Impact Factor ISSN: 1449-1907
- Issue 9; 2026
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Volume 23; 2026
- Past Issues
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Conclusion
Abbreviations
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2025; 22(13):3477-3489. doi:10.7150/ijms.112248 This issue Cite
Research Paper
NDUFS1 upregulates ENaCα by NAD+ to promote alveolar fluid clearance in acute lung injury
Mengmeng Wang1#, Mengting Chen1#, Jianping Zhu1, Yu Zhang1, Jian Lu3, Zhiying Yue2, Zhengfeng Yang1,2 ![]() , Ruilan Wang1
, Ruilan Wang1 ![]()
1. Department of Critical Care Medicine, Shanghai General Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai 201620, China.
2. Precision Research Center for Refractory Diseases, Shanghai Jiao Tong University Pioneer Research Institute for Molecular and Cell Therapies, Shanghai General Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai, 201620, China; State Key Laboratory of Innovative Immunotherapy, School of Pharmaceutical Sciences, Shanghai Jiao Tong University, Shanghai, 200240, China.
3. Department of Critical Care Medicine, Shanghai United Family Hospital, Shanghai, 200335, China.
# These authors contributed equally to this work.
Received 2025-2-15; Accepted 2025-6-23; Published 2025-7-28
Abstract

Alveolar edema and following respiratory distress results in the aggravation of epithelial damage and the progression of acute lung injury (ALI), however, with unclear molecular mechanism remained to be elucidated. Through proteomic screening and scRNA-seq mining analysis, we detected the decline expression of NDUFS1 in epithelial cells in lungs from paraquat/LPS-induced ALI models. NDUFS1 deficiency in alveolar epithelial cells reduced ENaCα expression, which impaired alveolar fluid clearance (AFC) and led to alveolar edema. Mechanistically, NDUSF1 deficiency in alveolar epithelial cells leads to mitochondrial dysfunction such as reduced complex I activity, impaired NAD+ production and increased ROS, these contributed to the decline of ENaCα. Supplementing NAD+ via Olaparib treatment alleviated the reduction of ENaCα abundance raised by NDUFS1 deficiency, improved AFC, and suppressed the progression of ALI. In summary, our study suggests that NDUFS1 promotes AFC by regulating ENaCα via NAD+ in pulmonary epithelial cells during ALI.
Keywords: acute lung injury, NDUFS1, mitochondrial dysfunction, NAD+, ENaCα
Introduction
Acute lung injury (ALI) presents diffuse alveolar damage caused by multiple types of insults, including toxins and microbes' infection [1]. The pathological changes of ALI mainly include damage to the integrity of alveolar epithelial cells, gas exchange disorders, alveolar edema, and the formation of transparent membranes [2]. The progressive ALI can be attributed to moderate or severe acute respiratory distress syndrome (ARDS) [3]. Although supportive therapies, including mechanical ventilation, pharmacological intervention, and prone positioning ventilation, can improve the symptoms of ARDS patients, these strategies cannot effectively decrease the mortality rate of ALI/ARDS [4, 5]. Therefore, it is still required to understand the precise pathogenesis of ALI to develop improved effective strategies.
Epithelial sodium channel (ENaC) is a structure composed of α, β, and γ subunits and α subunit is essential for the functional ENaC channels [6]. Under physiological conditions, the ENaC on the apical membrane of alveolar epithelial cells (AECs) transports sodium ions from the alveolar cavity into the cells, then the Na-K-ATPase on the basement membrane side of AECs transfers them into the lung interstitium [7], which maintains sodium concentration gradient in AECs and thus provide the force to drive fluid from the alveolar space into the pulmonary interstitium. This process is called alveolar fluid clearance (AFC), which keeps the alveolar cavity dry and facilitates air exchange. However, in the early stage of ALI with ENaC deficiency, excess fluid was accumulated in the alveolar space that impairs air exchange and aggravates hypoxia, leading to a further damage to the alveolar epithelium and forming a vicious cycle that promotion of ALI progression [8]. Indeed, most of ARDS patients present severe disorders in alveolar fluid clearance, leading to acute respiratory failure together with a high mortality rate [9]. Clearing excess fluid from the alveolar cavity and promoting air exchange thus would be the watershed to determine the prognosis of ARDS patients [10]. Consistently, several studies show that reducing alveolar edema in the initial stage of ALI alleviates pulmonary inflammation and subsequent injury [11, 12], including indirect pharmacotherapies for AFC via reducing systemic inflammation, however, little strategies developed to directly target AFC via ENaC [7]. A combination of anti-inflammatory and promoting alveolar fluid clearance would expect to disturb the vicious progression of ALI.
Multiple factors have been reported to mediate the expression and activity of ENaC [13], including aldosterone, Angiotensin II, and phospholipids, etc. Though each agent induces the expression of ENaC via different signaling cascades, these stimulators share a common route to mediate energy metabolism and the production of reactive oxidative species, which has been well recognized to be dependent on mitochondrial activity [14, 15]. The importance of mitochondria in modulation of AFC during ALI has not been well characterized. However, it has been reported that mitochondria play a crucial role in maintaining lung health and modulation of respiratory diseases [16], making it reasonable to assume a potential regulation of ENaC activation and following AFC by mitochondria. Mitochondrial activity can be contributed by the activation of respiratory chain complexes, from which complex I is a rate-limiting step in the overall electron transfer and essential for mitochondrial oxidative respiration and following energy production [17]. NDUFS1 is the largest core subunit of mitochondrial respiratory chain complex I and regulates the generation of NAD+ [18]. NDUFS1 has been reported to modulate tissue or cell injury in certain scenarios, including cardiac dysfunction and myocardial fibrosis after myocardial infarction [19], and the enhancement of radiation sensitivity to cancer cells for suppressing tumor progression [20, 21], etc. Here, we further identified the importance of NDUFS1 in regulation of paraquat (PQ)/lipopolysaccharide (LPS)-induced acute lung injury via modulation of ENaCα production, expanding the understanding of mitochondrial activity in AFC and acute lung injury.
Materials and Methods
Animals and treatments
For PQ-induced ALI model, 8-12 weeks old C57BL/6 male mice were intraperitoneally injected with 40 mg/kg PQ one time [22] and sacrificed at day 3. For LPS-induced ALI model, 6-8 weeks old male mice were treated with 5mg/kg LPS via the non-surgical intratracheal instillation. For additional NAD supplementation, 5mg/kg Olaparib were intraperitoneally injected in the ALI mice model. For inhibiting NDUFS1 in mice lung tissues, 3mg/kg Rotenone were intraperitoneally injected in mice. PQ and LPS were purchased from Sigma-Aldrich (St. Louis, MO, USA). Rotenone and Olaparib were bought from Med Chem Express (NJ, USA). All of the mice were fed at the Shanghai General Hospital Laboratory Animal Center. The mice were housed with a 12h day/night cycles and free to water and food for 72 hours before this study. The animal experiments have been approved by the Animal Ethics Committee of the Shanghai General Hospital.
Cell culture
The A549 human epithelial cell line was bought from Cell Bank of the Chinese Academy of Sciences (Shanghai, China), and cultured in DMEM medium (Gibco, USA) containing 10% fetal bovine serum (Gibco, USA) at 37 °C incubator with 5% carbon dioxide. The mediums of cells were changed every 2-3 days. To mimic PQ-induced epithelial cell damage, A549 cells were treated with PQ at a final concentration of 400 µM for 24 hours and then we collected cells for subsequent experiments. To mimic LPS-induced epithelial cell damage, we used LPS at a final concentration of 10 µg/ml to stimulate A549 cells for 12 hours.
Western blotting
Cells and mice tissues were collected and the proteins were extracted with RIPA lysis buffer (Beyotime, P0013C) on the ice. Then we use the BCA protein assay kit (Thermo Fisher Scientific, 23225) to detect protein concentration. Equal amounts of protein (20-40μg) were separated by 7.5% or 10% SDS-PAGE gels (Epizyme Biotech, PG111; Epizyme Biotech, PG112) and transferred to nitrocellulose filter membranes (Pall, 66485). The membranes were blocked with 5% skimmed milk at room temperature (RT) for 1-2 hours. Then we incubated these membranes with primary antibodies at 40 °C overnight, including anti-E-cadherin (Proteintech, 60335-1-IG-50UL,1:2000), anti-Vimentin (Cell Signaling Technology, 5741S, 1:1000), anti-ENaCα (Proteintech, 28707-1AP, 1:1000), anti-SFTPC (Abcam, ab90716, 1:1000), anti-NDUFS1antibody (Abcam, ab169540, 1:2000), anti-GAPDH (Affinity, AF7021, 1:3000). We washed these membranes with Tris-buffered saline with Tween 20 (TBST) for about 6 minutes for 6 times in total, after which horseradish peroxidase-labeled secondary antibody (Beyotime A0216, A0208;1:2000) was incubated with membranes at RT for 1 hour. The membranes were washed for about 6 minutes for 6 times with TBST. Target proteins were detected by chemiluminescence detection system (Thermo Fisher Scientific, USA).
Quantitative reverse-transcription (qRT)-PCR assays
Cells and mice tissues were harvested and total RNA were extracted with RNA-easyTM Isolation Reagent (Vazyme, Nanjing, China). The HiScript III RT SuperMix kit (Vazyme Biotech Co, R323) was used to reverse transcribe the total RNA to cDNA. Cham Q SYBR Color qPCR Master Mix (Vazyme Biotech Co, Q431-02) was used to perform qRT-PCR to detect the relative mRNA expression of b-actin, Ndufs1, Ndufs2, Ndufs3, Ndufs7, Ndufs8, Ndufv1, Ndufv2 and ENaCα. We used β-actin to normalize the data and calculated using 2 [- (Ct target gene-Ct β-actin)]. The sequences of the PCR primers are available in Table S1.
Immunofluorescence staining
The lung tissues from LPS-induced ALI mice model were fixed within 4% paraformaldehyde, embedded in paraffin, and sliced into 4-μm-thick sections. The slices were then placed in an incubator at 60℃ for 1 hour, followed by deparaffinization, rehydration, and antigen retrieval by microwave oven (high fire 6 minutes and low fire 10 minutes) in EDTA antigen retrieval solution (BBI, E673003-0250). Next, we used 5% bovine serum albumin to block the slices at RT for 1 hour, then these slices were incubated overnight at 4 °C with primary antibodies, followed by the incubation of indicated secondary antibodies (1:2000) for 1 hour at RT. The primary antibodies utilized included rat anti-SFTPC (Abcam, ab90716, 1:1000) and mouse anti-NDUFS1 (Proteintech, 68253-1-IG, 1:600). The secondary antibodies included Goat anti Mouse IgG (H+) Alexa Fluor 488 (Invitrogen, A32723) and Goat anti Rabbit IgG (H+) Alexa Fluor 594 (Invitrogen, A32740). After washing 5 minutes with PBS for 3 times, these slides were mounted with DAPI Fluoromount-GTM (Yeasen, China). The samples were observed and images were captured with laser scanning confocal microscopy (Leica TCS SP8, Leica).
Lentivirus generation and cell infection
Plasmids
Human NDUFS1 cloned in pLV-mPuro-C-GFPSpark was purchased from Sino Biological.
Lentivirus generation and cell infection
HEK293T cells were seeded in 6 cm dishes, then we added a mixture of 2 µg shRNA targeting NDUFS1 (5'- GCAGTAGAGGAACCATCCATA-3'; 5'-TAACCTTTGTGACGAACATAA-3'), 2 µg packaging assistance plasmid (psPAX2), 2 µg envelope plasmid (pMD2.G) and Polyjet transfection reagent in to cells. After 6-8h, the medium was removed and cells were cultured with fresh medium for 24 hours. Then lentivirus supernatant was collected to infect A549 cells. A549 cells were seeded in 6-well plates, cultured with 1.5 ml lentivirus supernatant and 1 ml DMEM containing 10% FBS for 24h for infection, after which the infected cells were selected with 7 µg/ml puromycin for 48-72h. The stable transient cell lines were maintained by adding 5 µg/ml puromycin in culture medium. Similar procedures were performed to generate NDUFS1 overexpression A549 cells.
Histological staining and assessment of lung injury
The left lung tissues were fixed at 4% paraformaldehyde after mice sacrificed, embedded in paraffin and sliced 4-6 µm for histological staining. These sections were dewaxed and rehydrated before staining. We followed standard procedures to make Hematoxylin and eosin (H&E) staining with H&E staining kits (Servicebio, China). Subsequently, the sections were imaged in a 40x optical microscope. Lung injury was assessed with semiquantitative lung injury scoring system [23].
Intracellular ROS generation
Infected A549 cells were plated in 24-well plates with a density of 3 × 104 cells per well and cultured overnight. The attached cells were added with 500 µl fresh DMEM culture medium containing 10 µM DCFH-DA (Beyotime, S0033S) and incubated at 37 °C for 30 minutes. Immediately after removing DCFH-DA working solution, we used PBS to wash cells for 3 times and imaged them with a fluorescence microscope (Leica).
Mitochondrial complex I activity assay
We detected the activity of mitochondrial complex I in NDUFS1 deficient epithelial cells with a complex I enzymatic activity assay kit (Abbkine, Wuhan, China). The protein content was determined by a BCA protein assay kit (Thermo Fisher Scientific, 23225).
NAD+/NADH assay
We measured the NAD+/NADH levels in cells with NAD+/NADH Assay Kit (Beyotime, S0175) following the manufacturer's instructions. A549 cells were plated in 6-well plates and added with 200 µL extracting solution. We used a multifunctional microplate reader to measure and record 450nm absorbance. Then we detected protein content using a BCA protein assay kit (Thermo Fisher Scientific, 23225).
SRB assay
A549 cells infected with shRNA (NC) were plated in 96-well plates with a density of 0.5 × 104 cells per well. Then 0, 3.12, 6.25, 12.5, 25, or 50 µM Olaparib were added to stimulate the cells for 24h. The cells were fixed with 10% TCA at 4 ºC for 1h and washed with flowing water 5 times. Next, the cells were stained with 0.4% SRB for 10 minutes at room temperature. Immediately, we used 1% acetic acid to wash cells for 5 times and dried out 96 well plates. Then we dissolved the SRB in cells with 50 µl of 10 mM Tris base buffer per well. Absorbance at 515nm was measured using a multifunctional microplate reader.
Wet/dry ratio (W/D) and total lung water content of lung tissues
To assess the severity of pulmonary edema, we weighed a part of lung tissue recorded as the wet weight. Then we transferred the lung tissues to an oven at 65 ºC until the lung weight is no longer changing, we recorded the weight as the dry weight. The ratio of wet weight to dry weight was calculated as W/D ratios. Total lung water content is calculated using the following formula: (wet weight - dry weight)/dry weight.
Single-cell data processing
The scRNA-seq dataset for LPS-induced acute lung injury was obtained from the GEO database (GSE276682).
Statistics
Data were stated as the mean ± standard deviation. We used student's t-test to analyze the differences between two groups. We used one-way analysis of variance (ANOVA) to analyze the differences between multiple groups. P < 0.05 was considered as a significant difference.
Results
NDUFS1 expression is declined in alveolar epithelial cells during acute lung injury progression by proteomics combined with single-cell RNA sequencing analysis
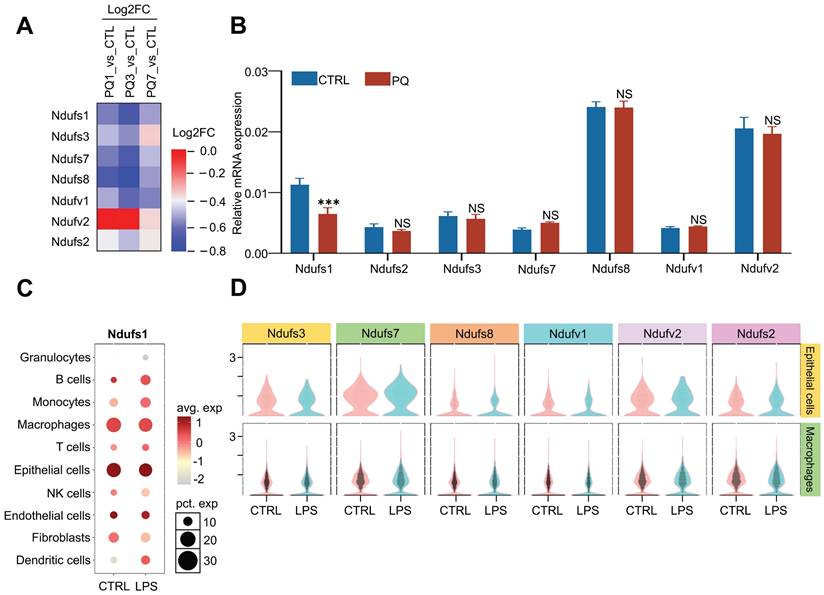
A proteomics screening found that the abundance of multiple subunits in the mitochondrial membrane respiratory chains was dynamically regulated during PQ- induced acute lung injury (ALI) progression (Fig.1A). Specifically, the protein abundance of NDUFS1, NDUFS3, NDUFS7, NDUFS8, and NDUFV1 were significantly reduced at day 3 and recovered at day 7 after PQ treatment. Considering day 3 after PQ treatment present severe acute lung injury symptom while mice survived after 7 days with PQ treatment were gradually recovered after lung injury, mitochondrial respiratory chains were probably activated in response to ALI progression. We next monitored the mRNA expression of these seven core subunits in A549 cells, the pulmonary epithelial cells, stimulated with or without PQ (Fig.1B). Interestingly, only the mRNA expression of NDUFS1 was specifically and significantly decreased after PQ stimulation. Given that NDUFS1 is the largest subunit of complex I and it plays an important role in complex I function [18], we assumed that NDUFS1-mediated mitochondrial respiratory chain complex I would be most important for mitochondria activity in pulmonary epithelial cells in response to ALI. To further reveal the specificity and potential importance of NDUFS1 in pulmonary epithelial cells, we performed single-cell data mining analysis on LPS-induced ALI mice (GSE276682) and identified that NDUFS1 was mainly enriched in epithelial cells and macrophages, with the expression of NDUFS1 decreased in epithelial cells with LPS stimulation (Fig.1C). Next, we analyzed the expression of other six core subunits in epithelial cells and macrophages on LPS-induced ALI mice (GSE276682) and discovered that they were enriched in epithelial cells (Fig.1D). However, compared with the control group, their expression changes in the LPS group were slight. Even the changes of some submits were opposite to those in proteomics and in vitro experiments before. These data indicated that NDUFS1 expression decreased in ALI and the change is relatively stable.
Proteomics combined with single-cell RNA sequencing analysis identifies that NDUFS1 declined in alveolar epithelial cells during acute lung injury progression. (A) Proteomic analysis of the expression of seven core subunits of complex I in lung tissues from PQ-induced ALI mice model. Mice were treated with PQ for 1 day (ALI 1), 3days (ALI 3) and & 7days (ALI 7), respectively. n=3 mice per group. PQ, paraquat. (B) The mRNA expression levels of Ndufs1, Nudfs2, Ndufs3, Ndufs7, Ndufs8, Ndufv1 and Ndufv2 in A549 cells stimulated with 400 µM PQ for 24 h. (C) Bubble plots indicating the presence of Ndufs1 in 10 cell types from lung tissues of CTRL and LPS-induced ALI mice model derived from the single cell-sequencing data of GSE276682. LPS, lipopolysaccharide. (D) Violin plots show the expression of other core submits in epithelial cells and macrophages of CTRL and LPS-induced ALI mice model derived from the single cell-sequencing data of GSE276682. Data were presented as mean ± SD. **p < 0.01, ***p < 0.001. NS, not statistically significant.
The NDUFS1 in pulmonary epithelial cells is further confirmed to decrease in PQ/LPS-induced acute lung injury models
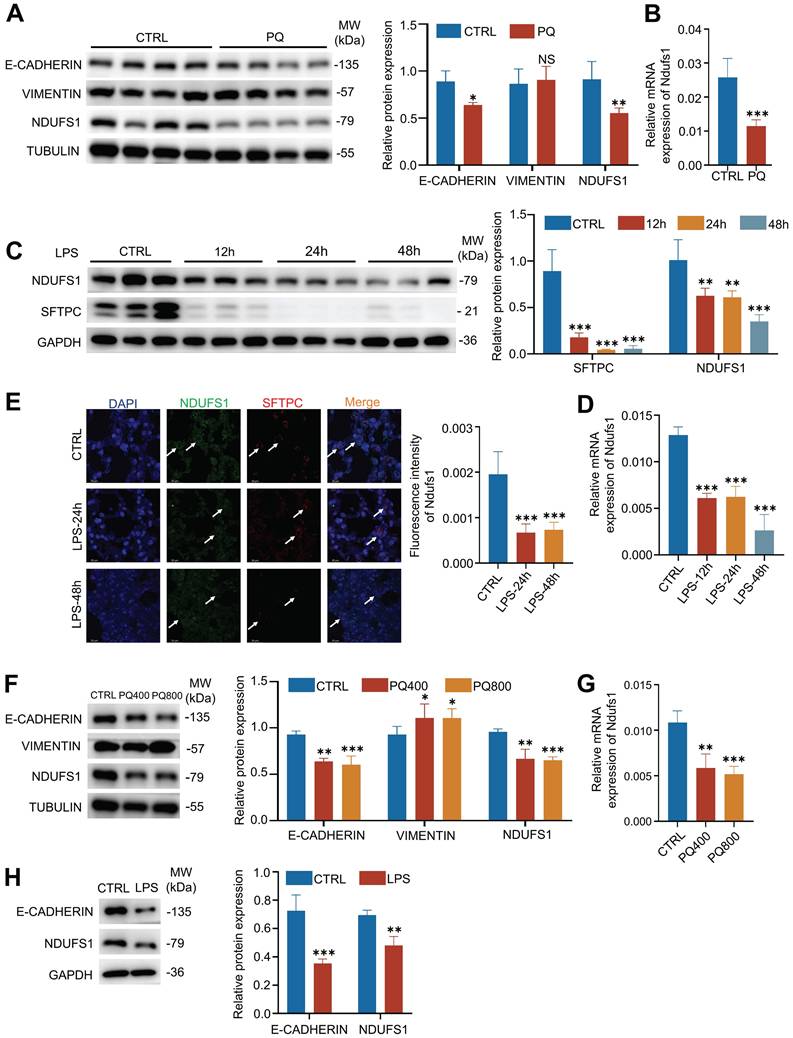
Next, we used PQ- or LPS-induced ALI models to verify the expression of NDUFS1 during lung injury. Firstly, we established the PQ-induced ALI mice model, indicated by the abundance of E-cadherin. Consistent with the proteomics results, the protein expression of NDUFS1 after lung injury in PQ-treated lung tissues was largely decreased compared with the control group (Fig.2A). Also, the mRNA expression level of NDUFS1 in lung tissues treated with PQ is significantly lower than that in the control group (Fig.2B). Similar results were observed in LPS-induced ALI mice model. Specifically, both protein and mRNA levels of NDUFS1 were decreased in the lung tissues with LPS treatment (Fig.2C-D). Of note, pulmonary surfactant-associated protein C (SFTPC), a surface marker of alveolar type II epithelial cells, was decreased after LPS stimulation, indicating the damage of alveolar epithelial cells. To further confirm that the expression of NDUFS1 was specifically decreased in alveolar type II epithelial cells in ALI, we performed immunofluorescence analysis with observation of the co-staining signal of NDUFS1 and SFTPC. The SFTPC signal is much weaker in the LPS group compared to the control group, indicating that alveolar type II epithelial cells were damaged with LPS stimulation. Importantly, the expression of NDUFS1 in SFTPC+ alveolar type II epithelial cells was decreased in the LPS treated group (Fig.2E), with other types of cells little affected, indicating the specific role of NDUFS1 in alveolar type II cells in modulation of ALI. A further in vitro analysis confirmed the potential importance of NDUFS1 in pulmonary epithelial cells. Particularly, both PQ and LPS stimulation reduced the abundance of NDUFS1 in A549 cells, the pulmonary epithelial cells (Fig.2F-H). Collectively, our data suggested that NDUFS1 was specifically decreased in pulmonary epithelial cells during PQ/LPS-induced ALI.
The expression of NDUFS1 is confirmed to decrease during PQ/LPS-induced acute lung injury. (A-B) Western blot (A) and QPCR (B) analysis of NDUFS1 expression in lung tissues from PQ-induced ALI mice model. Mice were treated with PQ for 3 days (ALI 3). n=4 mice per group. (C-D) Western blot (C) and QPCR (D) analysis of NDUFS1 expression in lung tissues from LPS-induced ALI mice model. Mice were treated with LPS for 12h, 24h and 48h. n=3 mice per group. (E) Immunofluorescence analysis of the colocalization signal between SFTPC and NDUFS1 in lung tissues from LPS-induced ALI mice model. Mice were treated with LPS for 24h and 48h. n=3 mice per group. Scale bars: 20 µm. (F-G) Western blot (F) and QPCR (G) analysis of E-cadherin, Vimentin or NDUFS1 abundance, as indicated, in A549 cells treated with or without 400 or 800 µM PQ for 24 h. (H) Western blot analysis of E-cadherin and Ndufs1 abundance in A549 cells treated with or without 10 µg/ml LPS for 12 h. Data were presented as mean ± SD. *p < 0.05, **p < 0.01, ***p < 0.001. NS, not statistically significant. Unless indicated, data were the representative of two independent experiments.
NDUFS1 deficiency in alveolar epithelial cells leads to alveolar fluid clearance dysfunction by reducing ENaCα
The specific reduction of NDUFS1 in pulmonary epithelial cells during ALI indicated the potential role of NDUSF1 in modulation of epithelial cells and ALI progression. Currently, most of ALI/ARDS patients present disorders in alveolar fluid clearance (AFC) and lack effective therapeutic, leading to acute respiratory failure together with a high mortality rate [7, 9]. Exploring the mechanisms of AFC may provide new targets for the treatment of ALI. We wondered whether NDUFS1 is involved in the process of AFC. Since the epithelial sodium channel (ENaC) plays a critical role in AFC and the ENaCα is the most important subunit of ENaC [6, 8], we established the NDUFS1 deficient epithelial cells and detect the expression of ENaCα. Our data found that NDUFS1 deficiency alone was sufficient to reduce ENaCα expression (Fig.3A-B). Furthermore, rotenone, the inhibitor of NDUFS1 [24], also largely reduced the expression of ENaCα (Fig.3C).
NDUFS1 mediates the expression of ENaCα in vitro and in vivo. (A-B) Western blot (A) and QPCR (B) analysis of NDUFS1 and ENaCα abundance in scrambled shRNA (NC) or NDUFS1 shRNA infected A549 cells. (C) Western blot analysis of NDUFS1 and ENaCα abundance in A549 cells treated with or without rotenone with indicated concentrations for 24 h. (D) Western blot analysis of NDUFS1 and ENaCα abundance in NC or NDUFS1 overexpression A549 cells treated with or without 400 µM PQ for 24 h. (E-F) Western blot (E) and QPCR (F) analysis of NDUFS1 and ENaCα abundance in lung tissues from control (CTRL) and rotenone treated mice. Mice were treated with rotenone for 24h. n=3 mice per group. (G) Hematoxylin and eosin staining of lung tissues from control (CTRL) and rotenone treated mice. And the lung injury score of the tissues. Mice were treated with rotenone for 24h. n=3 mice per group. Scale bars: 15 µm. Data were presented as mean ± SD. *p < 0.05, **p < 0.01, ***p < 0.001. Unless indicated, data were the representative of two independent experiments.
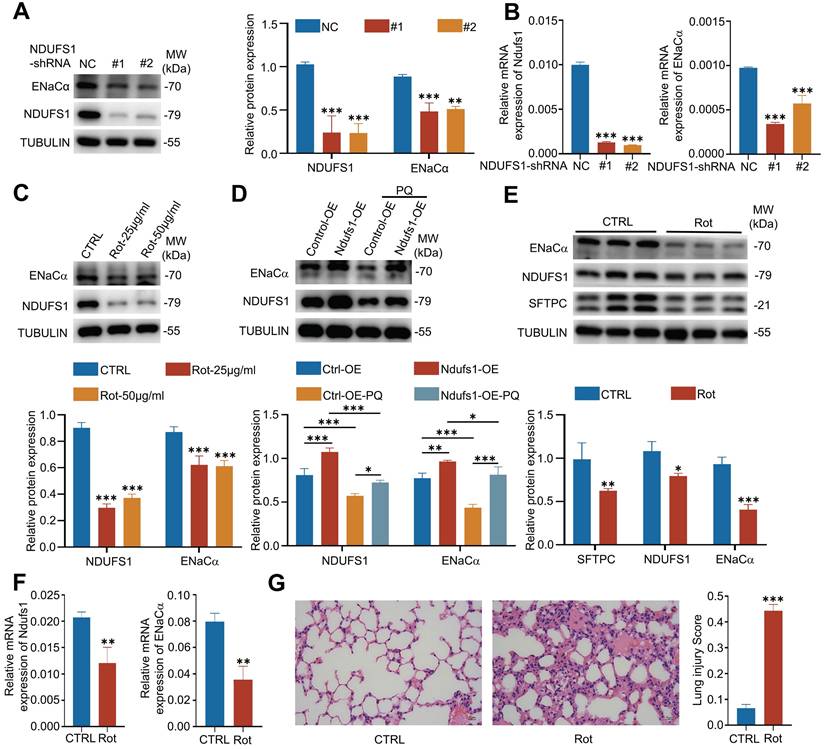
In contrast, NDUFS1 overexpression in pulmonary epithelial cells promoted the expression of ENaCα in the physiological condition and further rescued the downregulated ENaCα expression by PQ stimulation (Fig.3D). These results together suggested that NDUFS1 was required for maintaining the expression of ENaCα in pulmonary epithelial cells. We then further confirmed the correlated expression alteration of NDUFS1 and ENaCα in vivo. We injected rotenone in mice intraperitoneally to suppress the expression of NUDFS1 in lung tissues [25]. Our data further suggested that the NDUFS1 deficiency in mice lung tissues reduced the expression of ENaCα (Fig.3E-F). Indeed, HE staining of pathological sections revealed that NDUFS1 deficiency led to severe lung injury accompanied with alveolar flooding in the physiological condition (Fig.3G). These data together suggested that NDUFS1 deficiency led to AFC dysfunction via impairment of ENaCα in pulmonary epithelial cells, and further promoted the pulmonary edema.
NDUFS1 gene knockdown reduces the expression of ENaCα in alveolar epithelial cells via mitochondrial dysfunction
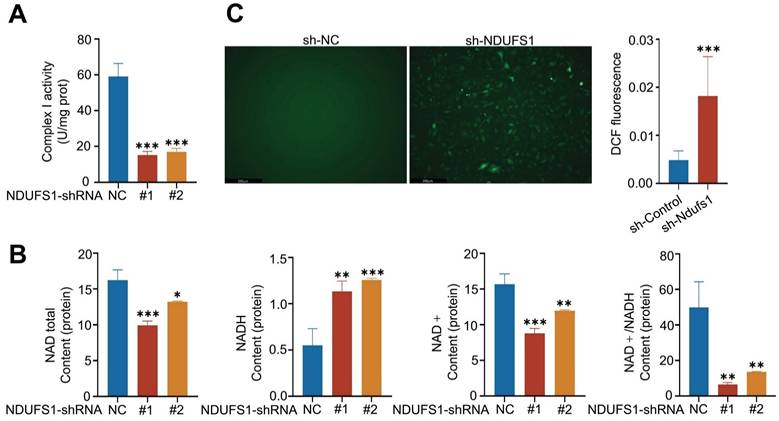
We wanted to investigate the mechanisms by which NDUFS1 modulated the expression of ENaCα. Research has demonstrated that mitochondrial dysfunction such as respiratory chain inhibition and increased ROS generation inhibited ENaC expression in collecting duct principal cells [26]. Since NDUFS1 is essential for the activation of mitochondrial respiratory complex I that maintains mitochondrial functions [18]. We speculated NDUFS1 may regulate ENaCα by mediating mitochondrial function. Firstly, we detected the activity of complex I in pulmonary epithelial cells. NDUFS1 deficiency largely reduced the activity of mitochondrial complex I in pulmonary epithelial cells (Fig.4A), which led to the dysfunction of the mitochondrial respiratory chain. Since the primary role of NDUFS1 in respiratory complex I is to regulate the generation of NAD+ [27], we next detected the abundance of total NAD, NADH, NAD+ and ratio of NAD+/NADH. Our data confirmed that the production of total NAD and NAD+ was significantly decreased while NADH was dramatically increased in NDUFS1 deficient pulmonary epithelial cells compared to control cells, leading to a great reduction of the ratio of NAD+/NADH (Fig.4B). Respiratory complex I dysfunction and NAD+ reduction led to increased ROS production [28, 29]. We further examined the ROS expression levels in NDUFS1 deficient epithelial cells via DHE staining and found a significantly increase in total ROS levels in NDUFS1 deficiency epithelial cells (Fig.4C). Taken together, these results suggested that NDUSF1 deficiency in lung epithelial cells led to mitochondrial dysfunction such as reduced complex I activity, impaired NAD+ production and increased ROS, these may contribute to the decline of ENaCα.
NDUFS1 deficiency in pulmonary epithelial cells leads to mitochondrial dysfunction. (A) Examination of the mitochondrial complex I activity in scrambled shRNA (NC) or NDUFS1 shRNA infected A549 cells. (B) Measurement of the abundance of total NAD, NADH, NAD+ and NAD+/NADH ratio in scrambled shRNA (NC) or NDUFS1 shRNA infected A549 cells. (C) Fluorescence imaging analysis of mitochondrial ROS in scrambled shRNA (NC) or NDUFS1 shRNA infected A549 cells. Scale bars: 250μm. Data were presented as mean ± SD. *p < 0.05, **p < 0.01, ***p < 0.001. Unless indicated, data were the representative of two independent experiments.
Mitochondrial dysfunction is correlated with mitophagy and cell apoptosis, the two processes be well recognized to mediate the damage of pulmonary epithelial cells during ALI [30, 31]. Our results confirmed the increased levels of PINK1 (a mitophagy marker) and cleaved caspase-3 (an apoptotic marker) in the PQ-induced lung injury model (Supplementary Fig.2A-B), underscoring the importance of both processes in mediating lung injury progression. Moreover, NDUFS1-deficient cells showed PINK1 upregulation but reduced cleaved caspase-3 levels, suggesting NDUFS1 is primarily essential for maintaining mitochondrial homeostasis but not sufficiently leading to cell death. Additional PQ stimulation further amplified PINK1 increases in NDUFS1-deficient cells, whereas NDUFS1 deficiency-mediated reduction of cleaved caspase-3 showed no significant change after PQ treatment (Supplementary Fig.2B). Collectively, these observations indicate that NDUFS1 deficiency primarily exacerbates mitochondrial damage in alveolar epithelial cells.
NDUFS1 promotes ENaCα expression and alveolar fluid clearance in acute lung injury through mediating NAD+
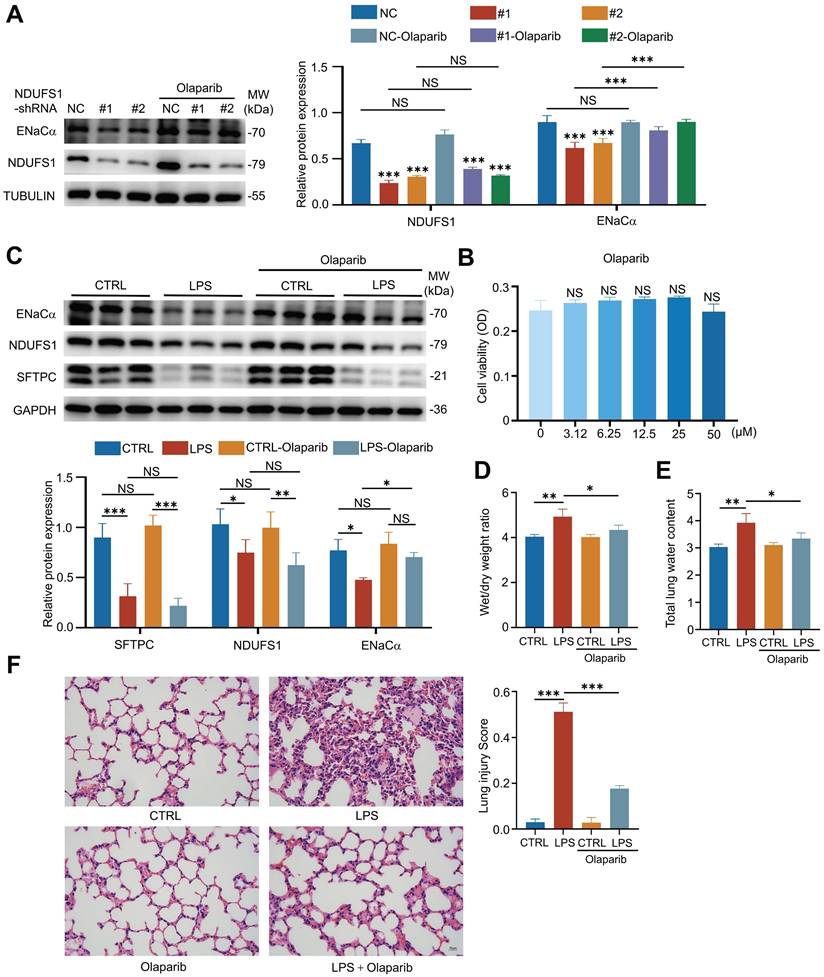
Literature has reported that NDUFS1 was mainly to regulate NAD generation in respiratory complex I [27]. Next, we wondered whether NDUFS1 maintains ENaCα expression during ALI via NAD+ production. It has been reported that NAD+ could be utilized by three major routes [32], including PARP family members, SIRT family members and CD38. Several clinical available PARP inhibitors have been reported to be effective in treating cancer progression [33], including Olaparib, Niraparib and Rucaparib, which promote the accumulation of NAD+ in cells. We therefore examined the effect of PARP inhibitors on ENaCα expression in NDUFS1 deficient pulmonary epithelial cells. Unexpectedly, we found that only Olaparib treatment specifically normalized the expression of ENaCα in NDUFS1 deficient cells at the concentration not showing large impairment on cell viability (Fig.5A-B, Supplementary Fig.1A-B). We then treated LPS- or PQ-induced ALI mice model with Olaparib and found that the downregulated expression of ENaCα in mice lung tissues under either LPS or PQ stimulation was largely recovered (Fig.5C, Supplementary Fig.3A). Importantly, Olaparib treatment also relieved the symptom of pulmonary edema in LPS or PQ stimulated mice model, indicated by Wet/dry weight ratio and total lung water content (Fig.5D-E, Supplementary Fig.3B-C). Consistently, pathological analysis further revealed that LPS-induced prominent thickening of the alveolar wall, alveolar cavity collapse and alveolar flooding in lung tissues could be ameliorated with addition of Olaparib (Fig.5F). Taken together, these findings provided evidence that NDUFS1 regulated the ENaCα via NAD+ to promotes alveolar fluid clearance, and facilitated the recovery of ALI.
NDUFS1 regulates ENaCα expression by modulating NAD+. (A) Western blot analysis of NDUFS1 and ENaCα abundance in scrambled shRNA (NC) or NDUFS1 shRNA infected A549 cells with or without 50 µM Olaparib treatment for 24 h. (B) SRB analysis of cell viability with Olaparib treatment, as indicated, in shRNA (NC) affected A549 cells for 24 h. (C) Western blot analysis of NDUFS1 and ENaCα abundance in lung tissues from LPS-induced ALI mice model with or without Olaparib treatment. Mice were treated with LPS for 2h. n=3 mice per group. (D) Wet/dry weight ratio analysis in lung tissues from LPS-induced ALI mice model with or without Olaparib treatment. Mice were treated with LPS for 2h. n=3 mice per group. (E) Total lung water content in lung tissues from LPS-induced ALI mice model with or without Olaparib treatment. Mice were treated with LPS for 2h. n=3 mice per group. (F) Hematoxylin and eosin staining of lung tissues from LPS-induced ALI mice model with or without Olaparib treatment and the lung injury score of the tissues. Mice were treated with LPS for 2h. Scale bars: 15 µm. n=3 mice per group. Data were presented as mean ± SD. *p < 0.05, **p < 0.01, ***p < 0.001. NS, not statistically significant. Unless indicated, data were the representative of two independent experiments.
Discussion
Acute lung injury (ALI) is caused by various pulmonary and extrapulmonary factors, including direct damage of intrinsic cells like epithelial cells, which leads to the accumulation of fluid in the alveoli and intractable hypoxemia [34, 35]. Acute respiratory distress syndrome (ARDS) is a severe stage of ALI and claims millions of lives worldwide each year [36]. Currently, the main treatment for ALI/ARDS is supportive therapies, including lung protective ventilation, prone positioning, and a fluid conservative strategy, together with anti-inflammation therapies. However, these therapies are not available to reduce the mortality rate [37, 38]. An insufficient understanding of the pathological mechanism on ALI/ARDS limits the development of potential precise strategies for improving ALI/ARDS. In this study, we identified the importance of NDUFS1 in preventing the vicious cycle of acute lung injury via maintaining the expression of ENaCα for alveolar fluid clearance that improves the gas exchange function of pulmonary epithelial cells. Importantly, NDUFS1 is well-known to convert NADH into NAD+ while accumulation of NAD+ in pulmonary epithelial cells by treatment with Olaparib, the clinical available PARP inhibitor promotion of NAD+ accumulation, normalized the reduced expression of ENaCα and lung damage during ALI. Olaparib has also reported to ameliorate the inflammatory responses during ALI [39]. Therefore, Olaparib would be a strategy of kill two birds with one stone, which is worth to examine to treat ARDS in clinic.
In physiological conditions, alveolar fluid clearance (AFC) relies on the synergistic transport of Na+ by epithelial sodium channel (ENaC) and Na-K-ATPase in alveolar epithelial cells [40]. In the early stages of ALI, ENaC or Na-K-ATPase deficiency impairs alveolar fluid absorption [41]. ENaC is the main driving force of AFC that facilitates the clear of excess fluid from alveoli. Mice lacking ENaCα were unable to remove edema fluid from alveolar spaces and died within 40 h of birth [42]. Studies have shown that ENaCα is regulated by multiple factors, such as cholinergic, Urokinase-like plasminogen activator (uPA), steroid hormone, and Protein Kinase C [40]. In this study, we further identified that either pharmacological or genetic inhibition of NDUFS1 significantly decreased the expression of ENaCα whereas NDUFS1 overexpression reversed the reduced ENaCα abundance during ALI. This indicated the potential role of NDUFS1 in enhancing alveolar fluid clearance in ALI. ENaC distributes widely in the epithelial cells of organs throughout the body and has been involved in the occurrence and development of many diseases [43]. ENaC in epidermis promotes skin epidermal differentiation and ENaC deficiency in epithelial cells can inhibit wound healing [44, 45]. ENaC dysfunction in hair cells of inner ears leads to disturbances in inner ear fluid balance and hearing loss [46]. In addition, ENaC also participates the progression of diseases such as cystic fibrosis and diabetic kidney disease [47, 48]. Our study first discovered that NDUFS1 played an important role in regulating ENaCα, which may provide a new idea for therapy of acute lung injury and other diseases.
Mitochondrial complex I is composed of 45 subunits and it is the beginning part of the respiratory chain [27]. NDUFS1 subunit is the largest part of the functional N-module of mitochondrial complex I and is responsible for the oxidation of NADH to NAD+ [18, 49]. Importantly, NDUFS1 plays a vital role in preserving the stability and function of the mitochondrial complex I, regulating the generation of ATP and ROS [50]. A prior study showed that NDUFS1 knockdown in C. elegans resulted in neuronal and mitochondrial functional damage, reduced ATP generation, and increased ROS generation [51]. In diabetic cardiomyopathy, AKAP1 deficiency in cardiomyocytes blocked the translocation of NDUFS1 from the cytosol to mitochondria, which reduced OXPHOS and raised the production of ROS [52]. These observations raised the importance of NDUFS1-mediated mitochondrial activity in maintaining organ functions. Our study further revealed a reduced expression of NDUFS1 in mouse lung tissues and alveolar epithelial cells after exposure to LPS or PQ, two insults well-known to raise ALI. Of note, we detected downregulated NDUFS1 protein and mRNA levels in murine lung tissues 2 hours post-LPS intratracheal instillation. LPS instillation only minimally activates pulmonary apoptosis within 2 hours [53], making less possibility of cell death in directly reducing NDUSF1 expression. Furthermore, PQ stimulation reduced NDUFS1 abundance even in NDUFS1-overexpressing cells, and NDUFS1 deficiency correlated with PQ-induced mitochondrial damage, which NDUFS1 deficiency in alveolar epithelial cells led to a decrease in mitochondrial complex I activity, increased ROS generation, and impairment of ENaCα expression. Together, these results indicate that NDUFS1 reduction during lung injury is mainly attributable to mitochondrial damage. As NDUFS1 deficiency in lung tissues also resulted in decreased ENaCα expression, impaired AFC and alveolar edema, we speculated that NDUFS1-mediated mitochondrial homeostasis is important for the regulation of ENaC and clearance of alveolar fluid and the recovery of acute lung injury.
NAD (Nicotinamide Adenine Dinucleotide) was first discovered in 1906 and described as an ingredient that could improve the fermentation rate of yeast [54]. NAD is the chemical backbone without charge, which is necessary for oxidation-reduction [55]. NAD+ and NADH (Nicotinamide adenine dinucleotide hydrogen) refer to the oxidized and reduced forms of NAD, respectively. NAD+ was identified as a cofactor associated with energy metabolism and diverse metabolic pathways such as glucolipid metabolism and tricarboxylic acid cycle [56]. NAD+ also plays a vital role in different physiological processes, including redox homeostasis, genomic stability, immunity and inflammation [57, 58]. NAD+ synthesis includes three classical pathways: the Preiss-Handler pathway (PHP), the de novo synthesis pathway (DNP) and the salvage pathway (SP) [59]. In addition, NAD+ can be cleaved by NAD+-consuming enzymes such as sirtuins, SARM1, polysaccharide polymerase (PARP) and cyclic ADP-ribose (cADPr) synthases CD38 and CD157 [60]. The abnormal metabolism of NAD+ thus leads to multiple diseases, including infection, apoptosis, tumorigenesis, age-associated neurodegeneration disorders, etc. In our research, increasing NAD+ via Olaparib treatment upregulated ENaCα protein expression in NDUFS1 deficient cells and improved alveolar fluid clearance (AFC) in LPS-induced ALI. We further identified the potential role of NAD+ for maintaining the expression of ENaCα and thus AFC to ameliorate ALI. Considering NDUFS1 mainly facilitates the production of NAD+ and NAD+ is widely required in different cell types, NAD+ would be the critical factor to maintain ENaCα expression in physiological conditions as well as in pathological conditions with no matter what insults are. Our study and other researches therefore provided the possibility that the homeostasis of ENaCα abundance would be largely dependent on NAD+ mediated cellular metabolism, including energy supplement and protein translational modification, which requires further efforts to elucidate. However, our work is the first to establish a direct link between NDUFS1, NAD+, and ENaC, providing a comprehensive framework for understanding how these components interact to maintain alveolar fluid clearance.
ALI involves complex multicellular interactions among alveolar epithelial cells, macrophages, fibroblasts, and other cell types. Our single-cell RNA sequencing mining analysis revealed that NDUFS1 expression is predominantly enriched in epithelial cells, justifying our focus on this population. NDUFS1 promotes alveolar fluid clearance specifically through ENaCα regulation in alveolar epithelial cells. Whether NDUFS1 regulates the activity of macrophages, fibroblasts, or other cell types by mediating ENaCα is yet to be elucidated. While our study did not directly examine other cell types, existing literature indicates mitochondria critically regulate macrophage polarization, immune responses, and fibroblast activation [61, 62]. Given NDUFS1's essential role in mitochondrial complex I function and homeostasis [18], it would be reasonable to investigate the importance of the NDUFS1-mitochondrial homeostasis-ENaCα axis in other cell types during ALI in future studies.
Conclusion
In conclusion, our research found that NDUFS1 was declined in alveolar epithelial cells during PQ/LPS-induced acute lung injury, which impaired ENaCα and alveolar fluid clearance. We further demonstrated that NDUFS1 maintained ENaCα expression and following alveolar fluid clearance via generation of NAD+, which provides a potential new theoretical basis for the treatment of acute lung injury.
Abbreviations
PQ: paraquat; LPS: lipopolysaccharide; ALI: acute lung injury; ARDS: acute respiratory distress syndrome; AFC: alveolar fluid clearance; ENaC: epithelial sodium channel; NAD: nicotinamide adenine dinucleotide; NADH: nicotinamide adenine dinucleotide hydrogen.
Supplementary Material
Supplementary figures and table.
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (82472218 to Ruilan Wang, 82272645 to Zhengfeng Yang), the National Key Clinical Specialist Construction Project (No. Z155080000004), the Key Supporting Discipline of Shanghai Healthcare System (NO. 2023ZDFC0102), Shanghai Hospital Development Center (SHDC22023239), the Shanghai Municipal Health Commission (2022YQ065). The Graphical Abstract was created in BioRender.
Availability of data
Relevant scientific data of this article are available from the corresponding author upon reasonable requests.
Author contributions
Ruilan Wang and Zhengfeng Yang conceived the research, interpreted results of experiments, revised manuscript and provided fundings. Mengmeng Wang performed the experiments, analyzed the data and wrote the original draft. Mengting Chen helped conduct the experiments and revised the manuscript. Jianping Zhu and Yu Zhang revised the manuscript. Jian Lu and Zhiying Yue revised the manuscript and provided fundings. All of the authors approved the final manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Long ME, Mallampalli RK, Horowitz JC. Pathogenesis of pneumonia and acute lung injury. Clin Sci (Lond). 2022;136:747-69
2. Matthay MA, Zemans RL, Zimmerman GA, Arabi YM, Beitler JR, Mercat A. et al. Acute respiratory distress syndrome. Nat Rev Dis Primers. 2019;5:18
3. Matuschak GM, Lechner AJ. Acute lung injury and the acute respiratory distress syndrome: pathophysiology and treatment. Mo Med. 2010;107:252-8
4. Bos LDJ, Ware LB. Acute respiratory distress syndrome: causes, pathophysiology, and phenotypes. Lancet. 2022;400:1145-56
5. Zhang J, Guo Y, Mak M, Tao Z. Translational medicine for acute lung injury. J Transl Med. 2024;22:25
6. Noreng S, Posert R, Bharadwaj A, Houser A, Baconguis I. Molecular principles of assembly, activation, and inhibition in epithelial sodium channel. Elife. 2020;9:e59038
7. Taenaka H, Matthay MA. Mechanisms of impaired alveolar fluid clearance. Anat Rec (Hoboken). 2025;308:1026-1039
8. Vadász I, Raviv S, Sznajder JI. Alveolar epithelium and Na,K-ATPase in acute lung injury. Intensive Care Med. 2007;33:1243-51
9. Ware LB, Matthay MA. Alveolar fluid clearance is impaired in the majority of patients with acute lung injury and the acute respiratory distress syndrome. Am J Respir Crit Care Med. 2001;163:1376-83
10. Fei X, Ziqian Y, Bingwu Y, Min L, Xinmiao X, Zhen M. et al. Aldosterone alleviates lipopolysaccharide-induced acute lung injury by regulating epithelial sodium channel through PI3K/Akt/SGK1 signaling pathway. Mol Cell Probes. 2021;57:101709
11. Mutlu GM, Machado-Aranda D, Norton JE, Bellmeyer A, Urich D, Zhou R. et al. Electroporation-mediated gene transfer of the Na+,K+ -ATPase rescues endotoxin-induced lung injury. Am J Respir Crit Care Med. 2007;176:582-90
12. Dumasius V, Sznajder JI, Azzam ZS, Boja J, Mutlu GM, Maron MB. et al. beta(2)-adrenergic receptor overexpression increases alveolar fluid clearance and responsiveness to endogenous catecholamines in rats. Circ Res. 2001;89:907-14
13. Rotin D, Staub O. Function and Regulation of the Epithelial Na(+) Channel ENaC. Compr Physiol. 2021;11:2017-45
14. de Cavanagh EM, Ferder M, Inserra F, Ferder L. Angiotensin II, mitochondria, cytoskeletal, and extracellular matrix connections: an integrating viewpoint. Am J Physiol Heart Circ Physiol. 2009;296:H550-8
15. Tsai CH, Chen ZW, Lee BC, Liao CW, Chang YY, Tsai YR. et al. Aldosterone, mitochondria and regulation of cardiovascular metabolic disease. J Endocrinol. 2024;263:e230350
16. Larson-Casey JL, He C, Carter AB. Mitochondrial quality control in pulmonary fibrosis. Redox Biol. 2020;33:101426
17. Sharma LK, Lu J, Bai Y. Mitochondrial respiratory complex I: structure, function and implication in human diseases. Curr Med Chem. 2009;16:1266-77
18. Ni Y, Hagras MA, Konstantopoulou V, Mayr JA, Stuchebrukhov AA, Meierhofer D. Mutations in NDUFS1 Cause Metabolic Reprogramming and Disruption of the Electron Transfer. Cells. 2019;8:1149
19. Qi B, Song L, Hu L, Guo D, Ren G, Peng T. et al. Cardiac-specific overexpression of Ndufs1 ameliorates cardiac dysfunction after myocardial infarction by alleviating mitochondrial dysfunction and apoptosis. Exp Mol Med. 2022;54:946-60
20. Sharanek A, Burban A, Laaper M, Heckel E, Joyal JS, Soleimani VD. et al. OSMR controls glioma stem cell respiration and confers resistance of glioblastoma to ionizing radiation. Nat Commun. 2020;11:4116
21. Ren L, Meng L, Gao J, Lu M, Guo C, Li Y. et al. PHB2 promotes colorectal cancer cell proliferation and tumorigenesis through NDUFS1-mediated oxidative phosphorylation. Cell Death Dis. 2023;14:44
22. Zhu Y, Meng X, Yang W, Wang J, Zhang J, Tian R. et al. HIF-1α promotes paraquat induced acute lung injury and implicates a role NF-κB and Rac2 activity. Toxicology. 2023;483:153388
23. Matute-Bello G, Downey G, Moore BB, Groshong SD, Matthay MA, Slutsky AS. et al. An official American Thoracic Society workshop report: features and measurements of experimental acute lung injury in animals. Am J Respir Cell Mol Biol. 2011;44:725-38
24. Shi Y, Wang Y, Jiang H, Sun X, Xu H, Wei X. et al. Mitochondrial dysfunction induces radioresistance in colorectal cancer by activating [Ca(2+)](m)-PDP1-PDH-histone acetylation retrograde signaling. Cell Death Dis. 2021;12:837
25. Zmijewski JW, Lorne E, Zhao X, Tsuruta Y, Sha Y, Liu G. et al. Mitochondrial respiratory complex I regulates neutrophil activation and severity of lung injury. Am J Respir Crit Care Med. 2008;178:168-79
26. Dizin E, Olivier V, Roth I, Sassi A, Arnoux G, Ramakrishnan S. et al. Activation of the Hypoxia-Inducible Factor Pathway Inhibits Epithelial Sodium Channel-Mediated Sodium Transport in Collecting Duct Principal Cells. J Am Soc Nephrol. 2021;32:3130-45
27. Pagniez-Mammeri H, Loublier S, Legrand A, Bénit P, Rustin P, Slama A. Mitochondrial complex I deficiency of nuclear origin I. Structural genes. Mol Genet Metab. 2012;105:163-72
28. Murphy MP. How mitochondria produce reactive oxygen species. Biochem J. 2009;417:1-13
29. Chen Q, Vazquez EJ, Moghaddas S, Hoppel CL, Lesnefsky EJ. Production of reactive oxygen species by mitochondria: central role of complex III. J Biol Chem. 2003;278:36027-31
30. Yang HH, Jiang HL, Tao JH, Zhang CY, Xiong JB, Yang JT. et al. Mitochondrial citrate accumulation drives alveolar epithelial cell necroptosis in lipopolysaccharide-induced acute lung injury. Exp Mol Med. 2022;54:2077-91
31. Xu X, Xu X, Zhong K, Wu Z, Wang C, Ding Z. et al. Salecan ameliorates LPS-induced acute lung injury through regulating Keap1-Nrf2/HO-1 pathway in mice. Int Immunopharmacol. 2024;128:111512
32. Conlon N, Ford D. A systems-approach to NAD+ restoration. Biochem Pharmacol. 2022;198:114946
33. Chan CY, Tan KV, Cornelissen B. PARP Inhibitors in Cancer Diagnosis and Therapy. Clin Cancer Res. 2021;27:1585-94
34. Thompson BT, Chambers RC, Liu KD. Acute Respiratory Distress Syndrome. N Engl J Med. 2017;377:562-72
35. Meyer NJ, Gattinoni L, Calfee CS. Acute respiratory distress syndrome. Lancet. 2021;398:622-37
36. Fan E, Brodie D, Slutsky AS. Acute Respiratory Distress Syndrome: Advances in Diagnosis and Treatment. Jama. 2018;319:698-710
37. Williams GW, Berg NK, Reskallah A, Yuan X, Eltzschig HK. Acute Respiratory Distress Syndrome. Anesthesiology. 2021;134:270-82
38. Wick KD, McAuley DF, Levitt JE, Beitler JR, Annane D, Riviello ED. et al. Promises and challenges of personalized medicine to guide ARDS therapy. Crit Care. 2021;25:404
39. Martins V, Santos SS, Rodrigues L, Salomao R, Liaudet L, Szabo C. Efficacy of Clinically Used PARP Inhibitors in a Murine Model of Acute Lung Injury. Cells. 2022;11:3789
40. Matalon S, Bartoszewski R, Collawn JF. Role of epithelial sodium channels in the regulation of lung fluid homeostasis. Am J Physiol Lung Cell Mol Physiol. 2015;309:L1229-38
41. Matthay MA. Resolution of pulmonary edema. Thirty years of progress. Am J Respir Crit Care Med. 2014;189:1301-8
42. Hummler E, Barker P, Gatzy J, Beermann F, Verdumo C, Schmidt A. et al. Early death due to defective neonatal lung liquid clearance in alpha-ENaC-deficient mice. Nat Genet. 1996;12:325-8
43. Chen Y, Yu X, Yan Z, Zhang S, Zhang J, Guo W. Role of epithelial sodium channel-related inflammation in human diseases. Front Immunol. 2023;14:1178410
44. Chifflet S, Hernandez JA. The Epithelial Sodium Channel and the Processes of Wound Healing. Biomed Res Int. 2016;2016:5675047
45. Charles RP, Guitard M, Leyvraz C, Breiden B, Haftek M, Haftek-Terreau Z. et al. Postnatal requirement of the epithelial sodium channel for maintenance of epidermal barrier function. J Biol Chem. 2008;283:2622-30
46. Chen J, He J, Luo J, Zhong S. Association of αENaC p. Ala663Thr Gene Polymorphism With Sudden Sensorineural Hearing Loss. Front Genet. 2021;12:659517
47. Veiras LC, Shen JZY, Bernstein EA, Regis GC, Cao D, Okwan-Duodu D. et al. Renal Inflammation Induces Salt Sensitivity in Male db/db Mice through Dysregulation of ENaC. J Am Soc Nephrol. 2021;32:1131-49
48. Lin J, Gettings SM, Talbi K, Schreiber R, Taggart MJ, Preller M. et al. Pharmacological inhibitors of the cystic fibrosis transmembrane conductance regulator exert off-target effects on epithelial cation channels. Pflugers Arch. 2023;475:167-79
49. Distelmaier F, Koopman WJ, van den Heuvel LP, Rodenburg RJ, Mayatepek E, Willems PH. et al. Mitochondrial complex I deficiency: from organelle dysfunction to clinical disease. Brain. 2009;132:833-42
50. Hirst J. Mitochondrial complex I. Annu Rev Biochem. 2013;82:551-75
51. Maglioni S, Schiavi A, Melcher M, Brinkmann V, Luo Z, Laromaine A. et al. Neuroligin-mediated neurodevelopmental defects are induced by mitochondrial dysfunction and prevented by lutein in C. elegans. Nat Commun. 2022;13:2620
52. Qi B, He L, Zhao Y, Zhang L, He Y, Li J. et al. Akap1 deficiency exacerbates diabetic cardiomyopathy in mice by NDUFS1-mediated mitochondrial dysfunction and apoptosis. Diabetologia. 2020;63:1072-87
53. Vernooy JH, Dentener MA, van Suylen RJ, Buurman WA, Wouters EF. Intratracheal instillation of lipopolysaccharide in mice induces apoptosis in bronchial epithelial cells: no role for tumor necrosis factor-alpha and infiltrating neutrophils. Am J Respir Cell Mol Biol. 2001;24:569-76
54. Harden A, Young WJ, Martin CJ. The alcoholic ferment of yeast-juice. Part II.—The coferment of yeast-juice. Proceedings of the Royal Society of London Series B, Containing Papers of a Biological Character. 1906;78:369-75
55. Warburg O, Christian W. Pyridin, der wasserstoffübertragende Bestandteil von Gärungsfermenten. Helvetica Chimica Acta. 1936;19:E79-E88
56. Yaku K, Okabe K, Nakagawa T. NAD metabolism: Implications in aging and longevity. Ageing Res Rev. 2018;47:1-17
57. Xie N, Zhang L, Gao W, Huang C, Huber PE, Zhou X. et al. NAD(+) metabolism: pathophysiologic mechanisms and therapeutic potential. Signal Transduct Target Ther. 2020;5:227
58. Amjad S, Nisar S, Bhat AA, Shah AR, Frenneaux MP, Fakhro K. et al. Role of NAD(+) in regulating cellular and metabolic signaling pathways. Mol Metab. 2021;49:101195
59. Rajman L, Chwalek K, Sinclair DA. Therapeutic Potential of NAD-Boosting Molecules: The In Vivo Evidence. Cell Metab. 2018;27:529-47
60. Zapata-Pérez R, Wanders RJA, van Karnebeek CDM, Houtkooper RH. NAD(+) homeostasis in human health and disease. EMBO Mol Med. 2021;13:e13943
61. Bueno M, Calyeca J, Rojas M, Mora AL. Mitochondria dysfunction and metabolic reprogramming as drivers of idiopathic pulmonary fibrosis. Redox Biol. 2020;33:101509
62. Nassef MZ, Hanke JE, Hiller K. Mitochondrial metabolism in macrophages. Am J Physiol Cell Physiol. 2021;321:C1070-c81
Author contact
![]() Corresponding authors: Ruilan Wang, M.D., Department of Critical Care Medicine, Shanghai General Hospital, Shanghai 201620, China, Email: wangruilangedu.cn; Zhengfeng Yang, Ph.D., Precision Research Center for Refractory Diseases, Shanghai General Hospital, Shanghai 201620, China, Email: zhengfeng.yangcn.
Corresponding authors: Ruilan Wang, M.D., Department of Critical Care Medicine, Shanghai General Hospital, Shanghai 201620, China, Email: wangruilangedu.cn; Zhengfeng Yang, Ph.D., Precision Research Center for Refractory Diseases, Shanghai General Hospital, Shanghai 201620, China, Email: zhengfeng.yangcn.