Impact Factor ISSN: 1449-1907
- Issue 9; 2026
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Volume 23; 2026
- Past Issues
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Tumor Antigens: Targets for...
Platforms for Cancer Vaccine...
Combination Therapies:...
Conclusion
Abbreviations
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2025; 22(16):4406-4417. doi:10.7150/ijms.120868 This issue Cite
Review
Advances in Tumor Antigen Vaccines: A New Frontier in Cancer Immunotherapy
Wanqi Feng1,2,3*, Yifan Zhao4*, Dongran Yu1,2,3*, Wenyu Jia5*, Hui Cao1,2, Yuling Zhang1,2, Jie Cao4 ![]() , Zequn Li1,2
, Zequn Li1,2 ![]()
1. Department of Gastrointestinal Surgery, The Affiliated Hospital of Qingdao University, Qingdao, 266000, China.
2. Gastrointestinal Tumor Translational Medicine Research Institute of Qingdao University, Qingdao, 266000, China.
3. Qingdao Medical College, Qingdao University, Qingdao, 266000, China.
4. Department of Pharmaceutics, School of Pharmacy, Qingdao University, Qingdao, 266071, China.
5. Department of Endocrinology, Qingdao Municipal Hospital, Qingdao, 266071, China.
* These authors contributed equally to the manuscript.
Received 2025-7-3; Accepted 2025-10-6; Published 2025-10-24
Abstract

With the increasing prominence of cancer immunotherapy, therapeutic tumor vaccines have emerged as a promising strategy to enhance antitumor immunity by increasing tumor immunogenicity and activating the patient's immune system to inhibit tumor growth. However, their clinical efficacy is often limited due to insufficient immune cell infiltration, low antigen immunogenicity, and tumor immune escape mechanisms. To address these challenges, various innovative approaches have been explored, including the optimization of tumor antigen selection, the development of advanced vaccine platforms, and the combination of vaccines with other treatment strategies such as radiotherapy, chemotherapy, immune checkpoint inhibitors (ICIs), cytokine therapy, and adoptive T-cell transfer. This review provides a comprehensive summary of the mechanisms underlying tumor antigen vaccines, discusses recent advancements in vaccine design and combinatorial strategies, and assesses their potential to enhance therapeutic outcomes. We also highlight the ongoing challenges and future directions, underscoring the importance of interdisciplinary efforts to realize the full potential of tumor vaccines as a foundation of personalized cancer immunotherapy.
Keywords: Tumor antigen vaccines, Tumor-associated antigens, Tumor-specific antigens, Vaccine platforms, Combination therapy.
Introduction
Cancer remains a major global health challenge, with conventional therapies like surgery, chemotherapy, and radiotherapy often yielding limited efficacy in advanced stages due to systemic toxicity and the development of resistance [1]. Immunotherapy has revolutionized oncology by harnessing the body's immune system to combat cancer. Among these strategies, ICIs and adoptive cell therapy (ACT) have achieved remarkable clinical success. ICIs function by blocking inhibitory pathways to “release the brakes” on pre-existing tumor-specific T cells, while ACT involves the ex vivo expansion and reinfusion of autologous immune cells to directly mediate tumor killing. As shown in Fig. 1, tumor vaccines deliver tumor antigens (proteins, peptides, mRNA, etc.) to the immune system, activate initial T cells, and trigger a long-term immune response against specific antigens, which subsequently leading to an anti-tumor effect [2, 3].
Schematic diagram of the mechanism of therapeutic tumor vaccines
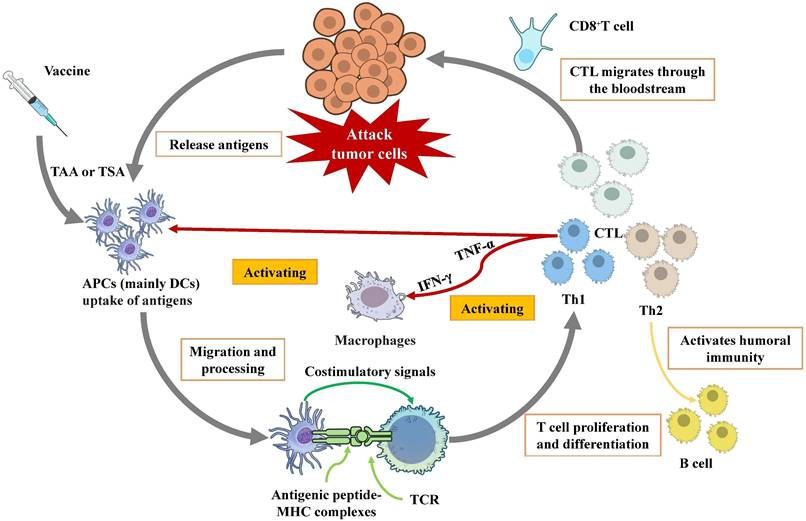
Tumor antigens, broadly categorized as tumor-associated antigens (TAAs) and tumor-specific antigens (TSAs), constitute the basis for vaccine design [4]. Following administration, vaccine antigens are primarily captured by antigen-presenting cells (APCs), with dendritic cells (DCs) being the most crucial type [5]. DCs internalize and process these antigens into short peptides, which are subsequently loaded onto major histocompatibility complex (MHC) molecules. The peptide-MHC complexes are presented on the DC surface [6]. T cell activation requires specific recognition of these complexes by the T-cell receptor (TCR), coupled with essential costimulatory signals. Once activated, T cells proliferate and differentiate into distinct functional subsets, primarily cytotoxic T lymphocytes (CTLs) and helper T cell [7]. CTL has the capacity to recognize and directly kill tumor cells that express corresponding antigens. This process can occur in three ways: (1) the secretion of pro-inflammatory cytokines, (2) the interaction between Fas ligands and Fas receptors, and (3) the secretion of perforin-containing cytolytic granules [8]. Th cells differentiate into Th1 and Th2 subsets. Th1 cells, characterized by IFN-γ and TNF-α secretion, are pivotal for activating and sustaining CTLs, enhancing APC function, and inducing specific antibodies that promote phagocytosis of infected or tumor cells. Th2 cells primarily support humoral immune responses [9].
However, tumors have developed various mechanisms, such as antigen modulation, abnormal expression of immune checkpoint molecules, immunosuppressive cell aggregation, cytokine environment modification, and tumor cell heterogeneity, which pose significant challenges to the efficacy of tumor antigen vaccines [10].
To overcome these challenges, considerable efforts are focused on optimizing antigen selection, developing novel vaccine platforms, and combining vaccines with other modalities. This review provides a comprehensive overview of the recent advances in tumor antigen vaccines, discussing the complexities of antigen selection, platform technologies, and combinatorial strategies that show promise for the future of cancer immunotherapy.
Tumor Antigens: Targets for Immunotherapy
During carcinogenesis, genetic alterations in normal tissues can lead to the aberrant expression of specific proteins. These tumor-associated antigens are differentially expressed between tumor and normal cells and are recognized by the immune system as foreign, thereby eliciting an immune response against cancer cells [11]. Such antigens serve as key targets for developing therapeutic cancer vaccines. However, most tumor antigens are endogenous in origin, which renders them less immunogenic compared to conventional exogenous antigens. Additionally, the heterogeneity of tumor antigens further challenges the identification of optimal targets for vaccination [12, 13]. As is shown in Table 1, tumor antigens are broadly classified into two categories: TAAs and TSAs.
The characteristics of TSAs and TAAs
| TSAs | TAAs | |
|---|---|---|
| Expression | Expressed in tumor cells while normal cells do not express Can be produced by carcinogenic viruses | Expressed in both tumor cells and some normal cells Usually highly expressed in tumor cells |
| Origination | Novel polypeptide chains with mutations primarily driven by cancer | Mainly from gene amplification or post-translational modifications |
| Example | HPV E6/E7 viral neoantigen | Cancer-testis antigens and oncofetal antigens |
TAAs: Characteristics and Clinical Challenges
Overexpressed antigens are self-proteins present at substantially elevated levels in tumor cells compared to normal tissues. Representative examples encompass carcinoembryonic antigen (CEA), prostate-specific antigen (PSA), melanoma antigen recognized by T cells 1 (MART-1), and gp100 [14-16]. New York esophageal squamous cell carcinoma 1 (NY-ESO-1) and melanoma-associated antigen family A (MAGE-A), exhibit expression largely confined to immune privileged sites (e.g., testes and ovaries), thereby rendering them attractive targets for vaccine design [17, 18]. Oncofetal antigens, normally expressed during fetal development and silenced in adults, can be re-expressed in certain cancers. Targeting strategies leveraging such antigens are exemplified by chimeric antigen receptor (CAR) T-cell therapy [19].
Nevertheless, immunotherapy targeting these distinctive TAAs faces several challenges. Clinical trials evaluating various cancer vaccines based on MAGE-A and NY-ESO-1 antigens across multiple malignancies (e.g., lung, bladder, and skin cancers) indicate that further investigation is necessary to establish their optimal clinical safety, efficacy, and tolerability [20, 21]. These findings highlight the necessity of more clearly elucidating the expression mechanisms of TAAs in both normal and malignant contexts. Moreover, additional clinical studies are essential to refine immunotherapeutic strategies and improve outcomes.
To enhance immunogenicity while mitigating autoimmune toxicity, attention is increasingly directed toward TAAs resulting from post-translational modifications (PTMs) or alternative splicing [22-24]. PTMs involve chemical alterations of amino acid residues, whereas alternative splicing generates unique protein isoforms through variant exon inclusion. Promising TAAs originating from these mechanisms include CD44v6, STn, and O-GD2 [25].
From TSAs to Personalized Vaccines
TSAs, also known as neoantigens, represent a distinct and highly advantageous class. A defining characteristic of TSAs is their exclusive expression in cancer cells and absence in normal tissues. This tumor-restricted expression classifies them as exogenous antigens, presenting high immunogenicity and capacity to stimulate CD4+ and CD8+ T cells, thereby eliciting potent immune responses. Crucially, TSAs do not typically induce autoimmune reactions-a key advantage over TAAs. Moreover, they evade central and peripheral immune tolerance mechanisms. Furthermore, central and peripheral immune tolerance does not affect TSAs. As a result, TSA-targeting vaccines hold strong potential for improved efficacy and safety profiles [26].
Emerging clinical evidence supports the promise of TSA vaccines. The TSA vaccine based on uridine mRNA-lipoplex nanoparticles in combination with atezolizumab and chemotherapy induced TSA-specific immune responses that significantly delayed the recurrence of pancreatic cancer. Of note, the personalized mRNA TSA vaccine could be completed within nine weeks, suggesting that mRNA cancer vaccines can be integrated into post-surgery clinical care [27]. Given the high heterogeneity of tumor cells, many cancers have been treated with tumor vaccines that target TSAs individually. For example, the synthetic long peptide vaccine for TSAs (#NCT03121677) for follicular lymphoma, the TSAs pulsed DC vaccine (#NCT02956551) for metastatic lung cancer [28, 29]. The employment of rapid and effective bioassays facilitates the selection of TSAs with high immunogenicity for each patient.
However, the development of personalized TSA vaccines remains costly and time-intensive. Advances in biomonitoring technologies, novel algorithms, and machine learning are urgently needed to accelerate mutation identification and enhance the screening of T-cell-recognizable epitopes. Beyond personalized approaches, the identification of “public” TSAs offers a promising path toward developing broadly effective, off-the-shelf TSA vaccines. Public TSAs often arise in essential driver genes regulating tumor growth, making them potent therapeutic targets [30, 31]. Leveraging pan-cancer proteo-genomics, refined MHC binding prediction, single-cell transcriptomics, and TCR sequencing, researchers are continuously expanding the repertoire of public TSAs. Examples include mutant KRAS peptides, altered driver gene products and RPL22 variants resulting from RNA splicing aberrations across diverse cancers [32-34].
Platforms for Cancer Vaccine Development
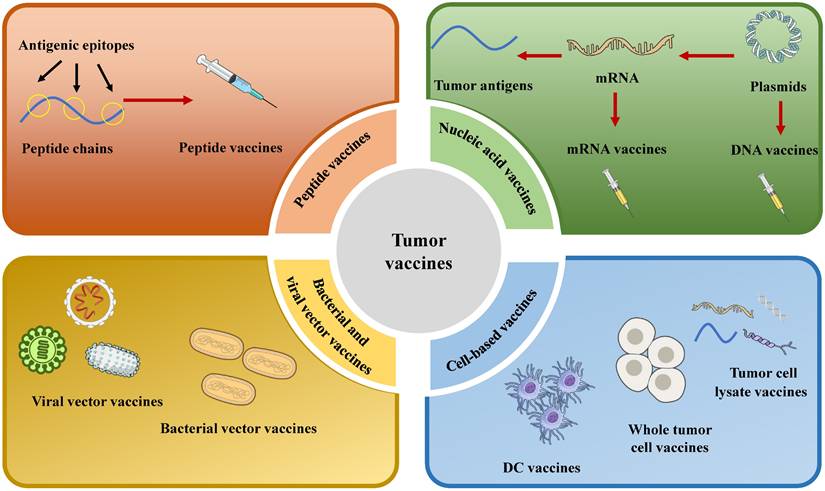
Selecting an optimal vaccine platform requires careful consideration of multiple factors. Moreover, it is essential to integrate a variety of tumor antigens with multiple vaccine platforms. Vaccine platforms are broadly classified into four types based on their fundamental design: cell-based cancer vaccines, peptide vaccines, viral and bacterial vector vaccines, and nucleic acid vaccines (Fig. 2).
Schematic diagram of the classification of therapeutic tumor vaccines
Peptide Vaccines: Precision and Limitations
Peptide vaccines typically consist of short sequences of approximately 25 amino acids. Shorter peptides exhibit excessively brief half-lives and are highly soluble in serum, which limits their efficacy. A primary objective of these vaccines is to activate CTLs, particularly CD8+ T cells, due to the crucial role of T-cell immunity in cancer immunotherapy. This activation primarily occurs through cross-presentation by APCs, a process crucial for mediating antitumor responses [35, 36].
Peptide vaccines offer several advantages, such as high specificity, a reduced risk of autoimmune reactions, and high safety. For example, the amphiphilic vaccine Amph-CpG-7909 (targeting mKRAS with a CpG adjuvant) was well-tolerated and induced mKRAS-specific T-cell responses in 84% of patients (21/25) [37]. Another first-in-human study of the peptide vaccine TAS0313 patients with advanced solid tumors confirmed its safety, tolerability, and ability to induce immune response [38]. Moreover, the direct binding of defined peptide epitopes to MHC molecules makes peptide vaccines valuable tools for identifying novel tumor-specific T cell epitopes, enabling efficient in vitro screening using APCs and T cells or peptide-MHC complex analyses.
However, peptide vaccines also face significant limitations, including complex manufacturing processes, high production costs, and a tendency to elicit weak immune responses that are often insufficient within the immunosuppressive tumor microenvironment [39, 40]. To address these limitations, self-assembling peptide vaccines have emerged as a promising advanced platform. By leveraging non-covalent interactions to form well-ordered nanostructures (e.g., nanofibers, nanoparticles, hydrogels), self-assembling peptides can present high densities of antigens in a multivalent manner, enhancing uptake by APCs and potently activating both cellular and humoral immunity without the need for additional adjuvants. This platform integrates the specificity of peptide vaccines with improved immunogenicity and stability, presenting a robust approach to cancer immunotherapy [41].
Nucleic Acid Vaccines: A Modern Modality
Nucleic acid vaccines, comprising both DNA and RNA platforms, function by delivering genetic material that encodes tumor antigens into host cells to elicit an immune response. The global response to the COVID-19 pandemic has accelerated significant advancements in nucleic acid vaccine technology, exemplified by the widespread clinical use of mRNA vaccines against SARS-CoV-2. This milestone highlights the translational potential of nucleic acid vaccines. Importantly, similar to other vaccine modalities, they demonstrate considerable promise for integration into cancer immunotherapy strategies [42].
DNA Vaccines: Challenges in Sustained Expression and Delivery
A key advantage of DNA vaccines is their capacity to enable sustained expression of tumor antigens, thereby inducing more prolonged immunogenicity compared to peptide platforms. Advancements in plasmid engineering have significantly enhanced this technology, with contemporary constructs incorporating strong promoters and immunostimulatory CpG motifs to enable the efficient and cost-effective expression of multiple antigens [43, 44].
Despite these advantages, the clinical application of DNA vaccines faces several limitations. A significant challenge is their suboptimal transfection efficiency, which is partly attributable to variations in the composition of cellular and nuclear membranes across different cell types. Plasmids must enter the cytoplasm via pinocytosis or endocytosis without being degraded-a process that remains inefficient. These delivery challenges contribute to issues such as low immunogenicity, potential autoimmune reactions, and the risk of host genomic integration [45]. Physical delivery methods, including electroporation, gene guns, and sonoporation, are commonly employed to enhance plasmid uptake; however, their efficacy remains limited [46-48]. To address these challenges, researchers are developing non-viral nanoparticle-based delivery systems utilizing lipid nanoparticles and cationic polymers [49].
Furthermore, certain adjuvants have proven effective for enhancing DNA vaccine efficacy, particularly nano-agonists targeting the stimulator of interferon genes (STING) pathway. The recognition of cytosolic DNA is mediated through the STING signaling axis, which induces type I interferon (IFN) production and contributes to the self-adjuvating properties of DNA plasmids. In preclinical studies, STING agonists, such as those based on manganese-doped silica nanoparticles, have been shown to enhance immune responses induced by plasmid DNA vaccines. These agents also facilitate DC activation and migration, thereby enhancing anti-tumor immunity [50].
RNA Vaccines: Rapid Development and Clinical Translation
Four primary classes of RNA have been employed in vaccine development: messenger RNA (mRNA), circular RNA (circRNA), trans-amplifying RNA (taRNA), and virus-derived self-amplifying RNA (saRNA) [51]. Among these, saRNA is particularly notable due to its ability to replicate within cells, leading to amplified antigen expression compared to conventional non-replicating mRNA. This property results in a prolonged intracellular half-life and lower required doses [52]. Conventional mRNA vaccines contain encoded antigen sequences flanked by 5' and 3' untranslated regions but do not self-replicate. mRNA vaccines exhibit a favorable safety profile-being non-infectious, non-integrating, and biodegradable-while also offering advantages in rapid, cost-effective development. These advantages-especially their rapid development timeline and manufacturing scalability-have established mRNA platforms as leading candidates for clinical translation, with numerous vaccine candidates currently progressing through clinical trials (Table 2).
Clinical trials on mRNA vaccines
| Sponsoring institution | Tumor types | Study phase | Clinical Trials. gov identifier |
|---|---|---|---|
| University of Florida | Recurrent adult glioblastoma | Phase I | NCT06389591 |
| University of Florida | Pediatric recurrent intracranial malignancies and other systemic solid tumors | Phase I | NCT05660408 |
| University of Florida | Pediatric high-grade gliomas and adult glioblastoma | Phase I | NCT04573140 |
| Chinese Academy of Medical Sciences | Advanced solid tumors | Phase I | NCT06610227 |
| Duke University | WHO grade IV malignant glioma | Phase I | NCT05283109 |
| BioNTech Cell & Gene Therapies GmbH | CLDN6-positive relapsed or refractory advanced solid tumors | Phase I | NCT04503278 |
| University of Florida | Medulloblastoma | Phase I | NCT06514898 |
| BioNTech SE | Head and neck cancer | Phase II | NCT04534205 |
mRNA vaccine technology has advanced rapidly in recent years. The carcinoembryonic antigen Claudin 6 (CLDN6), for instance, is highly expressed in a variety of solid tumors [53]. One study evaluated the efficacy of CAR T cells targeting CLDN6 in combination with an amplifying RNA vaccine (CARVac). The CARVac vaccine consists of a nucleoside-modified mRNA encoding CLDN6, packaged into lipid nanoparticles (LNPs) to enhance delivery efficiency and stability [54]. These LNPs protect mRNA from enzymatic degradation and facilitate its uptake by APCs. It has been shown that complexing mRNA with positively charged liposomes forms RNA-LPX, which shields the RNA from nucleases and promotes efficient entry into APCs. Moreover, LNPs enhance vaccine immunogenicity by improving antigen uptake and presentation. Clinical trials have corroborated these findings, indicating favorable safety and immunogenicity profiles. The BNT211-01 trial demonstrated that although the combination of CLDN6 CAR-T and CARVac was safe, the assessment of efficacy was limited by the small sample size, high patient heterogeneity, and the absence of control groups, thereby complicating the isolation of the vaccine's contribution. The delayed vaccination likely missed the optimal window for immune stimulation and was unable to compensate for lymphodepletion or counteract the immunosuppressive tumor microenvironment. The absence of predictive biomarkers further limits clinical applicability. Future trials should optimize dosing schedules, increase cohort sizes, and investigate the sequencing of combination therapies [55].
Despite these advances, mRNA vaccines face several challenges, including limited stability, intricate regulatory pathways, suboptimal immunogenicity, inefficient delivery, and difficulties in production and storage. To overcome these limitations, researchers are pursuing strategies such as optimizing mRNA sequence and structure, refining delivery systems, and elucidating underlying immune mechanisms. Efforts are also underway to improve manufacturing processes and storage conditions to enable safe and effective vaccine deployment [56, 57].
circRNAs represent a distinct class of single-stranded RNAs with covalently closed circular structures. This conformation confers resistance to exonuclease degradation and enables sustained protein expression in vivo. Preclinical studies of circRNA encapsulated in LNPs have demonstrated potent antitumor effects in mouse models, owing to improved cytosolic delivery and reduced innate immunogenicity compared to linear mRNA [58, 59].
Cell-Based Vaccines: Harnessing Cellular Immunity
Cell-based cancer vaccines represent a class of immunotherapies that utilize whole cells-either as carriers or as central immunogenic components-to elicit anti-tumor immune responses. This category primarily includes tumor cell vaccines and DC vaccines.
Tumor cell vaccines are prepared from autologous or allogeneic tumor cells. Whole-cell vaccines employ tumor cells that have been inactivated through physical, chemical, or genetic methods to eliminate tumorigenicity while preserving immunogenicity. Alternatively, tumor cell lysate vaccines utilize solubilized tumor materials containing a broad spectrum of TAAs, which may help overcome antigenic heterogeneity and immune escape [60-62].
DC vaccines leverage the potent antigen-presenting capacity of DC to stimulate anti-tumor immunity. These vaccines are produced by loading TAAs or tumor-specific antigens onto DCs ex vivo, which are then reinfused to activate T-cell responses against tumor cells. In 2010, the U.S. FDA approved Sipuleucel-T, the first therapeutic DC vaccine for metastatic prostate cancer, marking a milestone in cell-based immunotherapy [63]. Nonetheless, its clinical application remains limited, and no other DC vaccine has subsequently obtained FDA approval.
Nevertheless, DC vaccines have exhibited promising safety and efficacy in numerous studies, thereby supporting their assessment across an expanding spectrum of cancers. For example, a single-arm, dual-center pilot study (ChiCTR-ONC-16009100, NCT02956551) involving advanced lung cancer reported favorable outcomes following the use of a personalized TSA-pulsed autologous DC vaccine (Neo-DCVac) [29]. DC vaccines continue to face significant challenges in achieving consistent clinical efficacy. Other clinical trials have explored DC vaccines targeting single antigens such as WT1, HER2, and IL-13Rα2 in glioma, with some showing preliminary efficacy. To address tumor heterogeneity, multi-antigen DC vaccines such as ICT-107 have been developed and have demonstrated promising results in early-phase clinical trials [64, 65]. Moreover, in cancers such as colorectal carcinoma, the majority of patients have not experienced substantial survival benefits from DC vaccines, highlighting the necessity for enhanced strategies in vaccine design, delivery, and combination therapies to overcome immunosuppressive tumor microenvironments [66].
A significant technical challenge in the production of DC vaccines is the limited availability of peripheral blood DCs. Second-generation DC vaccines often use monocyte-derived DCs (Mo-DCs) as a more feasible source of APCs. However, Mo-DCs exhibit significant functional and genetic differences compared to natural DCs, including impaired cross-presentation, limited migratory capacity, and poor survival after infusion, resulting in inefficient lymph node homing and T-cell priming. The limited availability and low purity of other DC subsets, coupled with technical challenges in their isolation and culture, further constrain broader application [67, 68]. In addition, the researchers have developed a novel whole tumor cell vaccine platform, which successfully solves the two core bottlenecks of traditional whole tumor cell vaccines through intracellular gelation technology combined with cell surface engineering of CD47 blockade and damage-related molecular pattern exposure [69].
Viral and Bacterial Vectors: Engineered Delivery Platforms
Viral and bacterial vector vaccines employ engineered viruses or bacteria to deliver genes encoding pathogen antigens into host cells. Through infection, these vectors facilitate the expression of target antigens, thereby eliciting specific immune responses. A key advantage of such vaccines is their high immunogenicity, enabling efficient cellular entry and the induction of durable immunity.
Viruses attenuated to eliminate pathogenicity while maintaining infectivity enable highly efficient gene delivery. Among these, oncolytic viruses (OVs), which selectively replicate in cancer cells, have demonstrated considerable clinical potential. For instance, vaccinia virus (VV), a member of the Poxviridae family, has been utilized both as a vaccine vector and an oncolytic agent. Preclinical and early-phase clinical studies indicate that VV-based vaccines, particularly in combination with standard treatments such as ICIs, exhibit notable efficacy in advanced non-small cell lung cancer and metastatic breast cancer [70, 71]. Several OV therapies, including Rigvir and Oncorine, have already obtained regulatory approval [72, 73]. Nonetheless, selecting an optimal viral vector involves balancing safety and immunogenicity: vesicular stomatitis virus (VSV) offers high immunogenicity but raises biosafety concerns; parainfluenza virus (PIV) vectors exhibit improved safety profiles but relatively weaker immune stimulation; and adenoviral vectors (AdVs) strike a balance between immunogenicity and sustained antigen presentation [74].
Regarding bacterial vectors, gram-negative bacteria secrete outer membrane vesicles (OMVs), nanoscale bilayer structures enriched with immunostimulatory components such as lipopolysaccharides and proteins. Through genetic engineering, OMVs can be designed to display tumor antigens, forming customizable vaccine platforms. “Plug-and-Display” technology enables rapid antigen conjugation to ClyA proteins on OMV surfaces, facilitating simultaneous DC activation and antigen cross-presentation. In preclinical models of melanoma and colorectal cancer, OMV-based vaccines have been shown to induce robust T-cell responses, suppress tumor growth and metastasis, and establish long-term immunological memory. Although OMV-based approaches date back to early tuberculosis vaccine research in the 20th century, advances in genetic engineering have now unlocked their full therapeutic potential.
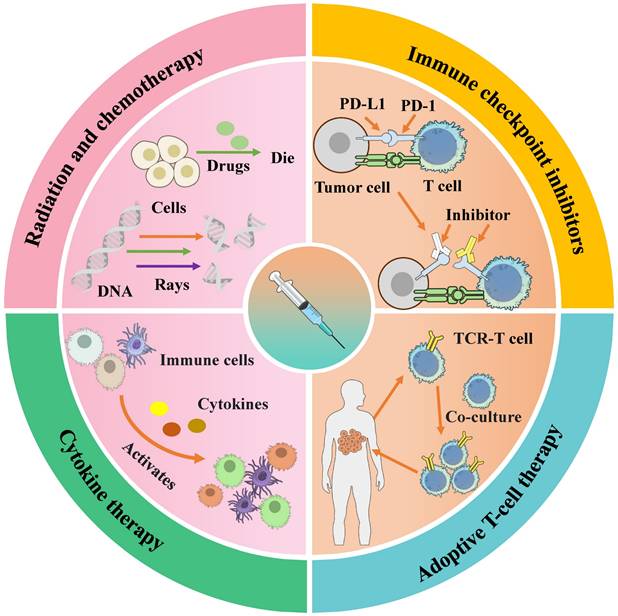
Combination Therapies: Synergistic Strategies
To overcome the inherent limitations of monotherapy, tumor antigen vaccines are increasingly being integrated with established and emerging cancer treatments. These combinations aim to remodel the tumor microenvironment and amplify antigen-specific immunity, as summarized in Fig. 3. The combination of tumor antigen vaccines with other modalities aims to overcome specific barriers in the cancer immunity cycle. The advantages, and limitations of these strategies are summarized in Table 3.
Comparison of combination strategies with tumor antigen vaccines
| Combination strategy | Key advantages | Limitations and challenges |
|---|---|---|
| Radiotherapy | Create an in situ vaccine effect; Reverse “cold” tumors to “hot” tumors; Provide localized and focused immune activation. | Abscopal effect is rare; Potential for systemic immunosuppression with high doses; Toxicity to healthy tissues. |
| Chemotherapy | Employ lymphodepletion to enhance homeostatic cytokine-driven T-cell expansion; Synergize with various vaccine platforms. | Cytotoxicity can deplete immune effectors; Triggers systemic toxicity; Careful timing required. |
| ICIs | Address a key mechanism of vaccine resistance; Generate durable, memory responses; “Rescue” dysfunctional T cells. | Risk of irAEs; High cost; Requires pre-existing T-cell infiltration. |
| Cytokine therapy | Directly amplify the immune response initiated by the vaccine; Enhance NK cell cytotoxicity. | Severe systemic toxicity; Short half-life; Pleiotropic effects. |
ICIs: Immune checkpoint inhibitors; irAEs: immune-related adverse events.
Schematic diagram of therapies used in combination with tumor vaccines
Radiation and Chemotherapy: Priming the Immune Landscape
Radiation therapy (RT) is a widely used modality for treating malignant tumors through the application of high-energy radiation. Its fundamental mechanism involves inducing DNA damage in tumor cells, thereby inhibiting their proliferation and survival. The primary goals of RT are to eradicate tumor cells and reduce tumor volume [75].
RT not only reduces tumor burden but also enhances the infiltration of effector immune cells into the tumor microenvironment, thereby addressing a key limitation of immunotherapy. Consequently, combining RT with vaccines represents a promising strategy to amplify anticancer immunity [76]. For instance, the novel adeno-associated virus (AAV)-based vaccine “meAAV”, when administered alongside RT, improves antigen presentation and sustains TSA-specific CTL responses. This combination therapy significantly enhances CTL activity, promotes their infiltration into tumors, and alleviates local immunosuppression [77]. In a 4T1 breast cancer mouse model, the combination of stereotactic ablative radiotherapy and a cancer vaccine targeting fibroblast-activating protein-alpha (FAP-α) effectively suppressed metastatic growth [78].
Chemotherapy, a fundamental component of cancer treatment, employs cytotoxic agents to systematically target and eliminate rapidly dividing tumor cells [79]. However, its efficacy is often compromised by tumor heterogeneity and the emergence of drug resistance. Growing evidence supports the combination of chemotherapy with immunotherapy to improve antitumor responses [80, 81]. For instance, the personalized TSA vaccine NEO-PV-01, administered in combination with chemotherapy and the anti-PD-1 antibody pembrolizumab as a first-line treatment for metastatic non-squamous non-small cell lung cancer, was well tolerated and elicited TSA-specific CD4+ T cell responses [82]. A phase II study involving patients with metastatic androgen-independent prostate cancer demonstrated that the combination of docetaxel (DTX) and a vaccine-based immunotherapy regimen was safe, did not compromise vaccine-induced T cell responses, and resulted in improved clinical outcomes [80].
The integration of chemoradiation with tumor antigen vaccines has been applied in clinical settings, demonstrating enhanced treatment efficacy. This multimodal approach helps overcome the lack of specificity associated with conventional chemoradiation and chemotherapy, thereby advancing the field of precision medicine.
ICIs: Releasing the Brakes on Immunity
Combining tumor antigen vaccines with ICIs represents a rational strategy to counteract tumor-induced T-cell exhaustion. As shown in Table 3, vaccines prime and expand antigen-specific T cells, ICIs remove inhibitory signals, enabling robust and durable cytotoxic responses. These inhibitory signals encompass not only the classical PD-1/PD-L1 pathway but also innate immune checkpoints mediated by macrophages, such as the CD47-SIRPα axis and the emerging CD24-Siglec10 axis. CD24, highly expressed on various tumor cells, binds to Siglec-10 on macrophages, transmitting a “don't eat me” signal that inhibits phagocytosis and facilitates tumor immune escape. Targeting this axis has become a new hotspot in cancer immunotherapy [2]. By interfering with inhibitory signals from tumor cells to immune cells, ICIs facilitate the targeting and destruction of tumors by activated T cells [83]. The combination of tumor antigen vaccines with ICIs has been shown to improve the immune system's ability to recognize and eliminate cancer cells.
Preclinical studies have demonstrated a potent synergistic effect between tumor antigen vaccines and ICIs, leading to substantial suppression of tumor growth. For example, in hepatocellular carcinoma (HCC) models, the combination of an alpha-fetoprotein (AFP) vaccine and anti-PD-L1 therapy led to significant inhibition of tumor progression in the majority of liver lesions, indicating that vaccine-primed T-cell responses can be effectively activated by checkpoint blockade [84]. In clinical settings, a phase II trial involving colorectal cancer patients revealed that the GVAX colon vaccine combined with cyclophosphamide and pembrolizumab induced biochemical responses (≥30% reduction in CEA levels) in 41% (7/17) of patients with mismatch repair-proficient tumors [85]. Although the trial did not meet its primary endpoint, the considerable biochemical response rate highlights the potential of combinatorial immunotherapy even in immunologically “cold” tumors, supporting further investigation of vaccine-ICI strategies beyond hypermutated cancers.
Beyond HCC and colorectal cancer, this combinatorial strategy has shown efficacy in various other malignancies. In metastatic castration-resistant prostate cancer (mCRPC), the integration of ICIs with androgen deprivation therapy (ADT) and tumor vaccines has exhibited promise not only for prostate cancer but also for melanoma and lung cancer [86]. Second-generation ICIs combined with Treg depletion strategies and murine cancer vaccines synergized in a CD8+ T cell-dependent manner, reducing tumor growth and improving survival [87]. In Mlh1-deficient mice with gastrointestinal tumors, combining a tumor vaccine with anti-PD-L1 therapy significantly extended survival, increased T cell infiltration, and reduced macrophages, neutrophils, and myeloid-derived suppressor cells (MDSCs) [88].
Overall, the combination of tumor antigen vaccines with ICIs holds considerable potential for enhancing treatment outcomes and advancing personalized and precision cancer medicine.
Cytokine Therapy: Amplifying Immune Signals
Cytokines are a diverse class of small, soluble polypeptides or glycoproteins that stimulate and regulate the activation and proliferation of immune cells. Although cytokines can enhance immune responses, they do not directly target tumors and rely on the host's pre-existing immune activity. Consequently, although cytokines demonstrate limited efficacy as monotherapies, they can substantially enhance antitumor immunity when used in conjunction with tumor vaccines [89].
Interleukin-15 (IL-15) has encountered obstacles in clinical application, including systemic toxicity, insufficient immune stimulation, and a short half-life, issues that are largely attributable to its widespread receptor distribution and rapid clearance. To overcome these challenges, a biomimetic nano-vaccine termed biNV-IL-15 has been developed. This platform consists of genetically engineered DC membrane vesicles that enable targeted delivery of both IL-15 and antigen/MHC complexes to antigen-specific CTLs, facilitating multivalent IL-15 self-presentation. Preclinical studies indicate that biNV-IL-15 effectively activates CD8+ T cells in vitro, extends systemic circulation, enhances accumulation in lymphoid organs, and promotes durable immune memory-all while exhibiting a favorable safety profile [90].
In summary, combining cytokine therapy (particularly IL-15) with cancer vaccines enhances antigen-specific immunity and reduces systemic toxicity. Furthermore, the integration of IL-15 with ICIs may also improve immunotherapeutic outcomes. Innovative delivery platforms, such as the biNV-IL-15 nano-vaccine, demonstrate that cytokine delivery can be precisely targeted to enhance efficacy and minimize systemic toxicity, paving the way for their clinical translation in combination regimens.
Conclusion
Despite significant progress in tumor antigen vaccines, key challenges persist. These include high research and manufacturing costs, limited access to personalized vaccines, tumor immune evasion mechanisms, and potential autoimmune reactions. Addressing these issues requires interdisciplinary collaboration among immunology, oncology, genetics, and engineering. Further clinical studies are also essential to improve long-term safety and efficacy.
In summary, therapeutic tumor antigen vaccines represent a major advance in cancer immunotherapy, offering a targeted and individualized treatment approach. By utilizing the immune system to eliminate tumor cells, they hold transformative potential for improving patient outcomes. Continued innovation in technology and deeper understanding of tumor biology and immunology are expected to drive future progress, potentially establishing these vaccines as a standard cancer treatment option.
Abbreviations
ICIs: immune checkpoint inhibitors; ACT: adoptive cell therapy; CAR-T cells: chimeric antigen receptor T cells; TAAs: tumor-associated antigens; TSAs: tumor-specific antigens; APCs: antigen-presenting cells; DCs: dendritic cells; MHC: major histocompatibility complex; TCR: T-cell receptor; CTLs: cytotoxic T lymphocytes; Th1: T helper 1 cells; IFN-γ: interferon-γ; TNF-α: tumor necrosis factor-α; CEA: carcinoembryonic antigen; PSA: prostate-specific antigen; MART-1: melanoma antigen recognized by T cells 1; NY-ESO-1: New York esophageal squamous cell carcinoma 1; MAGE-A: melanoma-associated antigen family A; PTMs: post-translational modifications; KRAS: Kirsten rat sarcoma viral oncogene homolog; RPL22: ribosomal protein L22; COVID-19: coronavirus disease-2019; SARS-CoV-2: severe acute respiratory syndrome coronavirus 2; CpG motifs: cytosine-phosphate-guanine motifs; STING: stimulator of interferon genes; mRNA: messenger RNA; circRNA: circular RNA; saRNA: self-amplifying RNA; CLDN6: claudin 6; LNPs: lipid nanoparticles; FDA: food and drug administration; HER2: human epidermal growth factor receptor 2; IL-13Rα2: interleukin-13 receptor alpha 2; Mo-DCs: monocyte-derived dendritic cells; Ovs: oncolytic viruses; VV: vaccinia virus; VSV: vesicular stomatitis virus; PIV: parainfluenza virus; AdVs: adenoviral vectors; OMVs: outer membrane vesicles; RT: radiation therapy; AAV: adeno-associated virus; FAP-α: fibroblast-activating protein-α; DTX: docetaxel; PD-1: programmed death protein 1; PD-L1: programmed death-ligand 1; HCC: hepatocellular carcinoma; AFP: α-fetoprotein; mCRPC: metastatic castration-resistant prostate cancer; ADT: androgen deprivation therapy; MDSCs: myeloid-derived suppressor cells; IL-15: interleukin-15.
Acknowledgements
Funding
This work was supported by Young Talent of Lifting engineering for Science and Technology in Shandong, China (No. SDAST2024QTA032), Wu Jieping Medical Foundation Clinical Research Special Fund (No. 320.6750.2024-17-41).
Availability of Data and Materials
All the related data are available upon request by contacting with the corresponding author.
Consent for publication
All authors read and approved the final manuscript.
Author Contributions
ZL and JC designed the study, ZL acquired fundings, WF, YZ and DY wrote the original draft, WJ contributed to the figures and tables, HC and YZ collected data, ZL, JC and YZ reviewed the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Liu B, Zhou H, Tan L, Siu KTH, Guan XY. Exploring treatment options in cancer: Tumor treatment strategies. Signal Transduct Target Ther. 2024;9:175
2. Fang J, Lin L, Cao Y, Tan J, Liang Y, Xiao X. et al. Targeting the CD24-Siglec10 Axis: A Potential Strategy for Cancer Immunotherapy. BIO Integration. 2024 5
3. Cao S, Jia W, Zhao Y, Liu H, Cao J, Li Z. A recent perspective on designing tumor vaccines for tumor immunology. Int Immunopharmacol. 2024;142:113090
4. Malacopol AT, Holst PJ. Cancer Vaccines: Recent Insights and Future Directions. Int J Mol Sci. 2024 25
5. Gardner A, Ruffell B. Dendritic Cells and Cancer Immunity. Trends Immunol. 2016;37:855-65
6. Palucka K, Banchereau J. Cancer immunotherapy via dendritic cells. Nat Rev Cancer. 2012;12:265-77
7. Wik JA, Skålhegg BS. T Cell Metabolism in Infection. Front Immunol. 2022;13:840610
8. Berke G. The CTL's kiss of death. Cell. 1995;81:9-12
9. Knutson KL, Disis ML. Tumor antigen-specific T helper cells in cancer immunity and immunotherapy. Cancer Immunol Immunother. 2005;54:721-8
10. Liu J, Fu M, Wang M, Wan D, Wei Y, Wei X. Cancer vaccines as promising immuno-therapeutics: platforms and current progress. J Hematol Oncol. 2022;15:28
11. Fan T, Zhang M, Yang J, Zhu Z, Cao W, Dong C. Therapeutic cancer vaccines: advancements, challenges, and prospects. Signal Transduct Target Ther. 2023;8:450
12. Leko V, Rosenberg SA. Identifying and Targeting Human Tumor Antigens for T Cell-Based Immunotherapy of Solid Tumors. Cancer Cell. 2020;38:454-72
13. Yang F, Yang XF. New concepts in tumor antigens: their significance in future immunotherapies for tumors. Cellular & molecular immunology. 2005;2:331-41
14. McNeel DG, Emamekhoo H, Eickhoff JC, Kyriakopoulos CE, Wargowski E, Tonelli TP. et al. Phase 2 trial of a DNA vaccine (pTVG-HP) and nivolumab in patients with castration-sensitive non-metastatic (M0) prostate cancer. J Immunother Cancer. 2023 11
15. Bhagat A, Lyerly HK, Morse MA, Hartman ZC. CEA vaccines. Human vaccines & immunotherapeutics. 2023;19:2291857
16. Verma S, Swain D, Kushwaha PP, Brahmbhatt S, Gupta K, Sundi D. et al. Melanoma Antigen Family A (MAGE A) as Promising Biomarkers and Therapeutic Targets in Bladder Cancer. Cancers (Basel). 2024 16
17. Alsalloum A, Shevchenko JA, Sennikov S. NY-ESO-1 antigen: A promising frontier in cancer immunotherapy. Clin Transl Med. 2024;14:e70020
18. Alsalloum A, Shevchenko JA, Sennikov S. The Melanoma-Associated Antigen Family A (MAGE-A): A Promising Target for Cancer Immunotherapy? Cancers (Basel). 2023 15
19. Heitzeneder S, Bosse KR, Zhu Z, Zhelev D, Majzner RG, Radosevich MT. et al. GPC2-CAR T cells tuned for low antigen density mediate potent activity against neuroblastoma without toxicity. Cancer Cell. 2022;40:53-69.e9
20. Fan C, Qu H, Wang X, Sobhani N, Wang L, Liu S. et al. Cancer/testis antigens: from serology to mRNA cancer vaccine. Semin Cancer Biol. 2021;76:218-31
21. Gordeeva O. Cancer-testis antigens: Unique cancer stem cell biomarkers and targets for cancer therapy. Semin Cancer Biol. 2018;53:75-89
22. Higgins JP, Bernstein MB, Hodge JW. Enhancing immune responses to tumor-associated antigens. Cancer biology & therapy. 2009;8:1440-9
23. Srivastava AK, Guadagnin G, Cappello P, Novelli F. Post-Translational Modifications in Tumor-Associated Antigens as a Platform for Novel Immuno-Oncology Therapies. Cancers (Basel). 2022 15
24. Matsushima S, Ajiro M, Iida K, Chamoto K, Honjo T, Hagiwara M. Chemical induction of splice-neoantigens attenuates tumor growth in a preclinical model of colorectal cancer. Sci Transl Med. 2022;14:eabn6056
25. Lodewijk I, Dueñas M, Paramio JM, Rubio C. CD44v6, STn & O-GD2: promising tumor associated antigens paving the way for new targeted cancer therapies. Front Immunol. 2023;14:1272681
26. Xie N, Shen G, Gao W, Huang Z, Huang C, Fu L. Neoantigens: promising targets for cancer therapy. Signal Transduct Target Ther. 2023;8:9
27. Cafri G, Gartner JJ, Zaks T, Hopson K, Levin N, Paria BC. et al. mRNA vaccine-induced neoantigen-specific T cell immunity in patients with gastrointestinal cancer. J Clin Invest. 2020;130:5976-88
28. Ramirez CA, Becker-Hapak M, Singhal K, Russler-Germain DA, Frenkel F, Barnell EK. et al. Neoantigen landscape supports feasibility of personalized cancer vaccine for follicular lymphoma. Blood advances. 2024;8:4035-49
29. Ding Z, Li Q, Zhang R, Xie L, Shu Y, Gao S. et al. Personalized neoantigen pulsed dendritic cell vaccine for advanced lung cancer. Signal Transduct Target Ther. 2021;6:26
30. Martinov T, Greenberg PD. Targeting Driver Oncogenes and Other Public Neoantigens Using T Cell Receptor-Based Cellular Therapy. Annual review of cancer biology. 2023;7:331-51
31. Pearlman AH, Hwang MS, Konig MF, Hsiue EH, Douglass J, DiNapoli SR. et al. Targeting public neoantigens for cancer immunotherapy. Nature cancer. 2021;2:487-97
32. Chandran SS, Ma J, Klatt MG, Dündar F, Bandlamudi C, Razavi P. et al. Immunogenicity and therapeutic targeting of a public neoantigen derived from mutated PIK3CA. Nat Med. 2022;28:946-57
33. Kwok DW, Stevers NO, Etxeberria I, Nejo T, Colton Cove M, Chen LH. et al. Tumour-wide RNA splicing aberrations generate actionable public neoantigens. Nature. 2025;639:463-73
34. Savage SR, Yi X, Lei JT, Wen B, Zhao H, Liao Y. et al. Pan-cancer proteogenomics expands the landscape of therapeutic targets. Cell. 2024;187:4389-407.e15
35. Abd-Aziz N, Poh CL. Development of Peptide-Based Vaccines for Cancer. J Oncol. 2022;2022:9749363
36. Speiser DE, Chijioke O, Schaeuble K, Münz C. CD4(+) T cells in cancer. Nature cancer. 2023;4:317-29
37. Pant S, Wainberg ZA, Weekes CD, Furqan M, Kasi PM, Devoe CE. et al. Lymph-node-targeted, mKRAS-specific amphiphile vaccine in pancreatic and colorectal cancer: the phase 1 AMPLIFY-201 trial. Nat Med. 2024;30:531-42
38. Kondo S, Shimizu T, Koyama T, Sato J, Iwasa S, Yonemori K. et al. First-in-human study of the cancer peptide vaccine TAS0313 in patients with advanced solid tumors. Cancer Sci. 2021;112:1514-23
39. Malonis RJ, Lai JR, Vergnolle O. Peptide-Based Vaccines: Current Progress and Future Challenges. Chemical reviews. 2020;120:3210-29
40. Liu Q, Wu P, Lei J, Bai P, Zhong P, Yang M. et al. Old concepts, new tricks: How peptide vaccines are reshaping cancer immunotherapy? Int J Biol Macromol. 2024;279:135541
41. Wu B, Wang HJBI. Peptide Assemblies as Promising Tumor Vaccines: Current Platforms and Progress. 2023.
42. Liao HC, Liu SJ. Advances in nucleic acid-based cancer vaccines. J Biomed Sci. 2025;32:10
43. Lopes A, Vandermeulen G, Préat V. Cancer DNA vaccines: current preclinical and clinical developments and future perspectives. J Exp Clin Cancer Res. 2019;38:146
44. Kozak M, Hu J. DNA Vaccines: Their Formulations, Engineering and Delivery. Vaccines (Basel). 2024 12
45. Pandya A, Shah Y, Kothari N, Postwala H, Shah A, Parekh P. et al. The future of cancer immunotherapy: DNA vaccines leading the way. Med Oncol. 2023;40:200
46. Ledesma-Feliciano C, Chapman R, Hooper JW, Elma K, Zehrung D, Brennan MB. et al. Improved DNA Vaccine Delivery with Needle-Free Injection Systems. Vaccines (Basel). 2023 11
47. Nishimura K, Ogawa K, Kawaguchi M, Fumoto S, Mukai H, Kawakami S. Suppression of Peritoneal Fibrosis by Sonoporation of Hepatocyte Growth Factor Gene-Encoding Plasmid DNA in Mice. Pharmaceutics. 2021 13
48. Gong X, Chen Z, Hu JJ, Liu C. Advances of Electroporation-Related Therapies and the Synergy with Immunotherapy in Cancer Treatment. Vaccines (Basel). 2022 10
49. Bayraktutan H, Symonds P, Brentville VA, Moloney C, Galley C, Bennett CL. et al. Sparsely PEGylated poly(beta-amino ester) polyplexes enhance antigen specific T-cell response of a bivalent SARS-CoV-2 DNA vaccine. Biomaterials. 2024;311:122647
50. Jia W, Chandra J, Teoh SM, Tolley L, Yang H, Tse BWC. et al. STING Nanoagonist Boosts Antitumor Immunity of Therapeutic DNA Vaccines. Nano letters. 2024;24:15588-97
51. Perenkov AD, Sergeeva AD, Vedunova MV, Krysko DV. In Vitro Transcribed RNA-Based Platform Vaccines: Past, Present, and Future. Vaccines (Basel). 2023 11
52. Maruggi G, Ulmer JB, Rappuoli R, Yu D. Self-amplifying mRNA-Based Vaccine Technology and Its Mode of Action. Current topics in microbiology and immunology. 2022;440:31-70
53. Du H, Yang X, Fan J, Du X. Claudin 6: Therapeutic prospects for tumours, and mechanisms of expression and regulation (Review). Mol Med Rep. 2021 24
54. Sato Y, Nakamura T, Yamada Y, Harashima H. The impact of, and expectations for, lipid nanoparticle technology: From cellular targeting to organelle targeting. J Control Release. 2024;370:516-27
55. Mackensen A, Haanen J, Koenecke C, Alsdorf W, Wagner-Drouet E, Borchmann P. et al. CLDN6-specific CAR-T cells plus amplifying RNA vaccine in relapsed or refractory solid tumors: the phase 1 BNT211-01 trial. Nat Med. 2023;29:2844-53
56. Pardi N, Hogan MJ, Porter FW, Weissman D. mRNA vaccines - a new era in vaccinology. Nature reviews Drug discovery. 2018;17:261-79
57. Yang R, Cui J. Advances and applications of RNA vaccines in tumor treatment. Mol Cancer. 2024;23:226
58. Li H, Peng K, Yang K, Ma W, Qi S, Yu X. et al. Circular RNA cancer vaccines drive immunity in hard-to-treat malignancies. Theranostics. 2022;12:6422-36
59. Niu D, Wu Y, Lian J. Circular RNA vaccine in disease prevention and treatment. Signal Transduct Target Ther. 2023;8:341
60. Diao L, Liu M. Rethinking Antigen Source: Cancer Vaccines Based on Whole Tumor Cell/tissue Lysate or Whole Tumor Cell. Adv Sci (Weinh). 2023;10:e2300121
61. Korbelik M. Optimization of Whole Tumor Cell Vaccines by Interaction with Phagocytic Receptors. Vaccines (Basel). 2021 9
62. Sondak VK, Sosman JA. Results of clinical trials with an allogenic melanoma tumor cell lysate vaccine: Melacine. Semin Cancer Biol. 2003;13:409-15
63. Cheever MA, Higano CS. PROVENGE (Sipuleucel-T) in prostate cancer: the first FDA-approved therapeutic cancer vaccine. Clin Cancer Res. 2011;17:3520-6
64. Hotchkiss KM, Batich KA, Mohan A, Rahman R, Piantadosi S, Khasraw M. Dendritic cell vaccine trials in gliomas: Untangling the lines. Neuro-oncology. 2023;25:1752-62
65. Bota DA, Piccioni DE, Duma CM, Kesari S, Carrillo JA, LaRocca RV. et al. Phase 2 trial of personal dendritic cell vaccines in newly diagnosed glioblastoma: 3-year follow-up and correlations with survival. Human vaccines & immunotherapeutics. 2025;21:2556591
66. Wooster AL, Girgis LH, Brazeale H, Anderson TS, Wood LM, Lowe DB. Dendritic cell vaccine therapy for colorectal cancer. Pharmacol Res. 2021;164:105374
67. Nava S, Lisini D, Frigerio S, Bersano A. Dendritic Cells and Cancer Immunotherapy: The Adjuvant Effect. Int J Mol Sci. 2021 22
68. Hato L, Vizcay A, Eguren I, Pérez-Gracia JL, Rodríguez J, Gállego Pérez-Larraya J. et al. Dendritic Cells in Cancer Immunology and Immunotherapy. Cancers (Basel). 2024 16
69. Gao C, Luo R, Kwong CHT, Liu J, Tang M, Xie B. et al. Cancer vaccine from intracellularly gelated tumor cells functionalized with CD47 blockage and damage-associated molecular pattern exposure. Cell Rep Med. 2025;6:102092
70. Gong N, Alameh MG, El-Mayta R, Xue L, Weissman D, Mitchell MJ. Enhancing in situ cancer vaccines using delivery technologies. Nature reviews Drug discovery. 2024;23:607-25
71. Viswanath DI, Liu HC, Huston DP, Chua CYX, Grattoni A. Emerging biomaterial-based strategies for personalized therapeutic in situ cancer vaccines. Biomaterials. 2022;280:121297
72. Liang M. Oncorine, the World First Oncolytic Virus Medicine and its Update in China. Curr Cancer Drug Targets. 2018;18:171-6
73. Alberts P, Tilgase A, Rasa A, Bandere K, Venskus D. The advent of oncolytic virotherapy in oncology: The Rigvir® story. Eur J Pharmacol. 2018;837:117-26
74. Wang S, Liang B, Wang W, Li L, Feng N, Zhao Y. et al. Viral vectored vaccines: design, development, preventive and therapeutic applications in human diseases. Signal Transduct Target Ther. 2023;8:149
75. Gong L, Zhang Y, Liu C, Zhang M, Han S. Application of Radiosensitizers in Cancer Radiotherapy. Int J Nanomedicine. 2021;16:1083-102
76. Zhang F, Zheng Z, Barman AK, Wang Z, Wang L, Zeng W. et al. Optimal combination treatment regimens of vaccine and radiotherapy augment tumor-bearing host immunity. Commun Biol. 2021;4:78
77. Huang KC, Lai CY, Hung WZ, Chang HY, Lin PC, Chiang SF. et al. A Novel Engineered AAV-Based Neoantigen Vaccine in Combination with Radiotherapy Eradicates Tumors. Cancer Immunol Res. 2023;11:123-36
78. Chen M, Xiao L, Jia H, Wang S, Jiang X, Lei X. et al. Stereotactic ablative radiotherapy and FAPα-based cancer vaccine suppresses metastatic tumor growth in 4T1 mouse breast cancer. Radiother Oncol. 2023;189:109946
79. Tilsed CM, Fisher SA, Nowak AK, Lake RA, Lesterhuis WJ. Cancer chemotherapy: insights into cellular and tumor microenvironmental mechanisms of action. Front Oncol. 2022;12:960317
80. Qin SY, Cheng YJ, Lei Q, Zhang AQ, Zhang XZ. Combinational strategy for high-performance cancer chemotherapy. Biomaterials. 2018;171:178-97
81. Li Z, Jia W, Niu J, Zhang L. Understanding the roles of negative immune regulator TIPE2 in different diseases and tumourigenesis. Histol Histopathol. 2018;33:919-28
82. Leung CSK, Van den Eynde BJ. Combining personalized neoantigen vaccination with chemotherapy and anti-PD-1 to treat NSCLC. Cancer Cell. 2022;40:903-5
83. Chhabra N, Kennedy J. A Review of Cancer Immunotherapy Toxicity: Immune Checkpoint Inhibitors. J Med Toxicol. 2021;17:411-24
84. Lu X, Deng S, Xu J, Green BL, Zhang H, Cui G. et al. Combination of AFP vaccine and immune checkpoint inhibitors slows hepatocellular carcinoma progression in preclinical models. J Clin Invest. 2023 133
85. Fan A, Wang B, Wang X, Nie Y, Fan D, Zhao X. et al. Immunotherapy in colorectal cancer: current achievements and future perspective. Int J Biol Sci. 2021;17:3837-49
86. Siewe N, Friedman A. Combination therapy for mCRPC with immune checkpoint inhibitors, ADT and vaccine: A mathematical model. PLoS One. 2022;17:e0262453
87. Becker W, Olkhanud PB, Seishima N, Moreno PA, Goldfarbmuren KC, Maeng HM. et al. Second-generation checkpoint inhibitors and Treg depletion synergize with a mouse cancer vaccine in accordance with tumor microenvironment characterization. J Immunother Cancer. 2024 12
88. Salewski I, Kuntoff S, Kuemmel A, Feldtmann R, Felix SB, Henze L. et al. Combined vaccine-immune-checkpoint inhibition constitutes a promising strategy for treatment of dMMR tumors. Cancer Immunol Immunother. 2021;70:3405-19
89. Conlon KC, Miljkovic MD, Waldmann TA. Cytokines in the Treatment of Cancer. Journal of interferon & cytokine research: the official journal of the International Society for Interferon and Cytokine Research. 2019;39:6-21
90. Wang K, Zhang X, Ye H, Wang X, Fan Z, Lu Q. et al. Biomimetic nanovaccine-mediated multivalent IL-15 self-transpresentation (MIST) for potent and safe cancer immunotherapy. Nat Commun. 2023;14:6748
Author contact
![]() Corresponding authors: Zequn Li (Email: lizequnedu.cn) or Jie Cao (Email: caojie0829edu.cn).
Corresponding authors: Zequn Li (Email: lizequnedu.cn) or Jie Cao (Email: caojie0829edu.cn).