1α-VEGF pathway, thereby mitigating LPS-induced ALI [107]. While the HIF-1α pathway demonstrates significant therapeutic potential, its efficacy in treating radiation pneumonia requires further experimental validation.
Radiotherapy induces DNA damage, including mitochondrial DNA damage, underscoring the therapeutic potential of targeting mitochondria. Esketamine activates mitophagy via the ULK1/FUNDC1 signaling pathway and exhibits beneficial effects in LPS-induced ALI animal models. It improves lung vascular permeability, reduces inflammatory responses, apoptosis, and oxidative stress [108]. The novel mitophagy inducer TJ0113 selectively targets damaged mitochondria, induces mitophagy, and suppresses the NF-κB pathway, thereby alleviating LPS-induced inflammation. However, its therapeutic potential for RP remains to be confirmed through further studies [109].
The Rap1 pathway is involved in cell proliferation, differentiation, and survival, and plays a significant role in immune regulation, angiogenesis, and cancer development. A research team has confirmed the therapeutic effects of anlotinib in RILI, and the authors additionally conducted KEGG enrichment analysis to reveal potential pathways, including Rap1, that may contribute to the treatment effects. However, this pathway lacks validation through in vitro and animal experiments [110]. Liu and Wang et al. validated through animal experiments that Potentilla anserina L. polysaccharide (PAP) significantly reduced LPS-induced ALI inflammation and inhibited M1 macrophage immune responses by activating the Rap1 signaling pathway [111]. Prostaglandins are an important group of lipid mediators with barrier-protective potential towards the vascular endothelium. Prostacyclin (PC) exhibits potent protective effects in ischemia-reperfusion and ventilator-induced lung injury. Experimental evidence shows that post-treatment with PC activates the Epac/Rap1/afadin-dependent mechanism, promoting endothelial barrier repair and reducing p38 MAPK and NF-κB inflammatory pathways, thereby accelerating lung recovery [112]. PC has been shown to effectively treat LPS-induced ALI in experimental models. Given its mechanisms, it may also hold therapeutic promise for RILI. However, its effectiveness in this setting needs to be rigorously evaluated.
Vasodilator-stimulated phosphoprotein (VASP) is a crucial controller of cytoskeletal dynamics and is essential for processes including cell migration, cytoskeletal restructuring, and the regulation of inflammatory responses. VASP demonstrates anti-inflammatory properties in liver tissues, but in lung tissues, its deletion significantly reduces symptoms in mice with LPS-induced ALI. VASP knockdown protects against LPS-induced ALI in mice by inhibiting M1 macrophage polarization, with its protective effects partially mediated by the cGMP-PKG signaling pathway [113]. The efficacy of targeting the cGMP-PKG signaling pathway in treating RILI remains unclear.
Macrophage efferocytosis is integral to immune system function, playing a vital role in preserving tissue equilibrium, mitigating inflammation, and facilitating healing. Research indicates that sorafenib, a selective ADAM9 inhibitor, can enhance ALI symptoms and is crucial in modulating inflammatory responses [114]. This inhibition leads to a decrease in macrophage and neutrophil counts in bronchoalveolar lavage, a reduction in pro-inflammatory cytokine levels, and an elevation of anti-inflammatory cytokines. Additionally, in vitro studies reveal that reducing ADAM9 expression boosts macrophage efferocytosis of apoptotic PMNs. The team also confirmed that ADAM9 in BMDMs binds to ITGAV in PMNs, and that inhibiting ITGAV enhances the macrophage efferocytosis mediated by ADAM9. Moreover, stifling the interaction between ADAM9 and ITGAV could potentially lead to the improvement of LPS-induced ALI by stimulating macrophage efferocytosis. Currently, there is a lack of validation regarding the efficacy of the ADAM9/ITGAV pathway in the treatment of RILI.
Currently, the burgeoning research into traditional Chinese medicine is fostering increased public recognition. Xuebijing Injection (XBJ), a preparation based on the Hematopoietic Blood Stasis Dispelling Soup formula, has exhibited potential in various therapeutic applications. The studies by Cui et al. have highlighted its capacity to significantly reduce plasma levels of endothelial cell damage-related biomarkers in LPS-induced sepsis and to mitigate LPS-induced lung injury by inhibiting the ACLY/MYB/RIG-I pathway [115]. Nevertheless, the question of whether XBJ can also effectively reduce endothelial damage in the context of RILI has yet to be answered, with its therapeutic promise in this domain requiring further exploration and validation.
While RP and LPS-induced pneumonitis diverge in etiology and therapeutic focus, their shared inflammatory endpoints (e.g., alveolar injury, cytokine storm) reveal a hidden therapeutic synergy. For example, TGF-β-targeted antifibrotics for RP could mitigate late-stage fibrosis in LPS-ALI, whereas TLR4 antagonists for LPS-ALI might attenuate radiation-induced bystander effects [116]. The treatments listed in Table 3, though currently used exclusively for either RP or LPS-ALI, may hold dual efficacy through overlapping pathways-a hypothesis awaiting experimental validation. In addition to the previously mentioned target pathways, we have also identified that drugs can alleviate LPS-induced lung injury in mice through mechanisms such as the STAT3 signaling pathway and miR-21/PTEN axis (Figure 3) [111, 117-121]. Whether these pathways could serve as potential therapeutic targets for RP remains to be further investigated. Furthermore, other pathways such as NAIP/NLRC4/ASC inflammasome autophagy, PINK1/PRKN-mediated mitophagy, and the TNKS1BP1/CNOT4/EEF2 axis have already been validated as effective targets for treating RP (Figure 3) [122-127].
Potential treatments for RP or LPS-induced pneumonitis. This figure lists pathways validated for treating LPS-induced and radiation pneumonitis, highlighting shared targets and emerging strategies.
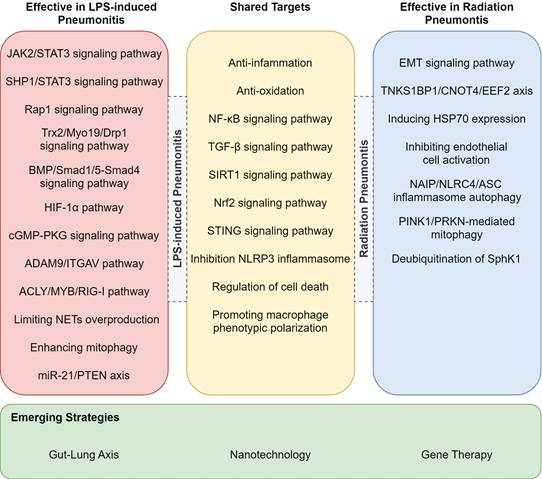
Potential mechanism or key pathway for RP or LPS-induced pneumonitis
| Mechanism of action | Treatment | Targets of action | References | |
|---|---|---|---|---|
| TGF-β signaling pathway | Ergothioneine | LPS-ALI* | Reported: TGF-β/smad/snail signaling pathway | [139] |
| RILI** | Not reported | |||
| Pirfenidone | LPS-ALI* | Reported: TGF-β/Smad signaling pathway | [28, 140] | |
| RILI** | Reported: TGF-β1/Smad3 pathway | |||
| Nicaraven | LPS-ALI* | Not reported | [141] | |
| RILI** | Reported: TGF-β/pSmad2 pathway | |||
| Nrf2 signaling pathway | Penehyclidine hydrochloride (PHC) | LPS-ALI* | Reported: mTOR / keap1 / Nrf2 signaling pathway | [142] |
| RILI** | Not reported | |||
| Anisodamine | LPS-ALI* | Not reported | [55] | |
| RILI** | Reported: Nrf2/ARE signaling pathway | |||
| Pyrroloquinoline quinone (PQQ) | LPS-ALI* | Not reported | [143] | |
| RILI** | Reported: the MOTS-c/Nrf2 signaling pathway | |||
| NF‑κB signaling pathway | Dihydroartemisinin | LPS-ALI* | Reported: NF‑κB signaling pathway | [144, 145] |
| RILI** | Reported: cGAS/STING/ NF‑κB signaling pathway | |||
| Ethyl caffeate | LPS-ALI* | Reported: TNF-α/NF-κB/MMP9 axis | [146] | |
| RILI** | Not reported | |||
| 3,3'-Diindolylmethane | LPS-ALI* | Not reported | [147] | |
| RILI** | Reported: TGF-β/Smad and NF-κB dual pathways | |||
| NLRP3 signaling pathway | Glycyrrhizin | LPS-ALI* & RILI** | Reported: Inhibiting the NLRP3 inflammasome | [148, 149] |
| Amentoflavone (AF) | LPS-ALI* | Reported: NLRP3/ASC/Caspase-1 axis | [89] | |
| RILI** | Not reported | |||
| Raspberry ketone | LPS-ALI* | Not reported | [150] | |
| RILI** | Reported: STAT2-P2X7r/NLRP3 pathway | |||
| AMPK signaling pathway | Hydrogen-rich solution | LPS-ALI* | Reported: ROS/AMPK/mTOR pathway | [151, 152] |
| RILI** | Reported: AMPK/mTOR/ULK1 signaling pathway | |||
| Nerolidol | LPS-ALI* | Reported: AMPK/Nrf-2/HO-1 pathway | [153] | |
| RILI** | Not reported | |||
| SIRT1 signaling pathway | L‑carnitine | LPS-ALI* | Not reported | [154] |
| RILI** | Reported: AMPK/SIRT1/TGF‑1ß pathway | |||
| Dapagliflozin (DPG) | LPS-ALI* | Reported: SIRT-1/PGC-1α pathway | [90] | |
| RILI** | Not reported | |||
| HIF-1α signaling pathway | Calcitonin gene-related peptide (CGRP) | LPS-ALI* | Reported: Inhibiting the HIF-1α pathway | [106] |
| RILI** | Not reported | |||
| Ophiopogonin D (OP-D) | LPS-ALI* | Reported: the HIF-1α-VEGF pathway | [107] | |
| RILI** | Not reported | |||
| Rap1 pathway | Potentilla anserina L. polysaccharide (PAP) | LPS-ALI* | Reported: the Rap1 signaling pathway | [111] |
| RILI** | Not reported | |||
| Prostacyclin (PC) | LPS-ALI* | Reported: the Epac/Rap1/afadin-dependent mechanism & reducing p38 MAPK and NF-κB inflammatory pathways | [112] | |
| RILI** | Not reported | |||
| cGMP-PKG signaling pathway | VASP knockdown | LPS-ALI* | Reported: Inhibiting M1 macrophage polarization mediated by the cGMP-PKG signaling pathway | [113] |
| RILI** | Not reported | |||
| ACLY/MYB/RIG-I pathway | Xuebijing Injection (XBJ) | LPS-ALI* | Reported: Inhibiting the ACLY/MYB/RIG-I pathway | [115] |
| RILI** | Not reported | |||
| Death of cells | Uridine | LPS-ALI* | Reported: Inhibiting ferroptosis of macrophage | [155] |
| RILI** | Not reported | |||
| Astragaloside IV | LPS-ALI* | Not reported | [156] | |
| RILI** | Reported: Suppressing ferroptosis | |||
| Anti-oxidation | Curcumin | LPS-ALI* | Reported: Enhancing anti-oxidant levels | [157, 158] |
| RILI** | Reported: Free radical scavenging and anti-oxidation | |||
| Alpinumisoflavone | LPS-ALI* | Reported: Anti-oxidation and anti-inflammation | [159] | |
| RILI** | Not reported | |||
| Suplatast tosilate | LPS-ALI* | Not reported | [160] | |
| RILI** | Reported: Suppression of oxidative stress | |||
| Anti-inflammatory | L-carnitine | LPS-ALI* | Reported: Mitochondria modulation and inflammation control | [154, 161] |
| RILI** | Reported: the AMPK/SIRT1/TGF‑1ß pathway | |||
| Diethylcarbamazine | LPS-ALI* | Reported: Apoptosis induction for inhibiting inflammatory cell accumulation | [162, 163] | |
| RILI** | Reported: Suppressing COX-2 and NF-kB pathway | |||
| Regulation of gut microbiota | Cordyceps militaris solid medium extract (CMME) | LPS-ALI* | Reported: Regulating intestinal flora and correcting metabolic disorders | [94] |
| RILI** | Not reported | |||
| Faecal microbiota transplantation (FMT) | LPS-ALI* | Not reported | [96] | |
| RILI** | Reported: Improving GI tract function and epithelial integrity | |||
| Limiting NETs overproduction | Epigallocatechin-3-gallate (EGCG) | LPS-ALI* | Reported: Downregulating CXCL2 | [104] |
| RILI** | Not reported | |||
| Enhancing mitophagy | Esketamine | LPS-ALI* | Reported: the ULK1/FUNDC1 signaling pathway | [108] |
| RILI** | Not reported | |||
| TJ0113 | LPS-ALI* | Reported: inducing mitophagy and suppressing the NF-κB pathway | [109] | |
| RILI** | Not reported | |||
| Macrophage efferocytosis | sorafenib | LPS-ALI* | Reported: the ADAM9/ITGAV pathway | [114] |
| RILI** | Not reported | |||
*: Including LPS-induced ALI, LPS-induced pneumonitis, LPS-induced ARDS.
**: Including RILI, RP, RIPF
Conclusion and perspectives
RP and LPS-induced pneumonitis exhibit similar manifestations, including alveolar damage and inflammatory infiltrates but they have different molecular mechanisms and clinical managements. RP primarily results from radiation-induced DNA damage, oxidative stress, and dysregulated TGF-β signaling, which collectively contribute to the development of pulmonary fibrosis. In contrast, LPS-induced pneumonitis is triggered by TLR4-dependent inflammatory signaling, characterized by a cytokine storm and a neutrophil- predominant immune response. Notably, both diseases share key pathways such as the release of certain inflammatory mediators and oxidative stress, suggesting that therapeutic strategies targeting these common nodes may be beneficial for both conditions, offering a potential for unified treatment approaches. In this comprehensive review, we have explored the therapeutic potential of RP by investigating a variety of approaches, including anti-inflammatory, antioxidant strategies, diverse modes of cell death, gut microbiota modulation, gene therapy, and nanotechnology. Our analysis has unveiled a subset of drugs that, while primarily employed in the treatment of LPS-induced pneumonitis, show considerable promise for RP by targeting shared mechanisms. These findings not only enrich our understanding of the therapeutic landscape for RP but also provides critical guidance for developing innovative drug repurposing strategies.
Abbreviations
OH: hydroxyl radicals; ACSL4: acyl-CoA synthetase long-chain family member 4; AF: Amentoflavone; ALI: acute lung injury; ALOXs: arachidonate lipoxygenases; ARDS: acute respiratory distress syndrome; ASMB: Au@mSiO2@Mn(CO)5Br; CMME: Cordyceps militaris solid medium extract; CTCAE: Common Terminology Criteria for Adverse Events; Cur@HMSN-BSA: Curcumin-loaded ROS-responsive hollow mesoporous silica nanoparticles; DAMPs: Damage-associated Molecular Patterns; DDR: DNA damage response; DPG: Dapagliflozin; DSBs: double-strand breaks; EMT: Epithelial-mesenchymal transition; EndMT: endothelial-mesenchymal transition; FMT: faecal microbiota transplantation; GPX4: glutathione peroxidase 4; GSH: glutathione; H₂O₂: hydrogen peroxide; IKKε: IκB Kinase ε; IRF3: Interferon Regulatory Factor 3; LBP: LPS binding protein; LOOHs: lipid hydroperoxides; LPS: lipopolysaccharide; MAPK: mitogen-activated protein kinase; MD2: Myeloid differentiation protein-2; MnSOD: Manganese superoxide dismutase; MODS: multiple organ dysfunction syndrome; MSCs: mesenchymal stem cells; mtDNA: mitochondrial DNA; mtROS: mitochondrial ROS; MyD88: myeloid differentiation primary response 88; NETs: neutrophil extracellular traps; NF-κB: nuclear factor κB; NLRP3: NLR Family Pyrin Domain-Containing 3; NOX: NADPH oxidase; Nrf2: Nuclear factor erythroid 2-related factor 2; NSAIDs: non-steroidal anti-inflammatory drugs; OB: Obacunone; PAR: poly ADP-ribose; PMNs: polymorphonuclear neutrophils; pMnSOD: plasmid-mediated MnSOD; PRRs: pattern recognition receptors; PUFAs: polyunsaturated fatty acids; RILI: radiation induced lung injury; RIPF: radiation induced pulmonary fibrosis; ROS: reactive oxygen species; RP: radiation pneumonitis; RTOG: Radiation Therapy Oncology Group; SASP: senescence-associated secretory phenotype; SOD: superoxide dismutase; SSBs: single-strand breaks; TBK1: TANK-Binding Kinase 1; TIR: toll/interleukin-1 receptor; TLR: Toll-like receptor; TRAF: TNF Receptor-Associated Factor; TRAM: Tumor Necrosis Factor Receptor-Associated Machinery; TRIF: TIR-domain-containing adapter inducing interferon-β.
Acknowledgements
We are grateful for the assistance provided by ChatGPT, which we used to refine the language in certain parts of our manuscript after independently completing the initial draft. Its use was solely aimed at improving the readability of the text.
Funding
This work was supported by the Shandong Provincial Natural Science Foundation [grant numbers ZR2022MH207] and Taishan Scholars [WX].
Consent for publication
All co-authors have read and agreed with the content of the manuscript.
Author contributions
S.N. and Z.L.: Writing - Original Draft, Investigation; N.W., G.X., H.X. and Q.H.: Review & Editing; S.N.: Visualization; W.X., Z.W., X.Y. and Q.X.: Supervision.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Yan Y, Fu J, Kowalchuk RO, Wright CM, Zhang R, Li X. et al. Exploration of radiation-induced lung injury, from mechanism to treatment: a narrative review. Transl Lung Cancer Res. 2022;11:307-22
2. Dasgupta Q, Jiang A, Wen AM, Mannix RJ, Man Y, Hall S. et al. A human lung alveolus-on-a-chip model of acute radiation-induced lung injury. Nat Commun. 2023;14:6506
3. Otani K, Seo Y, Ogawa K. Radiation-Induced Organizing Pneumonia: A Characteristic Disease that Requires Symptom-Oriented Management. International Journal of Molecular Sciences. 2017 18
4. Rajan Radha R, Chandrasekharan G. Pulmonary injury associated with radiation therapy - Assessment, complications and therapeutic targets. Biomed Pharmacother. 2017;89:1092-104
5. Lu L, Sun C, Su Q, Wang Y, Li J, Guo Z. et al. Radiation-induced lung injury: latest molecular developments, therapeutic approaches, and clinical guidance. Clin Exp Med. 2019;19:417-26
6. Yin C, Alam MZ, Fallon JT, Huang W. Advances in Development of Novel Therapeutic Strategies against Multi-Drug Resistant Pseudomonas aeruginosa. Antibiotics (Basel). 2024 13
7. Chen H, Bai C, Wang X. The value of the lipopolysaccharide-induced acute lung injury model in respiratory medicine. Expert Rev Respir Med. 2010;4:773-83
8. Nguyen N, Xu S, Lam TYW, Liao W, Wong WSF, Ge R. ISM1 suppresses LPS-induced acute lung injury and post-injury lung fibrosis in mice. Mol Med. 2022;28:72
9. Vernooy JH, Dentener MA, van Suylen RJ, Buurman WA, Wouters EF. Long-term intratracheal lipopolysaccharide exposure in mice results in chronic lung inflammation and persistent pathology. Am J Respir Cell Mol Biol. 2002;26:152-9
10. Li Y, Huang J, Foley NM, Xu Y, Li YP, Pan J. et al. B7H3 ameliorates LPS-induced acute lung injury via attenuation of neutrophil migration and infiltration. Sci Rep. 2016;6:31284
11. Kuipers ME, van Doorn-Wink KCJ, Hiemstra PS, Slats AM. Predicting Radiation-Induced Lung Injury in Patients With Lung Cancer: Challenges and Opportunities. Int J Radiat Oncol Biol Phys. 2024;118:639-49
12. Roy S, Salerno KE, Citrin DE. Biology of Radiation-Induced Lung Injury. Semin Radiat Oncol. 2021;31:155-61
13. Giridhar P, Mallick S, Rath GK, Julka PK. Radiation induced lung injury: prediction, assessment and management. Asian Pac J Cancer Prev. 2015;16:2613-7
14. Arroyo-Hernandez M, Maldonado F, Lozano-Ruiz F, Munoz-Montano W, Nunez-Baez M, Arrieta O. Radiation-induced lung injury: current evidence. BMC Pulm Med. 2021;21:9
15. Ozturk B, Egehan I, Atavci S, Kitapci M. Pentoxifylline in prevention of radiation-induced lung toxicity in patients with breast and lung cancer: a double-blind randomized trial. Int J Radiat Oncol Biol Phys. 2004;58:213-9
16. Lei X, Ma N, Liang Y, Liu J, Zhang P, Han Y. et al. Glucosamine protects against radiation-induced lung injury via inhibition of epithelial-mesenchymal transition. J Cell Mol Med. 2020;24:11018-23
17. Dehbanipour R, Ghalavand Z. Acinetobacter baumannii: Pathogenesis, virulence factors, novel therapeutic options and mechanisms of resistance to antimicrobial agents with emphasis on tigecycline. J Clin Pharm Ther. 2022;47:1875-84
18. Rhodes KA, Schweizer HP. Antibiotic resistance in Burkholderia species. Drug Resist Updat. 2016;28:82-90
19. Helgeson SA, Levitt JE, Festic E. Systemic and Inhaled Corticosteroids, with or without Beta Agonists, as Adjuvant Therapy in Community Acquired Pneumonia. Acta Med Acad. 2020;49:9-20
20. Zhou J, Peng Z, Wang J. Trelagliptin Alleviates Lipopolysaccharide (LPS)-Induced Inflammation and Oxidative Stress in Acute Lung Injury Mice. Inflammation. 2021;44:1507-17
21. Perkins SD, Smither SJ, Atkins HS. Towards a Brucella vaccine for humans. FEMS Microbiol Rev. 2010;34:379-94
22. Guerra MES, Destro G, Vieira B, Lima AS, Ferraz LFC, Hakansson AP. et al. Klebsiella pneumoniae Biofilms and Their Role in Disease Pathogenesis. Front Cell Infect Microbiol. 2022;12:877995
23. Karampatakis T, Tsergouli K, Behzadi P. Carbapenem-Resistant Klebsiella pneumoniae: Virulence Factors, Molecular Epidemiology and Latest Updates in Treatment Options. Antibiotics (Basel). 2023 12
24. Yamashita C, Moriyama K, Hasegawa D, Kato Y, Sakai T, Kawaji T. et al. In Vitro Study of Endotoxin Adsorption by a Polymyxin B-Immobilized Fiber Column. Blood Purif. 2018;46:269-73
25. Jiao J, Buzukela A, Li M, Baihetinisha T. [Advance in research on application of endotoxin adsorption membrane in treatment of coronavirus disease 2019]. Zhonghua Wei Zhong Bing Ji Jiu Yi Xue. 2022;34:538-41
26. Li X, Xue J, Lu Y. [Current situation and prospect of treatment for radiation-induced lung injury]. Sheng Wu Yi Xue Gong Cheng Xue Za Zhi. 2010;27:937-40
27. Tu J, Chen X, Li C, Liu C, Huang Y, Wang X. et al. Nintedanib Mitigates Radiation-Induced Pulmonary Fibrosis by Suppressing Epithelial Cell Inflammatory Response and Inhibiting Fibroblast-to-Myofibroblast Transition. Int J Biol Sci. 2024;20:3353-71
28. Ying H, Fang M, Hang QQ, Chen Y, Qian X, Chen M. Pirfenidone modulates macrophage polarization and ameliorates radiation-induced lung fibrosis by inhibiting the TGF-beta1/Smad3 pathway. J Cell Mol Med. 2021;25:8662-75
29. Tsoutsou PG, Koukourakis MI. Radiation pneumonitis and fibrosis: mechanisms underlying its pathogenesis and implications for future research. Int J Radiat Oncol Biol Phys. 2006;66:1281-93
30. Giuranno L, Ient J, De Ruysscher D, Vooijs MA. Radiation-Induced Lung Injury (RILI). Front Oncol. 2019;9:877
31. Beach TA, Groves AM, Williams JP, Finkelstein JN. Modeling radiation-induced lung injury: lessons learned from whole thorax irradiation. Int J Radiat Biol. 2020;96:129-44
32. Bledsoe TJ, Nath SK, Decker RH. Radiation Pneumonitis. Clin Chest Med. 2017;38:201-8
33. Jin H, Yoo Y, Kim Y, Kim Y, Cho J, Lee YS. Radiation-Induced Lung Fibrosis: Preclinical Animal Models and Therapeutic Strategies. Cancers (Basel). 2020 12
34. Feng Y, Yuan P, Guo H, Gu L, Yang Z, Wang J. et al. METTL3 Mediates Epithelial-Mesenchymal Transition by Modulating FOXO1 mRNA N(6) -Methyladenosine-Dependent YTHDF2 Binding: A Novel Mechanism of Radiation-Induced Lung Injury. Adv Sci (Weinh). 2023;10:e2204784
35. Rezaie F, Ghafouri Khosroshahi A, Larki-Harchegani A, Nourian A, Khosravi H. Hydroalcoholic Sumac Extract as a Protective Agent Against X-Ray-Induced Pulmonary Fibrosis. Rep Biochem Mol Biol. 2024;13:231-42
36. Hsieh PC, Wu YK, Yang MC, Su WL, Kuo CY, Lan CC. Deciphering the role of damage-associated molecular patterns and inflammatory responses in acute lung injury. Life Sci. 2022;305:120782
37. Kasmann L, Dietrich A, Staab-Weijnitz CA, Manapov F, Behr J, Rimner A. et al. Radiation-induced lung toxicity - cellular and molecular mechanisms of pathogenesis, management, and literature review. Radiat Oncol. 2020;15:214
38. Nova Z, Skovierova H, Calkovska A. Alveolar-Capillary Membrane-Related Pulmonary Cells as a Target in Endotoxin-Induced Acute Lung Injury. Int J Mol Sci. 2019 20
39. Tobias PS, Mathison J, Mintz D, Lee JD, Kravchenko V, Kato K. et al. Participation of lipopolysaccharide-binding protein in lipopolysaccharide-dependent macrophage activation. Am J Respir Cell Mol Biol. 1992;7:239-45
40. Shen B, Zhang H, Zhu Z, Ling Z, Zeng F, Wang Y. et al. Baicalin Relieves LPS-Induced Lung Inflammation via the NF-kappaB and MAPK Pathways. Molecules. 2023 28
41. Shen X, He L, Cai W. Role of Lipopolysaccharides in the Inflammation and Pyroptosis of Alveolar Epithelial Cells in Acute Lung Injury and Acute Respiratory Distress Syndrome. J Inflamm Res. 2024;17:5855-69
42. Zheng Y, Gao Y, Zhu W, Bai XG, Qi J. Advances in molecular agents targeting toll-like receptor 4 signaling pathways for potential treatment of sepsis. Eur J Med Chem. 2024;268:116300
43. Brooks D, Barr LC, Wiscombe S, McAuley DF, Simpson AJ, Rostron AJ. Human lipopolysaccharide models provide mechanistic and therapeutic insights into systemic and pulmonary inflammation. Eur Respir J. 2020 56
44. Hussain S, Johnson CG, Sciurba J, Meng X, Stober VP, Liu C. et al. TLR5 participates in the TLR4 receptor complex and promotes MyD88-dependent signaling in environmental lung injury. Elife. 2020 9
45. Guo J, Luo Y, Zuo J, Teng J, Shen B, Liu X. Echinacea Polyphenols Inhibit NLRP3-Dependent Pyroptosis, Apoptosis, and Necroptosis via Suppressing NO Production during Lipopolysaccharide-Induced Acute Lung Injury. J Agric Food Chem. 2023;71:7289-98
46. Chen M, Zhang J, Huang H, Wang Z, Gao Y, Liu J. miRNA-206-3p alleviates LPS-induced acute lung injury via inhibiting inflammation and pyroptosis through modulating TLR4/NF-κB/NLRP3 pathway. Sci Rep. 2024;14:11860
47. Li C, Qi X, Xu L, Sun Y, Chen Y, Yao Y. et al. Preventive Effect of the Total Polyphenols from Nymphaea candida on Sepsis-Induced Acute Lung Injury in Mice via Gut Microbiota and NLRP3, TLR-4/NF-kappaB Pathway. Int J Mol Sci. 2024 25
48. Rao X, Zhou D, Deng H, Chen Y, Wang J, Zhou X. et al. Activation of NLRP3 inflammasome in lung epithelial cells triggers radiation-induced lung injury. Respir Res. 2023;24:25
49. Bezerra FS, Lanzetti M, Nesi RT, Nagato AC, Silva CPE, Kennedy-Feitosa E. et al. Oxidative Stress and Inflammation in Acute and Chronic Lung Injuries. Antioxidants (Basel). 2023 12
50. Li Y, Song Q, Yao Y, Dong Y, Gao Y, Wu B. [Progression of Anti-oxygen Therapy in Radiation-Induced Lung Injury]. Zhongguo Fei Ai Za Zhi. 2019;22:579-82
51. Imai H, Matsuoka M, Kumagai T, Sakamoto T, Koumura T. Lipid Peroxidation-Dependent Cell Death Regulated by GPx4 and Ferroptosis. Curr Top Microbiol Immunol. 2017;403:143-70
52. Yang K, Han QT, Xing RX, Li ZY, Xu LT, Chen LZ. et al. Sphaeropsidin A covalently binds to Cys 151 of Keap1 to attenuate LPS-induced acute pneumonia in mice. Redox Biol. 2025;82:103621
53. Li G, Guan Y, Xu L, Peng G, Han Q, Wang T. et al. Total alkaloids from Thesium chinense inhibit lipopolysaccharide-induced respiratory inflammation by modulating Nrf2/NF-κB/NLRP3 signaling pathway. Chin J Nat Med. 2025;23:421-30
54. Drishya S, Dhanisha SS, Raghukumar P, Guruvayoorappan C. Amomum subulatum mitigates experimental thoracic radiation-induced lung injury by regulating antioxidant status and inflammatory responses. Food Funct. 2023;14:1545-59
55. Guo H, Chen J, Yu H, Dong L, Yu R, Li Q. et al. Activation of Nrf2/ARE pathway by Anisodamine (654-2) for Inhibition of cellular aging and alleviation of Radiation-Induced lung injury. Int Immunopharmacol. 2023;124:110864
56. Zhang M, Lan H, Jiang M, Yang M, Chen H, Peng S. et al. NLRP3 inflammasome mediates pyroptosis of alveolar macrophages to induce radiation lung injury. J Hazard Mater. 2025;484:136740
57. Zhang Y, Zhang H, Li S, Huang K, Jiang L, Wang Y. Metformin Alleviates LPS-Induced Acute Lung Injury by Regulating the SIRT1/NF-kappaB/NLRP3 Pathway and Inhibiting Endothelial Cell Pyroptosis. Front Pharmacol. 2022;13:801337
58. Yang C, Song C, Wang Y, Zhou W, Zheng W, Zhou H. et al. Re-Du-Ning injection ameliorates radiation-induced pneumonitis and fibrosis by inhibiting AIM2 inflammasome and epithelial-mesenchymal transition. Phytomedicine. 2022;102:154184
59. Li X, Gong Y, Li D, Xiang L, Ou Y, Jiang L. et al. Low-Dose Radiation Therapy Promotes Radiation Pneumonitis by Activating NLRP3 Inflammasome. Int J Radiat Oncol Biol Phys. 2020;107:804-14
60. Kumari M, Sharma A, Tirpude NV. Herbacetin ameliorates lipopolysaccharide-elicited inflammatory response by suppressing NLRP-3/AIM-2 inflammasome activation, PI3K/Akt/MAPKs/NF-kappaB redox inflammatory signalling, modulating autophagy and macrophage polarization imbalance. Mol Biol Rep. 2024;51:1159
61. Ma X, Xu D, Ai Y, Ming G, Zhao S. Fas inhibition attenuates lipopolysaccharide-induced apoptosis and cytokine release of rat type II alveolar epithelial cells. Mol Biol Rep. 2010;37:3051-6
62. Reap EA, Roof K, Maynor K, Borrero M, Booker J, Cohen PL. Radiation and stress-induced apoptosis: a role for Fas/Fas ligand interactions. Proc Natl Acad Sci U S A. 1997;94:5750-5
63. Chen T, Chen M, Chen J. Ionizing radiation potentiates dihydroartemisinin-induced apoptosis of A549 cells via a caspase-8-dependent pathway. PLoS One. 2013;8:e59827
64. Lin M, Deng K, Li Y, Wan J. Morphine enhances LPS-induced macrophage apoptosis through a PPARgamma-dependent mechanism. Exp Ther Med. 2021;22:714
65. Zhang Y, Zhang X, Rabbani ZN, Jackson IL, Vujaskovic Z. Oxidative stress mediates radiation lung injury by inducing apoptosis. Int J Radiat Oncol Biol Phys. 2012;83:740-8
66. Speir M, Tye H, Gottschalk TA, Simpson DS, Djajawi TM, Deo P. et al. A1 is induced by pathogen ligands to limit myeloid cell death and NLRP3 inflammasome activation. EMBO Rep. 2023;24:e56865
67. Gu Y, Lv L, Jin J, Hua X, Xu Q, Wu R. et al. STING mediates LPS-induced acute lung injury by regulating ferroptosis. Exp Cell Res. 2024;438:114039
68. Li X, Zhuang X, Qiao T. Role of ferroptosis in the process of acute radiation-induced lung injury in mice. Biochem Biophys Res Commun. 2019;519:240-5
69. Li L, Wu D, Deng S, Li J, Zhang F, Zou Y. et al. NVP-AUY922 alleviates radiation-induced lung injury via inhibition of autophagy-dependent ferroptosis. Cell Death Discov. 2022;8:86
70. Dixon SJ, Olzmann JA. The cell biology of ferroptosis. Nat Rev Mol Cell Biol. 2024;25:424-42
71. Xiang S, Yan W, Ren X, Feng J, Zu X. Role of ferroptosis and ferroptosis-related long non'coding RNA in breast cancer. Cell Mol Biol Lett. 2024;29:40
72. Lee YB, Shin HW, Shrestha S, Kim JK, Jung SJ, Shin MS. et al. Ferroptosis in neutrophils. J Leukoc Biol. 2025
73. Lee H, Gan B. Ferroptosis execution: Is it all about ACSL4? Cell Chem Biol. 2022;29:1363-5
74. Williams AB, Schumacher B. p53 in the DNA-Damage-Repair Process. Cold Spring Harb Perspect Med. 2016 6
75. Biechonski S, Olender L, Zipin-Roitman A, Yassin M, Aqaqe N, Marcu-Malina V. et al. Attenuated DNA damage responses and increased apoptosis characterize human hematopoietic stem cells exposed to irradiation. Sci Rep. 2018;8:6071
76. Sun Y, Aliyari SR, Parvatiyar K, Wang L, Zhen A, Sun W. et al. STING directly interacts with PAR to promote apoptosis upon acute ionizing radiation-mediated DNA damage. Cell Death Differ. 2025
77. Santivasi WL, Xia F. Ionizing radiation-induced DNA damage, response, and repair. Antioxid Redox Signal. 2014;21:251-9
78. Ou HL, Schumacher B. DNA damage responses and p53 in the aging process. Blood. 2018;131:488-95
79. Su L, Dong Y, Wang Y, Wang Y, Guan B, Lu Y. et al. Potential role of senescent macrophages in radiation-induced pulmonary fibrosis. Cell Death Dis. 2021;12:527
80. Rieber M, Rieber MS. Sensitization to radiation-induced DNA damage accelerates loss of bcl-2 and increases apoptosis and autophagy. Cancer Biol Ther. 2008;7:1561-6
81. Liu L, Yi G, Li X, Chen C, Chen K, He H. et al. IL-17A's role in exacerbating radiation-induced lung injury: Autophagy impairment via the PP2A-mTOR pathway. Biochim Biophys Acta Mol Cell Res. 2025;1872:119864
82. Wang L, Wang T, Li H, Liu Q, Zhang Z, Xie W. et al. Receptor Interacting Protein 3-Mediated Necroptosis Promotes Lipopolysaccharide-Induced Inflammation and Acute Respiratory Distress Syndrome in Mice. PLoS One. 2016;11:e0155723
83. Wang J, Luan Y, Fan EK, Scott MJ, Li Y, Billiar TR. et al. TBK1/IKKepsilon Negatively Regulate LPS-Induced Neutrophil Necroptosis and Lung Inflammation. Shock. 2021;55:338-48
84. Xiang Q, Tian Y, Yang K, Du Y, Xie J. Galphaq/11 aggravates acute lung injury in mice by promoting endoplasmic reticulum stress-mediated NETosis. Mol Med. 2025;31:67
85. Sha HX, Liu YB, Qiu YL, Zhong WJ, Yang NS, Zhang CY. et al. Neutrophil extracellular traps trigger alveolar epithelial cell necroptosis through the cGAS-STING pathway during acute lung injury in mice. Int J Biol Sci. 2024;20:4713-30
86. Liu S, Su X, Pan P, Zhang L, Hu Y, Tan H. et al. Neutrophil extracellular traps are indirectly triggered by lipopolysaccharide and contribute to acute lung injury. Sci Rep. 2016;6:37252
87. Teijeira A, Garasa S, Ochoa MC, Sanchez-Gregorio S, Gomis G, Luri-Rey C. et al. Low-Dose Ionizing γ-Radiation Elicits the Extrusion of Neutrophil Extracellular Traps. Clinical Cancer Research. 2024;30:4131-42
88. Denning NL, Aziz M, Gurien SD, Wang P. DAMPs and NETs in Sepsis. Front Immunol. 2019;10:2536
89. Sun Y, Luo M, Guo C, Gao J, Su K, Chen L. et al. [Amentoflavone alleviates acute lung injury in mice by inhibiting cell pyroptosis]. Nan Fang Yi Ke Da Xue Xue Bao. 2025;45:692-701
90. Savran M, Akin SE, Camas HE, Ilhan I, Arlioglu M, Zeynalov T. et al. Protective effect of dapagliflozin on lipopolysaccharide-induced acute lung injury via the SIRT-1/PGC-1alpha pathway. Mol Biol Rep. 2025;52:171
91. Zhang X, Fan H, Su L, Wang Y, Chen G. Mdivi-1 Attenuates Sepsis-Associated Acute Lung Injury by Inhibiting M1 Alveolar Macrophage Polarization and Pyroptosis. Mediators Inflamm. 2025;2025:3675276
92. Li J, Deng SH, Li J, Li L, Zhang F, Zou Y. et al. Obacunone alleviates ferroptosis during lipopolysaccharide-induced acute lung injury by upregulating Nrf2-dependent antioxidant responses. Cell Mol Biol Lett. 2022;27:29
93. Zhou X, Liao Y. Gut-Lung Crosstalk in Sepsis-Induced Acute Lung Injury. Front Microbiol. 2021;12:779620
94. Wang X, Zhang K, Zhang J, Xu G, Guo Z, Lu X. et al. Cordyceps militaris solid medium extract alleviates lipopolysaccharide-induced acute lung injury via regulating gut microbiota and metabolism. Front Immunol. 2024;15:1528222
95. Zhang B, Zhang M, Tian J, Zhang X, Zhang D, Li J. et al. Advances in the regulation of radiation-induced apoptosis by polysaccharides: A review. Int J Biol Macromol. 2024;263:130173
96. Cui M, Xiao H, Li Y, Zhou L, Zhao S, Luo D. et al. Faecal microbiota transplantation protects against radiation-induced toxicity. EMBO Mol Med. 2017;9:448-61
97. Wang X, Lu Y, Cheng X, Zhu X, Li D, Duan H. et al. Local Multiple-site Injections of a Plasmid Encoding Human MnSOD Mitigate Radiation-induced Skin Injury by Inhibiting Ferroptosis. Curr Drug Deliv. 2024;21:763-74
98. Chen HX, Xiang H, Xu WH, Li M, Yuan J, Liu J. et al. Manganese Superoxide Dismutase Gene-Modified Mesenchymal Stem Cells Attenuate Acute Radiation-Induced Lung Injury. Hum Gene Ther. 2017;28:523-32
99. Chu M, Wang H, Bian L, Huang J, Wu D, Zhang R. et al. Nebulization Therapy with Umbilical Cord Mesenchymal Stem Cell-Derived Exosomes for COVID-19 Pneumonia. Stem Cell Rev Rep. 2022;18:2152-63
100. Zhang HH, Kuo WS, Tu PY, Lee CT, Wang HC, Huang YT. et al. Enhancing Lung Recovery: Inhaled Poly(lactic-co-glycolic) Acid Encapsulating FTY720 and Nobiletin for Lipopolysaccharide-Induced Lung Injury, with Advanced Inhalation Tower Technology. ACS Nano. 2025;19:7634-49
101. Sun G, Wu X, Zhu H, Yuan K, Zhang Y, Zhang C. et al. Reactive Oxygen Species-Triggered Curcumin Release from Hollow Mesoporous Silica Nanoparticles for PM(2.5)-Induced Acute Lung Injury Treatment. ACS Appl Mater Interfaces. 2023;15:33504-13
102. Li Y, Zhou B, Liu D, Nie G, Yang F, Chen J. et al. Carbon monoxide gas molecules: Therapeutic mechanisms in radiation-induced lung injury. J Colloid Interface Sci. 2025;688:250-63
103. Thakur P, DeBo R, Dugan GO, Bourland JD, Michalson KT, Olson JD. et al. Clinicopathologic and Transcriptomic Analysis of Radiation-Induced Lung Injury in Nonhuman Primates. Int J Radiat Oncol Biol Phys. 2021;111:249-59
104. Wang X, Kong F, Liu Q, Liu X. EGCG Alleviates Lipopolysaccharide-Induced Septic Shock by Inhibiting NET-Mediated ROS Production by Regulating CXCL2 Expression. Biochemical Genetics. 2025
105. Yuan D, Yang F, Hou L, Zhang Y, Pang X, Du Y. et al. PFKFB2-Driven Glycolysis Promotes Dendritic Cell Maturation and Exacerbates Acute Lung Injury. Advanced Science. 2025
106. Zhang R, Zhong Y, Liu Q, Zhang M, Wang D, Li S. et al. CGRP alleviates lipopolysaccharide-induced ARDS inflammation via the HIF-1α signaling pathway. Clinical Science. 2025;139:373-87
107. Fang Y, Qiu J, Xu Y, Wu Q, Huo XC, Liu SH. Ophiopogonin D Alleviates Sepsis-Induced Acute Lung Injury Through Improving Microvascular Endothelial Barrier Dysfunction via Inhibition of HIF-1alpha-VEGF Pathway. Cell Biochem Biophys. 2025
108. Ding M, Pei P, Liu W, Cao Y, Weng Y, Yu W. Esketamine Regulates Mitophagy through ULK1/FUNDC1 Signaling Pathway to Improve LPS-induced Acute Respiratory Distress Syndrome. Curr Pharm Des. 2025
109. Liu Z, Fang D, Chen K, Dong L, Huang H, Chen Z. Novel mitophagy inducer TJ0113 alleviates pulmonary inflammation during acute lung injury. Frontiers in Pharmacology. 2025 16
110. Bai M, Liang G, Sun R, Dong Y, Geng C, Wang B. et al. Investigating the therapeutic role of anlotinib in radiation-induced lung injury. Naunyn-Schmiedeberg's Archives of Pharmacology. 2025
111. Liu H, Wang J, Zhao J, Gu S, Chen S, Jia W. et al. Potentilla anserina L. polysaccharide ameliorates LPS-induced acute lung injury and relevant intestinal mucosal barrier impairment. Int J Biol Macromol. 2025;305:140667
112. Birukova AA, Meng F, Tian Y, Meliton A, Sarich N, Quilliam LA. et al. Prostacyclin post-treatment improves LPS-induced acute lung injury and endothelial barrier recovery via Rap1. Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease. 2015;1852:778-91
113. Tang J, Ding Y, Chen W, Shi J, Zhang C, Zhao X. et al. VASP Knockdown Ameliorates Lipopolysaccharide-Induced Acute Lung Injury with Inhibition of M1 Macrophage Polarization Through the cGMP-PKG Signaling Pathway. Inflammation. 2025
114. Li LZ, Gu WJ, Yuan XK, Fan WM, Zhang LM, Qu ZK. et al. Blocking the ADAM9/ITGAV Pathway Ameliorates Sepsis-Induced Acute Lung Injury by Promoting Macrophage Efferocytosis. The FASEB Journal. 2025 39
115. Cui J, Deng Y, Li X, Gao L, Li J, Li Z. et al. Herbal-based Xuebijing injection ameliorated vascular endothelial dysfunction via inhibiting ACLY/MYB/RIG-I axis in sepsis-associated lung injury. Phytomedicine. 2025 140
116. Zhang Y, Tian R, Feng X, Xiao B, Yue Q, Wei J. et al. Overexpression of METTL3 in lung cancer cells inhibits radiation-induced bystander effect. Biochem Biophys Res Commun. 2025;761:151714
117. Shen Y, Ye X, Jiang L, Li H, Zhang Y, Wang W. et al. Inhibition of S100A12 Attenuates LPS-Induced Endothelial Barrier Dysfunction in HPMECs through the JAK2/STAT3 Signaling Pathway. Curr Mol Med. 2025
118. Wu S, He Y, Li J, Zhuang H, Wang P, He X. et al. TREM2 alleviates sepsis-induced acute lung injury by attenuating ferroptosis via the SHP1/STAT3 pathway. Free Radic Biol Med. 2025;229:111-26
119. Li Q, Ang Y, Zhou QQ, Shi M, Chen W, Wang Y. et al. Coral calcium hydride promotes peripheral mitochondrial division and reduces AT-II cells damage in ARDS via activation of the Trx2/Myo19/Drp1 pathway. J Pharm Anal. 2025;15:101039
120. Shi Q, Liu H, Wang H, Tang L, Di Q, Wang D. MFGE8 regulates the EndoMT of HLMECs through the BMP signaling pathway and fibrosis in acute lung injury. Respir Res. 2025;26:142
121. Zheng Z, Yang T, Cao H, Yu J, Fang X, He X. et al. Liang-Ge-San drives macrophages toward M2 polarization for alleviating lipopolysaccharide-induced acute lung injury via activating the miR-21/PTEN axis. Fitoterapia. 2025;184:106572
122. Zhu J, Ao X, Liu Y, Zhou S, Hou Y, Yan Z. et al. TNKS1BP1 mediates AECII senescence and radiation induced lung injury through suppressing EEF2 degradation. Respir Res. 2024;25:299
123. Kim JS, Son Y, Jung MG, Jeong YJ, Kim SH, Lee SJ. et al. Geranylgeranylacetone alleviates radiation-induced lung injury by inhibiting epithelial-to-mesenchymal transition signaling. Mol Med Rep. 2016;13:4666-70
124. English J, Dhanikonda S, Tanaka KE, Koba W, Eichenbaum G, Yang WL. et al. Thrombopoietin mimetic reduces mouse lung inflammation and fibrosis after radiation by attenuating activated endothelial phenotypes. JCI Insight. 2024 9
125. Deng S, Yang Y, He S, Chen Z, Xia X, Zhang T. et al. FlaA N/C attenuates radiation-induced lung injury by promoting NAIP/NLRC4/ASC inflammasome autophagy and inhibiting pyroptosis. J Transl Med. 2025;23:237
126. Zhang A, Luo S, Li P, Meng L, Huang L, Cheng H. et al. Urolithin A alleviates radiation pneumonitis by activating PINK1/PRKN-mediated Mitophagy. Int Immunopharmacol. 2025;156:114671
127. Epshtein Y, Mathew B, Chen W, Jacobson JR. UCHL1 Regulates Radiation Lung Injury via Sphingosine Kinase-1. Cells. 2023 12
128. Lim EY, Kim GD, Kim HJ, Eom JE, Song HJ, Shin DU. et al. Cirsium japonicum leaf extract attenuated lipopolysaccharide-induced acute respiratory distress syndrome in mice via suppression of the NLRP3 and HIF1alpha pathways. Phytomedicine. 2025;140:156601
129. Wang L, Lei W, Zhang S, Yao L. MCC950, a NLRP3 inhibitor, ameliorates lipopolysaccharide-induced lung inflammation in mice. Bioorg Med Chem. 2021;30:115954
130. Liu B, Wang Y, Han G, Zhu M. Tolerogenic dendritic cells in radiation-induced lung injury. Front Immunol. 2023;14:1323676
131. Zhang Y, Huang J, Zhang Y, Jiang F, Li S, He S. et al. The Mitochondrial-Derived Peptide MOTS-c Alleviates Radiation Pneumonitis via an Nrf2-Dependent Mechanism. Antioxidants (Basel). 2024 13
132. de Almeida L, Dorfleutner A, Stehlik C. In vivo Analysis of Neutrophil Infiltration during LPS-induced Peritonitis. Bio Protoc. 2016 6
133. Garcia AN, Casanova NG, Valera DG, Sun X, Song JH, Kempf CL. et al. Involvement of eNAMPT/TLR4 signaling in murine radiation pneumonitis: protection by eNAMPT neutralization. Transl Res. 2022;239:44-57
134. Cong C, Niu S, Jiang Y, Zhang X, Jing W, Zheng Y. et al. Renin-angiotensin system inhibitors mitigate radiation pneumonitis by activating ACE2-angiotensin-(1-7) axis via NF-kappaB/MAPK pathway. Sci Rep. 2023;13:8324
135. Wu H, Zhao G, Jiang K, Chen X, Zhu Z, Qiu C. et al. Plantamajoside ameliorates lipopolysaccharide-induced acute lung injury via suppressing NF-kappaB and MAPK activation. Int Immunopharmacol. 2016;35:315-22
136. Xia J, Li J, Deng M, Yin F, Liu J, Wang J. Diosmetin alleviates acute lung injury caused by lipopolysaccharide by targeting barrier function. Inflammopharmacology. 2023;31:2037-47
137. Wang L, Li N, Wang Y, Chen X. Esculin alleviates lipopolysaccharide (LPS)-induced pneumonia by regulating the USP7/MAPK14 axis. J Appl Toxicol. 2024;44:1949-61
138. Chen Y, Zhang Q, Zhou Y, Yang Z, Tan M. Inhibition of miR-182-5p attenuates pulmonary fibrosis via TGF-beta/Smad pathway. Hum Exp Toxicol. 2020;39:683-95
139. Iqbal S, Jabeen F, Aslam N, Manan M. Anti-EMT properties of ergothioneine attenuate lipopolysaccharide-induced oxidative stress-mediated acute lung injury via modulating TGF-beta/smad/snail signaling pathway. Hum Exp Toxicol. 2023;42:9603271231178015
140. Paik SS, Lee JM, Ko I-G, Kim SR, Kang SW, An J. et al. Pirfenidone Alleviates Inflammation and Fibrosis of Acute Respiratory Distress Syndrome by Modulating the Transforming Growth Factor-β/Smad Signaling Pathway. International Journal of Molecular Sciences. 2024 25
141. Xu Y, Zhai D, Goto S, Zhang X, Jingu K, Li TS. Nicaraven mitigates radiation-induced lung injury by downregulating the NF-kappaB and TGF-beta/Smad pathways to suppress the inflammatory response. J Radiat Res. 2022;63:158-65
142. Weng J, Chen Z, Weng S, Guo R, Shi B, Liu D. et al. Study on the mechanism of action of Penehyclidine hydrochloride on LPS-induced acute lung injury by regulating autophagy through the mTOR/Keap1/Nrf2 signaling pathway. J Pharm Biomed Anal. 2025;263:116938
143. Zhang Y, Huang J, Li S, Jiang J, Sun J, Chen D. et al. Pyrroloquinoline Quinone Alleviates Mitochondria Damage in Radiation-Induced Lung Injury in a MOTS-c-Dependent Manner. J Agric Food Chem. 2024;72:20944-58
144. Huang XT, Liu W, Zhou Y, Hao CX, Zhou Y, Zhang CY. et al. Dihydroartemisinin attenuates lipopolysaccharide-induced acute lung injury in mice by suppressing NF-kappaB signaling in an Nrf2-dependent manner. Int J Mol Med. 2019;44:2213-22
145. Wang C, Lin X, Guan S, Wu Q, Liang S. Dihydroartemisinin Attenuates Radiation-Induced Lung Injury by Inhibiting the cGAS/STING/NF-kappaB Signaling Pathway. Drug Dev Res. 2025;86:e70090
146. Huang Y, Li G, Li D, Liu C, Chen M, Cai L. et al. Ethyl caffeate alleviates inflammatory response and promotes recovery in septic-acute lung injury via the TNF-alpha/NF-kappaB/MMP9 Axis. Phytomedicine. 2025;141:156700
147. Zhou X, Bao WA, Zhu X, Lin J, Fan JF, Yang Y. et al. 3,3'-Diindolylmethane attenuates inflammation and fibrosis in radiation-induced lung injury by regulating NF-kappaB/TGF-beta/Smad signaling pathways. Exp Lung Res. 2022;48:103-13
148. Chai Y, Wang Z, Li Y, Wang Y, Wan Y, Chen X. et al. Glycyrrhizin alleviates radiation-induced lung injury by regulating the NLRP3 inflammasome through endoplasmic reticulum stress. Toxicol Res (Camb). 2024;13:tfae009
149. Wang J, Ren C, Bi W, Batu W. Glycyrrhizin mitigates acute lung injury by inhibiting the NLRP3 inflammasome in vitro and in vivo. J Ethnopharmacol. 2023;303:115948
150. Jiang YC, Zhao B, Jiang P, Wang SY, Ma L, Wu GL. et al. Raspberry ketone alleviates radiation-induced lung injury through the STAT2-P2X7r/NLRP3 signaling pathway. Phytomedicine. 2025;145:156984
151. Wang Y, Zhang J, Bo J, Wang X, Zhu J. Hydrogen-rich saline ameliorated LPS-induced acute lung injury via autophagy inhibition through the ROS/AMPK/mTOR pathway in mice. Exp Biol Med (Maywood). 2019;244:721-7
152. Yin Z, Xu W, Ling J, Ma L, Zhang H, Wang P. Hydrogen-rich solution alleviates acute radiation pneumonitis by regulating oxidative stress and macrophages polarization. J Radiat Res. 2024;65:291-302
153. Ni YL, Shen HT, Su CH, Chen WY, Huang-Liu R, Chen CJ. et al. Nerolidol Suppresses the Inflammatory Response during Lipopolysaccharide-Induced Acute Lung Injury via the Modulation of Antioxidant Enzymes and the AMPK/Nrf-2/HO-1 Pathway. Oxid Med Cell Longev. 2019;2019:9605980
154. Karakuyu NF, Ozseven A, Akin SE, Camas HE, Ozmen O, Cengiz C. L-carnitine protects the lung from radiation-induced damage in rats via the AMPK/SIRT1/TGF-1ss pathway. Naunyn Schmiedebergs Arch Pharmacol. 2024;397:8043-51
155. Lai K, Song C, Gao M, Deng Y, Lu Z, Li N. et al. Uridine Alleviates Sepsis-Induced Acute Lung Injury by Inhibiting Ferroptosis of Macrophage. Int J Mol Sci. 2023 24
156. Chen Y, Wu M. Exploration of molecular mechanism underlying protective effect of astragaloside IV against radiation-induced lung injury by suppressing ferroptosis. Arch Biochem Biophys. 2023;745:109717
157. Chen T, Zhuang B, Huang Y, Liu Y, Yuan B, Wang W. et al. Inhaled curcumin mesoporous polydopamine nanoparticles against radiation pneumonitis. Acta Pharmaceutica Sinica B. 2022;12:2522-32
158. Kumari A, Tyagi N, Dash D, Singh R. Intranasal curcumin ameliorates lipopolysaccharide-induced acute lung injury in mice. Inflammation. 2015;38:1103-12
159. Li PY, Liang YC, Sheu MJ, Huang SS, Chao CY, Kuo YH. et al. Alpinumisoflavone attenuates lipopolysaccharide-induced acute lung injury by regulating the effects of anti-oxidation and anti-inflammation both in vitro and in vivo. RSC Adv. 2018;8:31515-28
160. Izumi Y, Nakashima T, Masuda T, Shioya S, Fukuhara K, Yamaguchi K. et al. Suplatast tosilate reduces radiation-induced lung injury in mice through suppression of oxidative stress. Free Radic Biol Med. 2019;136:52-9
161. Wu D, He H, Chen J, Yao S, Xie H, Jiang W. et al. L-carnitine reduces acute lung injury via mitochondria modulation and inflammation control in pulmonary macrophages. Brazilian Journal of Medical and Biological Research. 2023 56
162. Fragoso IT, Ribeiro EL, Gomes FOdS, Donato MAM, Silva AKS, Oliveira ACOd. et al. Diethylcarbamazine attenuates LPS-induced acute lung injury in mice by apoptosis of inflammatory cells. Pharmacological Reports. 2017;69:81-9
163. Farzipour S, Amiri FT, Mihandoust E, Shaki F, Noaparast Z, Ghasemi A. et al. Radioprotective effect of diethylcarbamazine on radiation-induced acute lung injury and oxidative stress in mice. Journal of Bioenergetics and Biomembranes. 2019;52:39-46
Author contact
![]() Corresponding authors: Prof. Wei Xu (Email: weixuedu.cn), Prof. Zhigang Wei (Email: weizhigang321321com), Prof. Xin Ye (Email: yexintaian2020com; ORCID: 0000-0002-4906-7746) and Prof. Qi Xie (Email: xieqiedu.cn; ORCID: 0000-0002-3152-9834).
Corresponding authors: Prof. Wei Xu (Email: weixuedu.cn), Prof. Zhigang Wei (Email: weizhigang321321com), Prof. Xin Ye (Email: yexintaian2020com; ORCID: 0000-0002-4906-7746) and Prof. Qi Xie (Email: xieqiedu.cn; ORCID: 0000-0002-3152-9834).