Impact Factor ISSN: 1449-1907
- Issue 9; 2026
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Volume 23; 2026
- Past Issues
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
1. Introduction
2. Radiotherapy modulates the TME
3. Radiotherapy modulates tumor...
4. Radiotherapy induces ICD
5. Radiotherapy Combined with...
6. Conclusions and Perspectives
Abbreviations
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2025; 22(13):3277-3291. doi:10.7150/ijms.109515 This issue Cite
Review
Radiotherapy elicits immunogenic cell death and metabolic shifts in the tumor microenvironment: implications for immunotherapy
Zi-Shan Hang1#, Yue-Ying Huang1#, An Song2, ![]() , Zhi-Jun Sun1,
, Zhi-Jun Sun1, ![]()
1. State Key Laboratory of Oral & Maxillofacial Reconstruction and Regeneration, Key Laboratory of Oral Biomedicine Ministry of Education, Hubei Key Laboratory of Stomatology, School & Hospital of Stomatology, Frontier Science Center for Immunology and Metabolism, Taikang Center for Life and Medical Sciences, Wuhan University, Wuhan, China.
2. The Affiliated Stomatological Hospital, State Key Laboratory Cultivation Base of Research, Prevention and Treatment of Oral Diseases, Jiangsu Province Engineering Research Center of Stomatological Translational Medicine, Nanjing Medical University, Nanjing, China.
# Equal contribution.
Received 2024-12-28; Accepted 2025-7-5; Published 2025-7-11
Abstract

Radiotherapy, one of the most utilized strategies to combat malignancies, has been constantly explored for its effectiveness and optimized, and it is currently operating at the molecular level. The tumor microenvironment (TME), where complicated changes take place under radiotherapy and other treatments, inevitably draws our attention to metabolic alterations, immunogenic cell death (ICD) and immunological interactions. In response to radiotherapy, tumor metabolism promotes DNA and membrane repair processes and reduces oxidative stress, thereby collectively alleviating the occurrence of cell death. Moreover, the induction of pyroptosis, necroptosis and ferroptosis under radiotherapy has the potential to increase antitumor immunity. Therefore, comprehensive knowledge about how radiotherapy triggers these modalities of ICD mechanically is necessary for developing nanomedicines with more accurate targets. In addition, information on clinical advancements as well as the management of adverse events is important for investigating radiotherapy combined with immunotherapy. This review provides an overview of up-to-date findings on metabolic changes and ICD under radiotherapy and provides insight into the status of the TME.
Keywords: radiotherapy, tumor microenvironment, immunogenic cell death, metabolism, immunotherapy
1. Introduction
Radiation therapy stands as a cornerstone of cancer treatment, with estimates suggesting that approximately half to two-thirds of oncologic patients receive radiotherapy at some stage in their treatment, either as a monotherapy or in conjunction with surgery, chemotherapy, or immunotherapy [1]. However, although radiotherapy is effective in eradicating tumor cells, it also induces alterations in tumor metabolism and the tumor microenvironment (TME). The TME is classified into three different immunophenotypes, namely, immune-excluded, immune-desert, and immune-inflamed phenotypes [2]. In immune-excluded tumors, CD8+ T cells are localized at malignancy margins and fail to infiltrate the main tumor mass, whereas immune-desert tumors lack CD8+ T cells within the tumor and its surrounding periphery. Both phenotypes are regarded as “cold” because of their abundance in immunosuppressive cells and lack of immunogenicity. In contrast, immune-flamed tumors, described as “hot” tumors, exhibit high T-cell infiltration, high PD-L1 expression and high tumor mutational burden. “Hot” tumors are more responsive to immune checkpoint blockades (ICBs) than “cold” tumors are, which indicates that turning “cold” tumors “hot” can be crucial to boosting immunotherapy [3].
Postradiotherapy, tumors exhibit significant TME and metabolic modifications. Initially, the metabolism of tumor cells is modified to mitigate their cytotoxic effects, and these changes extend to the TME via metabolite secretion [4]. In addition, various nonneoplastic stromal cells within the TME are directly impacted by radiotherapy, leading to significant metabolic alterations. Alterations in the physical and chemical properties of the TME subsequently influence metabolic pathways. These metabolic reprogramming processes play pivotal roles in facilitating energy utilization, mitigating oxidative damage, and repairing DNA and the cellular membrane [5, 6]. This understanding allows for the design of metabolic inhibitors that target these modified sites, and there have been relevant reviews summarizing drugs that target tumor metabolism [6, 7]. Certain metabolic drugs that target tumors have demonstrated unique mechanisms of inducing cell death [8]. When single or combined cancer metabolic therapies fail, inducing cell death via the combination of radiotherapy and immunotherapy can be promising.
Radiotherapy induces cell death that is immunogenic and proinflammatory, including pyroptosis, necroptosis and ferroptosis, which can be an effective way to turn “cold” tumors “hot” and prepare the TME for immunotherapy. Pyroptosis is morphologically characterized by nuclear condensation, cell swelling and pore formation on the cell membrane [9]. Mechanistically, several pathways of pyroptosis can be initiated by stress, including radiotherapy, and lead to pore formation by different gasdermins via different cascades [10]. Necroptosis, perceived as regulated necrosis due to its morphological similarity to necrosis, is an indispensable part of cell death under radiotherapy [9]. Necroptosis exhibits immunogenic and proinflammatory properties and is partially responsible for tumor metastasis [11]. Ferroptosis, an iron- and lipid-dependent cell death modality, has been defined and molecularly described in the last decade [12]. The mitochondrion, as the key organelle involved in ferroptosis, is not only closely related to reactive oxygen species (ROS) generation and lipid metabolism mechanistically but also shrinks with membrane densification and cristae disappearance morphologically [9]. Currently, ferroptosis stands at the intersection of metabolic modulation and cell death induction in nanomedicine development.
This review focuses on the radiotherapy-induced TME, metabolic changes and cell death. Within the TME, complicated interactions triggered by radiotherapy are elucidated, which can enhance our understanding of the multitarget effects of cancer treatment and optimize its application in reshaping an inflammatory environment conducive to immunotherapy. Specifically, alterations in metabolic pathways, including glycolysis, glutaminolysis and lipid metabolism, are discussed. In addition to elucidating the relationship between altered metabolism and cell death, attention is given to radiotherapy-induced pyroptosis, necroptosis and ferroptosis at the molecular mechanism level, revealing potential nanomedicine targets. Additionally, the modulatory effects of these immunogenic cell death (ICD) modalities on antitumor immunity are described. From a clinical perspective, advancements in radiotherapy combined with immunotherapy are discussed. Furthermore, adverse events of radiotherapy have been noted, and preventive strategies have been explored to better synergize with immunotherapy and ultimately achieve a more satisfactory outcome. Radiotherapy, as a traditional treatment modality, presents opportunities and challenges when combined with other treatments, especially immunotherapy.
2. Radiotherapy modulates the TME
Radiotherapy can transform “cold” tumors into “hot” tumors to enhance therapeutic effects, recruiting more immune cells to infiltrate the TME, increasing the expression of biomarkers, and leading to the development of immunogenic tumors [13]. These changes are predominantly exemplified in the TME, which includes cancer associated with immune cells, blood vessels and noncellular factors such as cytokines, chemokines and metabolites. Its characteristics include acidity, hypoxia, immune cell recruitment, and increased lactate and reduced glucose concentrations, which change with distance from blood vessels (Fig. 1).
Several changes in the tumor microenvironment (TME) occur during radiotherapy. The acidity of the TME is attributed mainly to glycolysis in cancer cells and is sensed by macrophages, leading to macrophage polarization. ICD in tumors caused by radiotherapy releases DAMPs, including ATP, CRT and HMGB1, which help with the initiation of adaptive immunity. Radiotherapy has different effects on various types of immune cells, such as upregulating the expression of PD-L1 in MDSCs and increasing the number of NKALs specific for NKG2D. Radiotherapy also affects the pattern and efficiency of blood vessel development in a dose-dependent manner. ICD, immunogenic cell death; DAMPs, damage-associated molecular patterns; ATP, adenosine triphosphate; CRT, calreticulin; HMGB1, high-mobility group box-1 protein; DCs, dendritic cells; PD-L1, programmed death ligand 1; MDSCs, myeloid-derived suppressor cells; NKALs, NK cell-activating ligands; NKG2D, natural killer group 2 member D.
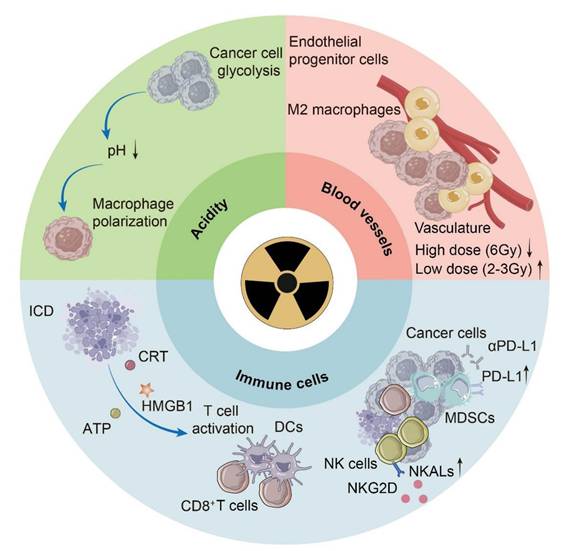
2.1 Radiotherapy calls for acidity reversion in the TME
Acidosis, a fundamental characteristic of the TME, is mostly attributed to glycolysis in tumor cells [14]. Acidification of the TME has long been blamed for poor prognosis, which is related to increased invasion ability and immune escape [15]. On the one hand, the acidic environment alters the expression of specific genes at the transcriptional level, contributing to tumor metastasis [15]. On the other hand, macrophages act as the medium for the disability of T cells and irresponsiveness to immunotherapy [16]. Conclusively, G protein-coupled receptors (GPCRs) on the surface of tumor-associated macrophages sense acidity, leading to an increase in cyclic adenosine monophosphate (cAMP), consequently increasing the expression of the transcriptional repressor inducible cAMP early repressor (ICER) and ultimately macrophage polarization toward a noninflammatory phenotype [16]. Although our knowledge about the effect of radiotherapy on acidity in the TME is still limited, neutralization of tumor acidity has been shown to promote the antitumor effect of radiotherapy, and the reversion of tumor acidity is now a key consideration in the development of antitumor nanomedicines [17].
2.2 Radiotherapy modulates immune cells in the TME
The effects of radiotherapy on immune cells play a dominant role in the remodeling of the TME, including both immune activation and immune suppression. For immune activation, radiotherapy causes ICD in tumors, which is characterized by the release of endogenous adjuvants named damage-associated molecular patterns (DAMPs), and fuels the release of DAMPs by increasing ROS, endoplasmic reticulum (ER) stress and, consequently, the unfolded protein response (UPR) [18]. DAMPs include surface-exposed calreticulin (CRT) and heat shock proteins (HSPs), which passively release high mobility group box-1 protein (HMGB1) and adenosine triphosphate (ATP), which are liberated into the extracellular space. Dendritic cells (DCs), as antigen-presenting cells, play a significant role in reacting to ICD and initiating adaptive immunity. CRT functions as an “eat me” signal to promote the uptake of dying tumor cells by DCs [19]. HMGB1, which binds to pattern recognition receptors (PRRs), facilitates the maturation of DCs and the release of proinflammatory cytokines, especially interferon-I (IFN-I) [20]. Tumor-infiltrating DCs play a major role in IFN-I induction via the cyclic GMP-AMP synthase (cGAS)-stimulator of interferon genes (STING) pathway due to the accumulation of mtDNA in the cytoplasm [21]. ATP acts as a “find me” signal for DC precursors and macrophages to facilitate the recruitment of myeloid cells [22]. Radiotherapy increases the number of tumor-associated macrophages (TAMs) and induces a phenotype transition from M2 to M1, which is proinflammatory [23]. Additionally, radiotherapy increases major histocompatibility complex class-I (MHC-I) expression on tumor cells, promoting the infiltration of CD8+ T cells [24]. It has been suggested that 12Gy radiation can significantly increase the secretion of C-X-C motif chemokine ligand 16 (CXCL16), which plays an important role in recruiting Th1 and cytotoxic T lymphocytes (CTLs) to all mouse and human breast cancer cells [25]. Additionally, radiotherapy can increase the expression of NK cell-activating ligands (NKALs) specific for natural killer group 2 member D (NKG2D), enhancing the killing effect on tumor cells [26]. In terms of immunosuppressive effects, radiation-induced lymphopenia (RIL) is reported as a potential adverse event, which is discussed later in this review [27]. Prolonged stimulation by IFN-I may cause immune exhaustion [28]. Additionally, radiotherapy helps with the mobilization of immunosuppressive cells. Radiotherapy increases C-C motif chemokine ligand 2 (CCL2) production, which facilitates C-C motif chemokine receptor 2 (CCR2)+ Treg recruitment [29]. Radiation can promote the infiltration of myeloid-derived suppressor cells (MDSCs) via the CCL2-CCR2 pathway [30]. Furthermore, radiotherapy can upregulate the expression of programmed death ligand 1 (PD-L1) in MDSCs, and combined treatment with PD-L1 blockade and immunogenic hypofractionation can help improve tumor immunity [31].
2.3 Radiotherapy modulates angiogenesis in the TME
In response to the demand for oxygen and nutrients for tumor growth and progression, blood vessels develop through either angiogenesis or vasculogenesis, which are responsible for hypoxia and acidosis in the TME [32]. This process is inevitably affected as radiotherapy is adopted. Under radiation, the formation of blood vessels switches from sprouting angiogenesis to intussusceptive angiogenesis as an adaptive mechanism, similar to the response to antiangiogenic therapies [33]. In general, radiotherapy damages endothelial cells to inhibit de novo and ongoing angiogenesis [34]. Because vasculogenesis relies on angioblast differentiation rather than endothelial cells, it is vital for tumor growth under radiotherapy [32]. The effect of radiotherapy on angiogenesis is time- and dose-dependent; high-dose (6 Gy) radiation causes a decrease in the tumor vasculature, whereas low-dose (2-3 Gy) radiation has a transient proangiogenic effect [35, 36]. In addition to the effects of radiotherapy on angiogenesis, the abnormal vasculature in tumors is responsible for resistance to radiotherapy [37]. As hypoxia exacerbates, the level of hypoxia-inducible factor-1α (HIF-1α) increases, increasing its downstream target stromal cell-derived factor-1 (SDF-1) and consequently recruiting proangiogenic macrophages as well as endothelial progenitor cells [38]. Therefore, the use of angiogenesis inhibitors in a sophisticated manner for blood vessel normalization has been adopted as a strategy [39].
3. Radiotherapy modulates tumor metabolism
Radiotherapy can eliminate tumor cells primarily by ROS-induced DNA damage and, secondarily, by destroying cellular structures such as the cell membrane. Despite numerous advances over the past few decades, radioresistance remains a challenge in radiotherapy, which can be attributed to changes in metabolism and the TME, as well as their interplay [40]. Therefore, understanding metabolic changes is particularly important for future radiotherapy, especially those related to energy utilization, redox balance, nucleic acid synthesis, and membrane repair (Fig. 2).
Radiotherapy-mediated metabolic shifts within the TME. Radiotherapy triggers tumor metabolic rewiring to enhance survival. Glycolysis fuels ATP, while PKM2 inactivation diverts intermediates to the PPP and SSP. Glutamine metabolism supports nucleotide and glutathione synthesis via GLS. Enhanced lipogenesis and lipid scavenging from stromal cells (e.g., CAFs and adipocytes) aid membrane repair. Tumor-secreted ROS and lactate suppresses immune activity, while stromal cells supply nutrients, fostering therapy resistance. Abbreviations: PKM2, pyruvate kinase M2; PPP, pentose phosphate pathway; SSP, serine synthesis pathway; GLS, glutaminolysis; CAF, cancer-associated fibroblast; ROS, reactive oxygen species.
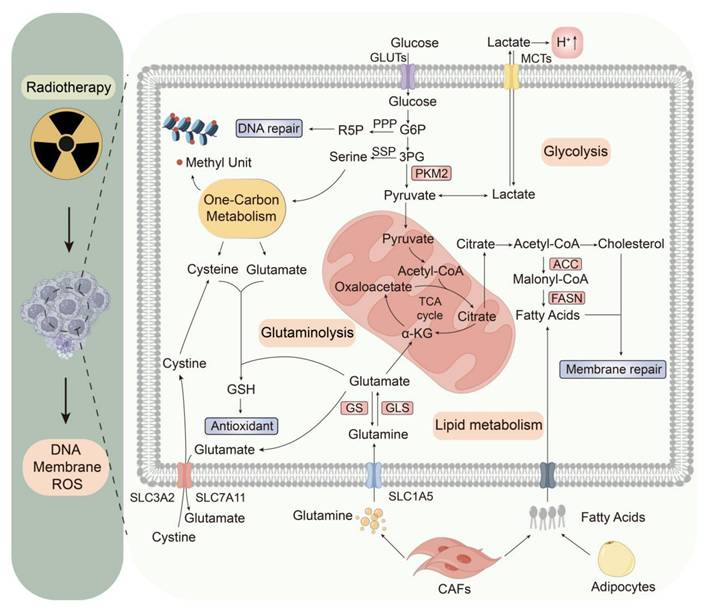
3.1 Radiotherapy modulates glycolysis
Glucose is one of the main nutrients (the other being glutamine) that support the survival and proliferation of cancer cells. More than 70% of human cancers exhibit amplified glycolytic gene expression [41], a metabolic feature further exacerbated by radiotherapy-induced glycolysis upregulation linked to malignant phenotypes and treatment resistance [5]. In the absence of radiation, most glucose is converted to pyruvate via lactate dehydrogenase (LDH). This process recycles nicotinamide adenine dinucleotide (NAD) to sustain glycolysis in cancer cells [42]. However, a decrease in the activity of the M2 isoform of pyruvate kinase (PKM2) is accompanied by an increase in ROS caused by radiation, implying that limiting pyruvate synthesis may increase tumor cell tolerance to oxidative stress [43, 44]. Paradoxically, despite reduced PKM2 enzymatic activity expected to decrease both pyruvate and lactate levels, the post-radiation lactate content significantly increases over time. This contradictory effect is presumably due to the oxidative decarboxylation of malate to pyruvate [45], increased expression of monocarboxylate transporter 1 (MCT1), which facilitates the export of lactate [46], and increased LDH activity in the anaerobic glycolytic state [44].
The inactivity of PKM2 reduces the synthesis of pyruvate and blocks glycolysis, which provides more upstream glycolytic intermediates to promote auxiliary glycolytic pathways related to the pentose phosphate pathway (PPP) and serine synthesis pathway (SSP) [47]. The up-regulated PPP metabolism can regenerate nicotinamide adenine dinucleotide phosphate (NADPH) to combat oxidative stress caused by ROS, produce ribose 5-phosphate (R-5-P) to facilitate nucleic acid synthesis, and supply glycerol-3-phosphate for the synthesis of membrane phospholipids [48, 49]. The overexpressed SSP metabolism contributes to NADPH generation, and via serine-driven one-carbon metabolism, it generates glutathione (GSH) precursors such as glycine and cysteine [50, 51]. Serine promotes an interconnected metabolic pathway network for carbon metabolism, promoting the biosynthesis of nucleotides, S-adenosylmethionine (SAM), NADPH, and GSH. Even when tumor cells increase the synthesis of serine, they still absorb a large amount of serine from the environment [52].
Multiple drugs, such as glycolytic inhibitors and pyruvate dehydrogenase (PDH) modulators, have been designed to customize cancer cell metabolism. Despite failures in clinical trials due to metabolic heterogeneity and reprogramming of the TME, combining radiotherapy, metabolism, and immunotherapy may have unexpected effects. Emerging evidence highlights the regulatory roles of miRNAs in modulating radioresistance. Specifically, miR-200a is inversely correlated with the expression of DNA repair enzymes, whereas microRNA-200c enhances the radiosensitivity of human cancer cells by modulating prosurvival signaling pathways and suppressing epithelial‒mesenchymal transition (EMT) [53, 54]. These findings underscore miRNAs as novel therapeutic targets to mitigate radioresistance, suggesting a promising strategy for combination therapy in cancer [55]. Additionally, biocompatible liposomes loaded with mannose and levamisole hydrochloride have been designed to regulate glucose and mitochondrial metabolic pathways in tumor cells and macrophages simultaneously. When combined with radiotherapy, this drug reverses the immunosuppressive TME by inhibiting M2 macrophages and further induces effective ICD and a systemic antitumor immune response [56].
3.2 Radiotherapy modulates glutaminolysis
Glutamine, the second major nutrient for tumor cells, is the most abundant free amino acid and plays an important role in tumor cell biology. In addition to being hallmarks of fast glucose metabolism, many types of tumors are characterized by elevated glutamine consumption [57]. Under normal circumstances, glutamine uptake is strongly regulated in cells. However, tumor cells require large amounts of glutamine and overcome this uptake and usage limitation by inducing the expression of glutamine transporters, glutamine synthetase (GS) and glutaminase (GLS) after radiation [44, 58, 59]. In the context of glutamine or glucose depletion, tumor cells might increase glutaminolysis to ensure the perpetuation of the tricarboxylic acid (TCA) cycle by stimulating α-ketoglutarate (α-KG) synthesis from glutamate, the end-product of glutaminolysis [60].
In addition to maintaining the TCA cycle, glutamine metabolism serves a multitude of pivotal physiological functions. Glutamine is an important precursor that contributes to purine and pyrimidine synthesis [61]. Because radiation damages DNA, tumor cells need to increase DNA synthesis to aid in repair. GS, which is transcriptionally regulated by signal transducer and activator of transcription 5 (STAT5) in response to radiation, facilitates nucleotide metabolism and subsequent DNA repair [62]. This damage is not fatal to some extent, but the disadvantages of the capacity for DNA repair and the efficiency of radiation damage cause cell death [63]. Interestingly, the decomposition of glutamine, the raw material of GSH, is postulated to regulate ferroptosis through the provision of α-KG in the TCA cycle [64]. However, high glutamine metabolic flux is converted into α-KG and enters the TCA cycle, which leads to the accumulation of a large amount of ROS, resulting in reduced expression of SLC7A11 and reduced synthesis of GSH after ionizing radiation. This results in oxidative stress injury to cells and ultimately leads to cell death [65]. Recent studies have shown that the inhibition of glutamine facilitates immunogenic tumor ferroptosis, further increasing the effectiveness of radiotherapy [8]. Considering that glutamine depletion has great adverse effects on the human body, the effect of glutamine supplementation in the tumor-bearing state needs further study.
3.3 Radiotherapy modulates lipid metabolism
Radiation not only damages DNA but also causes damage to the cell membrane. In addition to considerable changes in glucose and glutamine metabolism, lipid metabolism is also significantly altered in tumor cells to repair the cell membrane and meet energy demands for proliferation [59]. As mentioned earlier, the process of glycolysis provides more upstream glycolytic intermediates to promote auxiliary glycolytic pathways reelected in the PPP and SSP after radiation, and this regulation of the PPP provides more NADPH necessary for lipid synthesis. Fatty acids are integral components of cell membranes and are crucial for repairing damage caused by radiation exposure.
Tumor cells fulfill their requirements for fatty acids through two primary mechanisms: de novo synthesis via the enzymatic actions of acetyl-CoA carboxylase (ACC) and fatty acid synthase (FASN), as well as the uptake of lipids from the TME. Current research indicates that FASN overexpression in tumors is associated with radioresistance [66]. Uniformly, previous studies have shown that inhibiting fatty acid biosynthesis results in increased cytotoxicity and sensitivity to irradiation with FASN siRNA [59]. Mechanistically, FASN confers radioresistance via the Akt- and NF-κB-mediated signaling pathways because the levels of proteins upstream of FASN, such as Akt and NF-κB, are increased in radioresistant tumor cells [66]. Additionally, cholesterol is involved in cell membrane synthesis and angiogenesis, and dysregulation of cholesterol metabolism has been linked to radiation resistance, suggesting a potential target for enhancing the efficacy of radiotherapy [67].
Several studies have demonstrated that the TME can supply lipids to tumor cells, thereby facilitating their survival following radiotherapy [68]. Cancer-associated fibroblasts (CAFs) associated with pancreatic and colorectal cancers are known to secrete lipids, whereas tumor cells can induce the decomposition of fat in adipocytes [69]. Hypoxia, which is a common consequence of radiation therapy, can suppress the oxygen-dependent pathway of de novo lipid synthesis in cancer cells, leading to increased reliance on exogenous lipid uptake [70]. By enhancing the absorption and accumulation of fatty acids, tumor cells can develop resistance to ROS, thus promoting their survival.
3.4 Crosstalk between metabolism and cell death
Cell death may occur under various stress conditions, such as hypoxia, nutrient deprivation, and external stimuli [71]. In the hypoxic TME, the phosphatidylinositol 3-kinase (PI3K)/Akt/HIF-1α signaling axis is activated, which in turn modulates the glycolytic process [72]. The high rate of glycolysis in tumor cells leads to substantial lactate production, thereby creating an acidic TME that is inherently immunosuppressive [73]. Concurrently, the metabolite α-KG recruits procaspase-8 and gasdermin C (GSDMC), leading to the activation of pyroptosis in an acidic environment [74]. Apoptosis [75], necrosis [76], and pyroptosis [77] are linked to energy stress, suggesting that energy stress could be a major trigger of cell death. AMP-activated protein kinase (AMPK), a cell death regulator, also functions as a crucial intracellular energy sensor, playing a significant role in the coordination of metabolism, cell death, and inflammatory responses [71]. Upon sensing an elevated ADP/ATP ratio, AMPK activates downstream signaling pathways, facilitating energy generation [78]. Additionally, lipid metabolism is associated with apoptosis [79], pyroptosis [80], and ferroptosis [81]. In particular, the metabolites, key enzymes and transcription factors involved in lipid metabolism participate in the regulation of ferroptosis. Ferroptosis is closely related to lipid peroxidation, and glutaminolysis plays a crucial role in the death process [64]. The nexus between cell death induced by radiotherapy and cellular metabolism has not been fully elucidated. Future studies are warranted to explore the interplay between metabolic pathways and models of cell death within the TME.
4. Radiotherapy induces ICD
Radiotherapy elicits several modalities of cell death, among which pyroptosis, necroptosis and ferroptosis are the most common and well studied. Based on these molecular pathways, key points related to the response to irradiation have been identified, revealing the mechanisms of regulated cell death induced by radiotherapy (Fig. 3). Here, how pyroptosis, necroptosis and ferroptosis interact with antitumor immunity differently is also summarized and discussed.
Mechanisms of immunogenic cell death modalities under radiotherapy. The types of pyroptosis induced by radiotherapy include the inflammasome-caspase 1-GSDMD pathway and the caspase 3-GSDME pathway. Necroptosis elicited by radiotherapy mainly relies on a ZBP1-mediated pathway. Ferroptosis, which involves the executive and suppressive systems, is driven by lipid peroxidation, which can be facilitated by radiotherapy and the consequential increase in ROS. ACSL4 and GPX4 are known targets of radiotherapy. DAMPs, damage-associated molecular patterns; ROS, reactive oxygen species; AIM2, absent in melanoma 2; NLRP3, NOD-, LRR- and pyrin domain-containing protein 3; ZBP1, Z-DNA-binding protein 1; RIPK1, receptor-interacting protein kinase 1; RIPK3, receptor-interacting protein kinase 3; MLKL, mixed lineage kinase domain-like protein; TfR1, transferrin receptor 1; STEAP, six-transmembrane epithelial antigen of prostate; PUFA-PL, polyunsaturated fatty acid-containing phospholipid; ACSL4, acyl-CoA synthetase long chain family member 4; LPCAT3, lysophosphatidylcholine acyltransferase 3; GPX4, glutathione peroxidase 4; GSH, glutathione; GSSG, oxidized glutathione.
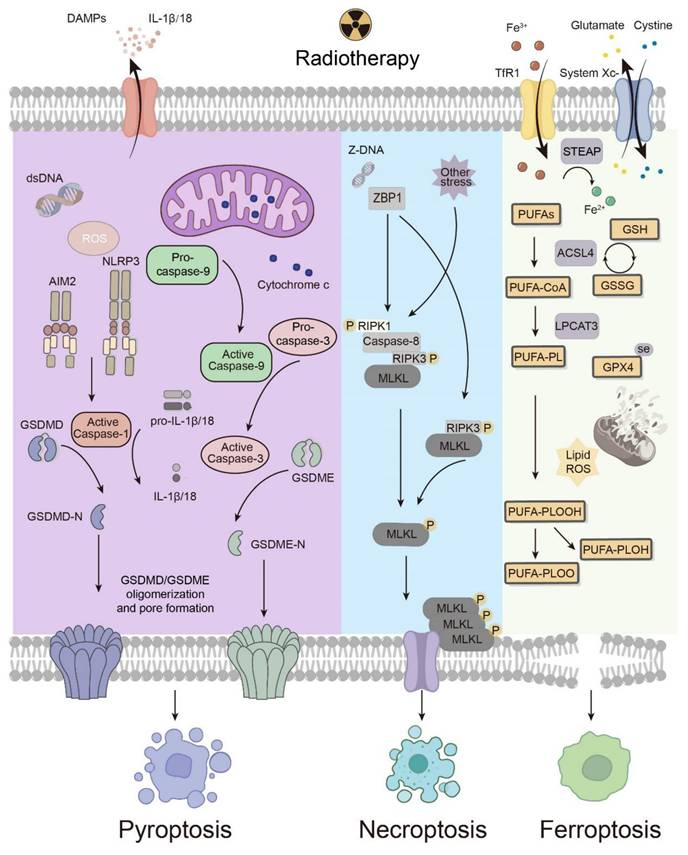
4.1 Pyroptosis
Pyroptosis, characterized by the activation of inflammatory caspases and proinflammatory cytokines as well as pore formation by gasdermins (GSDMs) on the plasma membrane, is a double-edged sword in antitumor immunity under radiotherapy [9]. The release of proinflammatory cytokines may favor tumorigenesis, whereas the immunogenic property of pyroptosis initiates adaptive immune responses to eliminate tumors. Pyroptosis can be evoked via parallel pathways, each of which is characterized by one of the GSDMs, including GSDMA, GSDMB, GSDMC, GSDMD, GSDME, and DFNB59 [10]. The N-terminus, which is exposed due to the cleavage of GSDMs, forms pores on the cell membrane, disrupting the integrity of the membrane [82]. Consequently, the cell contents flow out, and proinflammatory cytokines are released, leading to inflammatory cell death. Despite sharing this generally common mechanism, only two of these pathways, namely, the inflammasome-caspase 1-GSDMD pathway and the caspase 3-GSDME pathway, have been verified to be involved in how radiotherapy induces pyroptosis. To better understand how these cascades work under radiotherapy and identify potential targets for medical development, it is necessary to elucidate the key molecules involved.
The inflammasome-caspase 1-GSDMD pathway is considered the canonical pathway of pyroptosis, and its mechanism has been revealed at the molecular level. When the PRR is stimulated, the inflammasome produces mature caspase 1, which cleaves GSDMD into N- and C-terminal domains. Simultaneously, caspase 1 transforms the precursors of IL-1β and IL-18 into their activated forms [9]. Notably, NOD-, LRR- and pyrin domain-containing protein 3 (NLRP3) and absent in melanoma 2 (AIM2) have been shown to be stimulated by irradiation and then trigger the downstream pathway, leading to cell death. DNA damage and cell death caused by irradiation lead to an increase in dsDNA, which activates AIM2 inflammasomes [83]. Additionally, irradiation promotes the production of mitochondrial reactive oxygen species (mROS), which are regulated by Ca2+ influx and K+ efflux, activating the NLRP3 inflammasome [84]. Impressively, not only can the cleavage of GSDMD be facilitated via the activation of NLRP3 and AIM2 by radiotherapy, but the expression of GSDMD itself can also be upregulated by irradiation [85]. Caspase 1, which activates IL-1 and IL-18, can also be activated directly by radiotherapy [86]. IL-18 is dispensable for antitumor effects, whereas the IL-1 pathway in DCs facilitates the survival and activation of CD8+ T cells under irradiation through cross-priming [87].
With respect to the caspase 3-GSDME pathway, although its subsequent influence on antitumor immunity remains limited, the choice between pyroptosis and apoptosis requires further exploration [88]. The caspase 3-GSDME pathway is triggered by radiotherapy via the production of ROS, which leads to the oxidation and aggregation of Tom20 [89]. Oxidized Tom20 translocates Bax to mitochondria, promoting the release of cytochrome c from the mitochondria into the cytoplasm, which activates caspase 9 and then consequently activates caspase 3 [89]. When GSDME is expressed at a high level, caspase 3 cleaves GSDME, inducing pyroptosis; when GSDME is expressed at a low level, caspase 3 induces apoptosis [88]. Intriguingly, irradiation itself can promote the expression of GSDME in several tumor cell lines [90]. Overall, radiotherapy induces pyroptosis in high GSDME-expressing tumor cells, and hyperfractionated radiotherapy with specific chemotherapeutic agents potentially induces pyroptosis more extensively.
4.2 Necroptosis
Necroptosis, a proinflammatory and immunogenic modality of regulated cell death, is characterized by receptor-interacting protein kinase 3 (RIPK3)-dependent mixed lineage kinase domain-like protein (MLKL) phosphorylation [9]. Mechanically, upon extracellular or intracellular stress initiating death receptors, the state of receptor-interacting protein kinase 1 (RIPK1), ubiquitination or phosphorylation, determines the fate of a cell at the crossroads of survival and death [91]. Its interaction with caspase 8 and RIPK3 plays a critical role in leading the cell to undergo apoptosis or necroptosis, as caspase 8 enables apoptosis while inhibiting RIPK3-MLKL-mediated necroptosis [92]. In RIPK1-dependent or RIPK1-independent manners, phosphorylated RIPK3 activates MLKL, which is the primary executor of necroptosis. Phosphorylation of MLKL induces oligomerization and translocation of MLKL to the cell membrane, followed by cell membrane permeabilization, subsequently leading to pore formation and the release of DAMPs [93]. This immunogenic consequence, along with RIPK1 and NF-κB signaling, gives rise to cross-priming of CD8+ T cells, facilitating adaptive immunity [94].
Although RIPK1, RIPK3 and MLKL were discovered successively over a decade ago, necroptosis in tumors subjected to radiotherapy remained elusive until the Z-DNA-binding protein 1 (ZBP1)-mediated pathway was first described [95]. The activation of ZBP1 has been reported to initiate RIPK3-mediated MLKL activation, which is RIPK1 independent.
Immunologically, ZBP1-mediated necroptosis has been reported to interact with the cGAS-STING pathway, which results in the activation of innate immunity [96]. The cGAS-STING pathway is also activated by cytosolic DNA to initiate innate immunity. In the TME after radiation treatment, mtDNA is an emerging key to activating the cGAS‒STING pathway, and the ZBP1‒RIPK3‒MLKL cascade induces the release of mtDNA from mitochondria and leads to the accumulation of cytosolic mtDNA. In addition to the ZBP1-mediated cascade, elevated levels of mitochondrial ROS, which can be induced by radiotherapy, may spark off RIPK1-RIPK3-MLKL-mediated necroptosis [97]. Collectively, necroptosis, especially ZBP1-mediated necroptosis, contributes to both the innate and adaptive immune systems for postradiotherapy antitumor immunity.
4.3 Ferroptosis
Ferroptosis is an iron-dependent form of regulated cell death driven by lipid peroxidation [12, 98]. The molecular mechanism of ferroptosis can be summarized as suppressive systems, which include the glutathione peroxidase 4 (GPX4)-dependent pathway as well as GPX4-independent pathways, and the executive system, which relies on the synthesis of polyunsaturated fatty acid-containing phospholipids (PUFA-PLs) along with sufficient ROS and ferrous ions to initiate lipid peroxidation [99]. Several sections of the suppressive and executive systems can act as targets under irradiation to trigger ferroptosis.
In the executive system of ferroptosis, the biosynthesis of PUFA-PLs lays a foundation for lipid peroxidation. The peroxidation and accumulation of PUFA-PLs trigger the collapse of the cell membrane, leading to cell death [100]. With acyl-CoA synthetase long chain family member 4 (ACSL4) as a catalyst, long-chain PUFAs are ligated with CoA, whereupon PUFA-PLs are generated by lysophosphatidylcholine acyltransferase 3 (LPCAT3) [101]. Fe3+ enters the cytoplasm via transferrin receptor 1 (TfR1) and is reduced to Fe2+ by six-transmembrane epithelial antigen of the prostate (STEAP) [64]. The degradation of heme mediated by heme oxygenase-1 (HO-1) provides divalent iron as well [99]. Iron is involved in not only lipid peroxidation catalyzed by lipoxygenase (LOX) and cytochrome P450 oxidoreductase (POR) but also the Fenton reaction: PLOOHs react with ferric and ferrous ions to generate free radicals, which subsequently react with PUFA-PLs to multiply PLOOH [102]. Additionally, as iron is a component of the electron transport chain, iron homeostasis is closely related to the production of mitochondrial ROS. On the one hand, radiotherapy upregulates ACSL4 to facilitate PUFA-PL biosynthesis [99]. On the other hand, radiotherapy promotes iron release from heme and ferritin [99]. Moreover, irradiation damages the structure and function of mitochondria, increasing the production of reactive oxygen species (ROS), which are also key factors in the executive system of ferroptosis [99].
In terms of the suppressive systems of ferroptosis, the GPX4-dependent system, also known as the System Xc--GSH-GPX4 signaling axis, is prominent. System Xc- consists of SLC3A2 and SLC7A11, which export intracellular glutamate while importing extracellular cystine, which is later reduced to cysteine and subsequently serves as an ingredient in the synthesis of GSH [103]. GSH not only eliminates intracellular ROS but also provides electrons when GPX4 catalyzes the reduction of PLOOH, which elucidates how GPX4 disrupts the accumulation of lipid peroxides and consequently suppresses ferroptosis [99]. Radiation has been shown to lead to GSH depletion, which favors ferroptosis by weakening GPX4-mediated suppression [99]. Lei et al. reported that irradiation induces the expression of SLC7A11 and GPX4, inhibiting ferroptosis, as an adaptive response to the upregulation of ACSL4 [99].
In the TME, ferroptosis of tumor cells and various immune cells facilitates or inhibits overall antitumor immunity and the functions of each other. Recent research has proposed that cancer cell death from ferroptosis impedes the antigen-presenting function of DCs, which may be related to the earlier finding that several kinds of oxidized lipids can hinder the activation of CTLs through blocking antigen presentation by DCs [104]. Moreover, the maturation and function of DCs in the microenvironment can also be negatively impacted by the death of DCs from ferroptosis [105]. Similarly, polymorphonuclear myeloid-derived suppressor cells (PMN-MDSCs) reportedly impair the antigen-presentation function of DCs via myeloperoxidase-driven lipid peroxidation and accumulation [106]. Additionally, in the TME, PMN-MDSCs that die spontaneously from ferroptosis release oxygenated lipids, which limits the activity of T cells and induces an immunosuppressive TME. Subsequent experiments demonstrated that inhibition of ferroptosis can abolish the suppressive effect of PMN-MDSCs and synergize with immune checkpoint blockade to repress tumor progression [107]. CTLs release IFN-γ, which favors ferroptosis in tumor cells [108]. Overall, ferroptosis in tumor cells and intratumoral immune cells still needs further exploration, with a focus on the precise induction of ferroptosis and the maximized effect of immunotherapy.
5. Radiotherapy Combined with Immunotherapy
Currently, radiotherapy combined with immunotherapy has been widely employed clinically, and various clinical trials have made progress in optimizing radiation modalities and immunotherapy targets. However, because adverse events of radiotherapy may impair immunity and overall well-being, appropriate management can be important for positive outcomes.
5.1 Clinical advancements
Radiotherapy combined with immunotherapy has demonstrated significant potential in enhancing antitumor immune responses and inducing abscopal effects. First, combination therapy facilitates the infiltration and activation of immune-stimulating cells induced by radiotherapy [109]. Concurrently, radiotherapy overcomes immune tolerance, thereby increasing the clinical efficacy of ICB [110, 111]. Second, abscopal effects are rare with monotherapy, particularly in tumors with low immunogenicity. However, the abscopal effects increase when radiotherapy is combined with immunotherapy [112]. For example, a phase II trial (NCT02474186) in metastatic solid tumors reported that radiotherapy combined with granulocyte‒macrophage colony‒stimulating factor (GM-CSF) elicited abscopal responses in 26.8% of patients, with prolonged median overall survival. This review summarizes recent clinical trials that have investigated diverse combinatorial strategies pairing distinct radiation modalities with immunotherapeutic agents, aiming to amplify the immunostimulatory effects of RT while minimizing adverse outcomes (Table 1). Nevertheless, the therapeutic benefits remain constrained by tumor heterogeneity, immunosuppressive microenvironments, and interpatient variability. Future research should prioritize the optimization of radiation dose fractionation regimens, the identification of predictive biomarkers, and the exploration of novel immunomodulators to overcome current limitations and maximize synergistic potential. Besides, synergistic effects between radiotherapy and other antitumor therapies, like tumor treating fields (TTFields) for glioblastoma treatment, are also worth exploring for their clinical prospects [113, 114].
Summary of clinical trials investigating the combination of radiotherapy and immunotherapy
| Radiation | Drug | Targets | Type of cancer | Clinical trials | Phase | Status |
|---|---|---|---|---|---|---|
| RT | Pembrolizumab | PD-1 | Head and neck cancer | NCT03383094 | Phase 2 | Recruiting |
| SBRT | Relatlimab, Nivolumab | LAG-3, PD-1 | Uveal melanoma | NCT05077280 | Phase 2 | Recruiting |
| SBRT | CBI | CTLA-4, PD-1/PD-L1 | Metastatic cancer | NCT02843165 | Phase 2 | Active, not recruiting |
| IMRT | Nivolumab | PD-1 | Stage III non-small cell lung cancer | NCT04577638 | Phase 2 | Completed |
| SBRT | Gemcitabine, Penpulimab | PD-1 | Osteosarcoma | NCT06114225 | Phase 2 | Recruiting |
| SBRT | Atezolizumab | PD-L1 | Recurrent, persistent, or metastatic cervical cancer | NCT03614949 | Phase 2 | Recruiting |
| CIRT | Pembrolizumab | PD-1 | Solid tumors | NCT05229614 | Phase 2 | Recruiting |
| RT | Atezolizumab | PD-L1 | Penile cancer | NCT03686332 | Phase 2 | Completed |
| EBRT | Tremelimumab | CTLA-4 | Bladder cancer | NCT03601455 | Phase 2 | Active, not recruiting |
| RT | Bevacizumab, Tislelizumab | VEGF, PD-1 | Stage III non-small cell lung cancer | NCT05468242 | Phase 2 | Recruiting |
| Brachytherapy | TSR-042 | PD-1 | Endometrial cancer | NCT03955978 | Phase 1 | Active, not recruiting |
| RT | Bevacizumab | VEGF | Rectal cancer | NCT00113230 | Phase 2 | Completed |
| SBRT | Fresolimumab | TGF-β | Non-small cell lung cancer | NCT02581787 | Phase 1 Phase 2 | Completed |
| RT | Pembrolizumab, Axatilimab | PD-1, CSF-1R | Triple-negative breast cancer | NCT05491226 | Phase 2 | Active, not recruiting |
| TDLN-sparing RT | CBI | PD-1 | Esophageal squamous cell carcinoma | NCT06676449 | Phase 3 | Recruiting |
| RT | BMS-986205 | IDO1 | Glioblastoma | NCT04047706 | Phase 1 | Active, not recruiting |
| RT | CDX-1140 | CD-40 | Unresectable and metastatic solid tumors | NCT04616248 | Phase 1 | Recruiting |
| SABR | Atezolizumab | PD-1 | Metastatic tumors | NCT02992912 | Phase 2 | Active, not recruiting |
| SBRT | Nivolumab, Pembrolizumab | PD-1 | Metastatic non-small cell lung cancer | NCT03825510 | Phase 2 | Completed |
| RT | SD-101 | TLR9 | Low-grade B-cell non-Hodgkin lymphomas | NCT03410901 | Phase 1 | Recruiting |
| RT | SD-101 | TLR9 | Solid tumors and lymphoma | NCT03322384 | Phase 1 Phase 2 | Completed |
| RT | SD-101 | TLR9 | Low-grade follicular lymphoma | NCT02927964 | Phase 1 Phase 2 | Completed |
| RT | Nivolumab, SD-101 | PD-1, TLR9 | Metastatic pancreatic cancer | NCT04050085 | Phase 1 | Completed |
| SBRT | Pembrolizumab, SD-101 | PD-1, TLR9 | Prostate cancer | NCT03007732 | Phase 2 | Active, not recruiting |
| SBRT | Sintilimab | PD-1 | Hepatocellular carcinoma | NCT06313190 | Phase 2 | Recruiting |
| RT | Rituximab, Pembrolizumab | CD20, PD-1 | Follicular lymphoma | NCT02677155 | Phase 2 | Completed |
| RT | Pembrolizumab | PD-1 | Vulvar cancer | NCT04430699 | Phase 2 | Recruiting |
| SBRT | CDX-301 | FLT3 | Non-small cell lung cancer | NCT02839265 | Phase 2 | Completed |
| RT | CDX-301, CDX-1140 | FLT3, CD40 | Solid tumors | NCT04616248 | Phase 1 | Recruiting |
| RT | Toripalimab | PD-1 | Laryngeal and hypopharyngeal cancer | NCT06039631 | Phase 1 | Recruiting |
| RT | GM-CSF | GM-CSF | Solid metastatic tumors | NCT02474186 | Phase 2 | Completed |
| RT | GM-CSF, PD1 antibody | GM-CSF, PD-1 | Head and neck cancer | NCT05760196 | Phase 2 | Recruiting |
| RT | GM-CSF | GM-CSF | Thymoma | NCT05407649 | Phase 2 | Completed |
RT, radiation therapy; SBRT, stereotactic body radiation therapy; IMRT, intensity-modulated radiation therapy; CIRT, carbon ion radiotherapy; EBRT, external beam radiation therapy; TDLN-sparing RT, tumor draining lymph node-sparing radiotherapy; SABR, stereotactic ablative radiotherapy; CBI, checkpoint blockaded immunotherapy.
5.2 Adverse events and management
Although advancements in the combination of radiotherapy and immunotherapy are promising, the risks associated with radiotherapy should not be neglected, especially impairment of the immune system [115]. Adverse events caused by radiotherapy can be categorized into stochastic effects and tissue reactions. Stochastic effects refer to DNA mutations triggered by radiation, leading to secondary malignant neoplasms (SMNs). Tissue reactions, or deterministic effects, involve tissue and organ dysfunction attributed to cell death. This category can be further divided into short-term adverse effects, which occur during radiotherapy or within 3 months after radiotherapy, and long-term consequences, which are observed thereafter [116]. The former, such as mucositis and diarrhea, heal within weeks to months, whereas the latter can be irreversible and progressive. Therefore, RIL is worthy of extra attention [27]. RIL has been reported in various types of solid tumors, such as head and neck cancer, lung cancer and prostate cancer, and is associated with poor prognosis [117-119]. Given that a decreased lymphocyte count after radiotherapy has been proven to result in unsatisfactory efficacy of ICB, protection of the immune system should be emphasized when combining radiotherapy with immunotherapy [120].
To maximize patients' benefit from radiotherapy, measures need to be taken before, during and after treatment to prevent and address adverse events [115]. Generally, conversations should be conducted with patients before treatment to inform them of potential risks and achieve good patient compliance, including nutritional support as well as smoking and alcohol cessation. During the course of treatment, dose adaptation, scheduling and fractionation should be performed when necessary, according to monitoring of patients' responses. After treatment, follow-up inspection visits and interventions are important to resolve undesirable symptoms secondary to the treatment. In particular, extensive exploration has been conducted to prevent RIL. Because the severity of RIL is associated with the baseline lymphocyte count, screening patients with high RIL risk before radiotherapy can be meaningful [117]. In addition, the adoption of hypofractionated radiotherapy can minimize RIL [121]. Another focus in this area is the prevention of SMNs. Given that stochastic effects do not occur with a specific dose threshold, the management of radiation dose and fractionation may be inadequate to mitigate the pro-oncogenic effect [122]. Strategies have been developed to protect DNA from radiation damage and are also supposed to be normal-tissue selective so as not to impede the tumor-killing effect. For example, the clinically approved amifostine can be employed as both a topical and systemic strategy by scavenging radiation-induced free radicals, providing DNA protection and accelerating repair [123]. In addition, antioxidants, including tempol and manganese superoxide dismutase (MnSOD), are used to attenuate radiation-induced oxidative stress [124, 125]. Moreover, inhibitors of SRC family kinases (SFKs), which are crucial in the formation of the radiation-induced pro-tumorigenic microenvironment, have been employed recently [126]. SFK inhibitors have improved radiotherapy-elicited antitumor effects, indicating the potential of reshaping the microenvironment to prevent radiation-induced carcinogenesis.
6. Conclusions and Perspectives
This review focuses on the radiotherapy-induced TME, metabolic alterations, ICD and consequent changes in antitumor immunity. The radiation-induced effects on cell metabolic pathways and the mechanisms of pyroptosis, necroptosis and ferroptosis suggest potential targets that may be utilized in medical development to synergize with radiotherapy and immunotherapy. Investigating the crosstalk between cellular metabolism and cell death in the TME may help in understanding this process. However, the underlying relationship between radiation-induced alterations in metabolism and radiation-induced cell death requires further exploration. To date, ICD, especially ferroptosis, has been the intersection of radiotherapy and immunotherapy, playing a significant role in nanomedicines. Metabolic drugs on a postradiotherapy basis are expected to interact with immunology more in antitumor strategies in the future.
The revealed and unknown mechanisms of ICD modalities encapsulate potential targets for antitumor therapies and may contribute to connecting metabolism with cell death. Ferroptosis, as discussed above, plays a crucial role in radiotherapy-induced cell death and tumor suppression, mediating the synergy between radiotherapy and immunotherapy. With increasing knowledge about cell death and cellular metabolism, new medicines may be developed to combine radiotherapy and immunotherapy, maximizing the antitumor effects of multiple strategies.
The significant improvements in therapeutic effects indicate the exciting potential of the combination of radiotherapy and immunotherapy. Clinical trials conducted with different radiotherapy modalities have demonstrated encouraging synergistic effects with immunotherapies that have distinct targets. The expanded range of cancer types in clinical trials implies the broader use of radiotherapy combined with immunotherapy in the future. In addition, increased abscopal effects have been observed under this combined therapy, which may inspire subsequent attempts of this kind. In the future, the synergistic effects of radiotherapy and immunotherapy will play an increasingly important role in cancer treatment, with the potential to cure more patients with locally advanced tumors and to provide benefits beyond palliative care for patients with metastatic cancer.
Abbreviations
TME: tumor microenvironment; ICB: immune checkpoint blockade; ROS: reactive oxygen species; GPCRs: G protein-coupled receptors; cAMP: cyclic adenosine monophosphate; ICER: inducible cAMP early repressor; ICD: immunogenic cell death; DAMPs: damage-associated molecular patterns; ER: endoplasmic reticulum; UPR: unfolded protein response; CRT: calreticulin; HSPs: heat shock proteins; HMGB1: high-mobility group box-1 protein; ATP: adenosine triphosphate; DCs: dendritic cells; PRRs: pattern recognition receptors; IFN-I: interferon-I; cGAS: cyclic GMP-AMP synthase; STING: stimulator of interferon genes; TAMs: tumor-associated macrophages; MHC-I: major histocompatibility class-I; CXCL16: C-X-C motif chemokine ligand 16; CTL: cytotoxic T lymphocyte; NKALs: NK cell-activating ligands; NKG2D: natural killer group 2 member D; RIL: radiation-induced lymphopenia; CCL2: C-C motif chemokine ligand 2; CCR2: C-C motif chemokine receptor 2; PD-L1: programmed death ligand 1; MDSCs: myeloid-derived suppressor cells; HIF-1α: hypoxia-inducible factor-1α; SDF-1: stromal cell-derived factor-1; LDH: lactate dehydrogenase; NAD: nicotinamide adenine dinucleotide; PKM2: M2 isoform of pyruvate kinase; MCT1: monocarboxylate transporter 1; PPP: pentose phosphate pathway; SSP: serine synthesis pathway; NADPH: nicotinamide adenine dinucleotide phosphate; R-5-P: ribose 5-phosphate; GSH: glutathione; SAM: S-adenosylmethionine; PDH: pyruvate dehydrogenase; EMT: epithelial-mesenchymal transition; GS: glutamine synthetase; GLS: glutaminase; TCA: tricarboxylic acid cycle; α-KG: α-ketoglutarate; STAT5: signal transducer and activator of transcription 5; ACC: acetyl-CoA carboxylase; FASN: fatty acid synthase; CAFs: cancer-associated fibroblasts; PI3K: phosphatidylinositol 3-kinase; GSDMC: gasdermin C; AMPK: AMP-activated protein kinase; GSDMs: gasdermins; NLRP3: NOD-, LRR- and pyrin domain-containing protein 3; AIM2: absent in melanoma 2; mROS: mitochondrial reactive oxygen species; RIPK3: receptor-interacting protein kinase 3; MLKL: mixed lineage kinase domain-like protein; RIPK1: receptor-interacting protein kinase 1; ZBP1: Z-DNA-binding protein 1; GPX4: glutathione peroxidase 4; PUFA-PL: polyunsaturated fatty acid-containing phospholipid; ACSL4: acyl-CoA synthetase long chain family member 4; LPCAT3: lysophosphatidylcholine acyltransferase 3; TfR1: transferrin receptor 1; STEAP: six-transmembrane epithelial antigen of prostate; HO-1: heme oxygenase-1; LOX: lipoxygenase; POR: cytochrome P450 oxidoreductase; PMN-MDSCs: polymorphonuclear myeloid-derived suppressor cells; GM-CSF: granulocyte-macrophage colony-stimulating factor; TTFields: tumor treating fields; SMNs: secondary malignant neoplasms; MnSOD: manganese superoxide dismutase; SFKs: SRC family kinases.
Acknowledgements
Funding
This work is supported by the National Natural Science Foundation of China (82472818 and 82273202), the Fundamental Research Funds for the Central Universities (2042022dx0003), and the National Key Research and Development Program (2022YFC2504200, Z.-J.S.).
Author contributions
Zi-Shan Hang, An Song and Zhi-Jun Sun conceived and designed the review. Zi-Shan Hang and Yue-Ying Huang wrote the original draft. Zi-Shan Hang, Yue-Ying Huang and An Song drew the figures. Zi-Shan Hang, Yue-Ying Huang, An Song and Zhi-Jun Sun revised the manuscript. All the authors have read and approved the final version of the submitted manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Wang K, Tepper JE. Radiation therapy-associated toxicity: Etiology, management, and prevention. CA Cancer J Clin. 2021;71:437-54
2. Zhang J, Huang D, Saw PE, Song E. Turning cold tumors hot: from molecular mechanisms to clinical applications. Trends Immunol. 2022;43:523-45
3. Gerard CL, Delyon J, Wicky A, Homicsko K, Cuendet MA, Michielin O. Turning tumors from cold to inflamed to improve immunotherapy response. Cancer Treat Rev. 2021;101:102227
4. Arner EN, Rathmell JC. Metabolic programming and immune suppression in the tumor microenvironment. Cancer Cell. 2023;41:421-33
5. Mittal A, Nenwani M, Sarangi I, Achreja A, Lawrence TS, Nagrath D. Radiotherapy-induced metabolic hallmarks in the tumor microenvironment. Trends Cancer. 2022;8:855-69
6. Tang L, Wei F, Wu Y, He Y, Shi L, Xiong F. et al. Role of metabolism in cancer cell radioresistance and radiosensitization methods. J Exp Clin Cancer Res. 2018;37:87
7. Gong L, Zhang Y, Liu C, Zhang M, Han S. Application of radiosensitizers in cancer radiotherapy. Int J Nanomedicine. 2021;16:1083-102
8. Song A, Wu L, Zhang BX, Yang QC, Liu YT, Li H. et al. Glutamine inhibition combined with CD47 blockade enhances radiotherapy-induced ferroptosis in head and neck squamous cell carcinoma. Cancer Lett. 2024;588:216727
9. Galluzzi L, Vitale I, Aaronson SA, Abrams JM, Adam D, Agostinis P. et al. Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018;25:486-541
10. Shi J, Gao W, Shao F. Pyroptosis: Gasdermin-mediated programmed necrotic cell death. Trends Biochem Sci. 2017;42:245-54
11. Strilic B, Yang L, Albarrán-Juárez J, Wachsmuth L, Han K, Müller UC. et al. Tumour-cell-induced endothelial cell necroptosis via death receptor 6 promotes metastasis. Nature. 2016;536:215-8
12. Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE. et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149:1060-72
13. Charpentier M, Spada S, Van Nest SJ, Demaria S. Radiation therapy-induced remodeling of the tumor immune microenvironment. Semin Cancer Biol. 2022;86:737-47
14. Cairns RA, Harris IS, Mak TW. Regulation of cancer cell metabolism. Nat Rev Cancer. 2011;11:85-95
15. Rohani N, Hao L, Alexis MS, Joughin BA, Krismer K, Moufarrej MN. et al. Acidification of tumor at stromal boundaries drives transcriptome alterations associated with aggressive phenotypes. Cancer Res. 2019;79:1952-66
16. Bohn T, Rapp S, Luther N, Klein M, Bruehl TJ, Kojima N. et al. Tumor immunoevasion via acidosis-dependent induction of regulatory tumor-associated macrophages. Nat Immunol. 2018;19:1319-29
17. Qian R, Yi X, Liu T, Chen H, Wang Y, Hu L. et al. Regulation of ion homeostasis for enhanced tumor radio-immunotherapy. Adv Sci (Weinh). 2023;10:e2304092
18. Golden EB, Frances D, Pellicciotta I, Demaria S, Helen Barcellos-Hoff M, Formenti SC. Radiation fosters dose-dependent and chemotherapy-induced immunogenic cell death. Oncoimmunology. 2014;3:e28518
19. Obeid M, Tesniere A, Ghiringhelli F, Fimia GM, Apetoh L, Perfettini JL. et al. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat Med. 2007;13:54-61
20. Saenz R, Futalan D, Leutenez L, Eekhout F, Fecteau JF, Sundelius S. et al. TLR4-dependent activation of dendritic cells by an HMGB1-derived peptide adjuvant. J Transl Med. 2014;12:211
21. Zitvogel L, Galluzzi L, Kepp O, Smyth MJ, Kroemer G. Type I interferons in anticancer immunity. Nat Rev Immunol. 2015;15:405-14
22. Fucikova J, Kepp O, Kasikova L, Petroni G, Yamazaki T, Liu P. et al. Detection of immunogenic cell death and its relevance for cancer therapy. Cell Death Dis. 2020;11:1013
23. Ren J, Li L, Yu B, Xu E, Sun N, Li X. et al. Extracellular vesicles mediated proinflammatory macrophage phenotype induced by radiotherapy in cervical cancer. BMC Cancer. 2022;22:88
24. Zeng H, Zhang W, Gong Y, Xie C. Radiotherapy activates autophagy to increase CD8(+) T cell infiltration by modulating major histocompatibility complex class-I expression in non-small cell lung cancer. J Int Med Res. 2019;47:3818-30
25. Matsumura S, Demaria S. Up-regulation of the pro-inflammatory chemokine CXCL16 is a common response of tumor cells to ionizing radiation. Radiat Res. 2010;173:418-25
26. Weiss T, Schneider H, Silginer M, Steinle A, Pruschy M, Polić B. et al. NKG2D-dependent antitumor effects of chemotherapy and radiotherapy against glioblastoma. Clin Cancer Res. 2018;24:882-95
27. Prades-Sagarra E, Yaromina A, Dubois LJ. Understanding the impact of radiation-induced lymphopenia: Preclinical and clinical research perspectives. Clin Transl Radiat Oncol. 2024;49:100852
28. Boukhaled GM, Harding S, Brooks DG. Opposing roles of type I interferons in cancer immunity. Annu Rev Pathol. 2021;16:167-98
29. Mondini M, Loyher PL, Hamon P, Gerbé de Thoré M, Laviron M, Berthelot K. et al. CCR2-dependent recruitment of Tregs and monocytes following radiotherapy is associated with TNFα-mediated resistance. Cancer Immunol Res. 2019;7:376-87
30. Liang H, Deng L, Hou Y, Meng X, Huang X, Rao E. et al. Host STING-dependent MDSC mobilization drives extrinsic radiation resistance. Nat Commun. 2017;8:1736
31. Mao L, Zhou JJ, Xiao Y, Yang QC, Yang SC, Wang S. et al. Immunogenic hypofractionated radiotherapy sensitising head and neck squamous cell carcinoma to anti-PD-L1 therapy in MDSC-dependent manner. Br J Cancer. 2023;128:2126-39
32. Patan S. Vasculogenesis and angiogenesis. Cancer Treat Res. 2004;117:3-32
33. Hlushchuk R, Riesterer O, Baum O, Wood J, Gruber G, Pruschy M. et al. Tumor recovery by angiogenic switch from sprouting to intussusceptive angiogenesis after treatment with PTK787/ZK222584 or ionizing radiation. Am J Pathol. 2008;173:1173-85
34. Imaizumi N, Monnier Y, Hegi M, Mirimanoff RO, Rüegg C. Radiotherapy suppresses angiogenesis in mice through TGF-betaRI/ALK5-dependent inhibition of endothelial cell sprouting. PLoS One. 2010;5:e11084
35. Geng L, Donnelly E, McMahon G, Lin PC, Sierra-Rivera E, Oshinka H. et al. Inhibition of vascular endothelial growth factor receptor signaling leads to reversal of tumor resistance to radiotherapy. Cancer Res. 2001;61:2413-9
36. Sofia Vala I, Martins LR, Imaizumi N, Nunes RJ, Rino J, Kuonen F. et al. Low doses of ionizing radiation promote tumor growth and metastasis by enhancing angiogenesis. PLoS One. 2010;5:e11222
37. Brown JM. Vasculogenesis: a crucial player in the resistance of solid tumours to radiotherapy. Br J Radiol. 2014;87:20130686
38. Ceradini DJ, Kulkarni AR, Callaghan MJ, Tepper OM, Bastidas N, Kleinman ME. et al. Progenitor cell trafficking is regulated by hypoxic gradients through HIF-1 induction of SDF-1. Nat Med. 2004;10:858-64
39. Jiao L, Dong Q, Zhai W, Zhao W, Shi P, Wu Y. et al. A PD-L1 and VEGFR2 dual targeted peptide and its combination with irradiation for cancer immunotherapy. Pharmacol Res. 2022;182:106343
40. Barker HE, Paget JT, Khan AA, Harrington KJ. The tumour microenvironment after radiotherapy: mechanisms of resistance and recurrence. Nat Rev Cancer. 2015;15:409-25
41. Altenberg B, Greulich KO. Genes of glycolysis are ubiquitously overexpressed in 24 cancer classes. Genomics. 2004;84:1014-20
42. Faubert B, Li KY, Cai L, Hensley CT, Kim J, Zacharias LG. et al. Lactate metabolism in human lung tumors. Cell. 2017;171:358-71.e9
43. Anastasiou D, Poulogiannis G, Asara JM, Boxer MB, Jiang JK, Shen M. et al. Inhibition of pyruvate kinase M2 by reactive oxygen species contributes to cellular antioxidant responses. Science (New York, NY). 2011;334:1278-83
44. Zhang L, Bailleul J, Yazal T, Dong K, Sung D, Dao A. et al. PK-M2-mediated metabolic changes in breast cancer cells induced by ionizing radiation. Breast Cancer Res Tr. 2019;178:75-86
45. Mitsuishi Y, Motohashi H, Yamamoto M. The Keap1-Nrf2 system in cancers: stress response and anabolic metabolism. Front Oncol. 2012;2:200
46. Liao EC, Hsu YT, Chuah QY, Lee YJ, Hu JY, Huang TC. et al. Radiation induces senescence and a bystander effect through metabolic alterations. Cell Death Dis. 2014;5:e1255
47. Wang H, Nicolay BN, Chick JM, Gao X, Geng Y, Ren H. et al. The metabolic function of cyclin D3-CDK6 kinase in cancer cell survival. Nature. 2017;546:426-30
48. Tian T, Lu Y, Lin J, Chen M, Qiu H, Zhu W. et al. CPT1A promotes anoikis resistance in esophageal squamous cell carcinoma via redox homeostasis. Redox Biol. 2022;58:102544
49. Cosentino C, Grieco D, Costanzo V. ATM activates the pentose phosphate pathway promoting anti-oxidant defence and DNA repair. Embo J. 2011;30:546-55
50. Fan J, Ye J, Kamphorst JJ, Shlomi T, Thompson CB, Rabinowitz JD. Quantitative flux analysis reveals folate-dependent NADPH production. Nature. 2014;510:298-302
51. Shen L, Hu P, Zhang Y, Ji Z, Shan X, Ni L. et al. Serine metabolism antagonizes antiviral innate immunity by preventing ATP6V0d2-mediated YAP lysosomal degradation. Cell Metab. 2021;33:971-87.e6
52. Diehl FF, Lewis CA, Fiske BP, Vander Heiden MG. Cellular redox state constrains serine synthesis and nucleotide production to impact cell proliferation. Nat Metab. 2019;1:861-7
53. Koo T, Cho BJ, Kim DH, Park JM, Choi EJ, Kim HH. et al. MicroRNA-200c increases radiosensitivity of human cancer cells with activated EGFR-associated signaling. Oncotarget. 2017;8:65457-68
54. Bilski M, Ciesielka M, Orzechowska M, Jarosz B, Całka P, Bilska S. et al. miR-200 family as new potential prognostic factor of overall survival of patients with WHO G2 and WHO G3 brain gliomas. Sci Rep. 2024;14:29345
55. Oghabi BT, Esmaeili R. Effects of noncoding RNAs in radiotherapy response in breast cancer: a systematic review. Cell Cycle. 2022;21:883-93
56. Shen W, Liu T, Pei P, Li J, Yang S, Zhang Y. et al. Metabolic homeostasis-regulated nanoparticles for antibody-independent cancer radio-immunotherapy. Adv Mater. 2022;34:e2207343
57. Seki T, Yang Y, Sun X, Lim S, Xie S, Guo Z. et al. Brown-fat-mediated tumour suppression by cold-altered global metabolism. Nature. 2022;608:421-8
58. Gao P, Tchernyshyov I, Chang TC, Lee YS, Kita K, Ochi T. et al. c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature. 2009;458:762-5
59. Mims J, Bansal N, Bharadwaj MS, Chen X, Molina AJ, Tsang AW. et al. Energy metabolism in a matched model of radiation resistance for head and neck squamous cell cancer. Radiat Res. 2015;183:291-304
60. Moloughney JG, Kim PK, Vega-Cotto NM, Wu CC, Zhang S, Adlam M. et al. mTORC2 responds to glutamine catabolite levels to modulate the hexosamine biosynthesis enzyme GFAT1. Mol Cell. 2016;63:811-26
61. Ward IM, Chen J. Histone H2AX is phosphorylated in an ATR-dependent manner in response to replicational stress. J Biol Chem. 2001;276:47759-62
62. Fu S, Li Z, Xiao L, Hu W, Zhang L, Xie B. et al. Glutamine synthetase promotes radiation resistance via facilitating nucleotide metabolism and subsequent DNA damage repair. Cell Rep. 2019;28:1136-43.e4
63. Zhou W, Yao Y, Scott AJ, Wilder-Romans K, Dresser JJ, Werner CK. et al. Purine metabolism regulates DNA repair and therapy resistance in glioblastoma. Nat Commun. 2020;11:3811
64. Gao M, Monian P, Quadri N, Ramasamy R, Jiang X. Glutaminolysis and transferrin regulate ferroptosis. Mol cell. 2015;59:298-308
65. Yang P, Luo X, Li J, Zhang T, Gao X, Hua J. et al. Ionizing radiation upregulates glutamine metabolism and induces cell death via accumulation of reactive oxygen species. Oxid Med Cell Longev. 2021;2021:5826932
66. Chuang HY, Lee YP, Lin WC, Lin YH, Hwang JJ. Fatty acid inhibition sensitizes androgen-dependent and -independent prostate cancer to radiotherapy via FASN/NF-κB pathway. Sci Rep. 2019;9:13284
67. Souchek JJ, Baine MJ, Lin C, Rachagani S, Gupta S, Kaur S. et al. Unbiased analysis of pancreatic cancer radiation resistance reveals cholesterol biosynthesis as a novel target for radiosensitisation. Br J Cancer. 2014;111:1139-49
68. Balaban S, Shearer RF, Lee LS, van Geldermalsen M, Schreuder M, Shtein HC. et al. Adipocyte lipolysis links obesity to breast cancer growth: adipocyte-derived fatty acids drive breast cancer cell proliferation and migration. Cancer Metab. 2017;5:1
69. Gong J, Lin Y, Zhang H, Liu C, Cheng Z, Yang X. et al. Reprogramming of lipid metabolism in cancer-associated fibroblasts potentiates migration of colorectal cancer cells. Cell Death Dis. 2020;11:267
70. Matschke J, Wiebeck E, Hurst S, Rudner J, Jendrossek V. Role of SGK1 for fatty acid uptake, cell survival and radioresistance of NCI-H460 lung cancer cells exposed to acute or chronic cycling severe hypoxia. Radiat Oncol. 2016;11:75
71. Yang S, Hu C, Chen X, Tang Y, Li J, Yang H. et al. Crosstalk between metabolism and cell death in tumorigenesis. Mol Cancer. 2024;23:71
72. Jin Y, Bian S, Wang H, Mo J, Fei H, Li L. et al. CRMP2 derived from cancer associated fibroblasts facilitates progression of ovarian cancer via HIF-1α-glycolysis signaling pathway. Cell Death Dis. 2022;13:675
73. Huber V, Camisaschi C, Berzi A, Ferro S, Lugini L, Triulzi T. et al. Cancer acidity: An ultimate frontier of tumor immune escape and a novel target of immunomodulation. Semin Cancer Biol. 2017;43:74-89
74. Zhang JY, Zhou B, Sun RY, Ai YL, Cheng K, Li FN. et al. The metabolite α-KG induces GSDMC-dependent pyroptosis through death receptor 6-activated caspase-8. Cell Res. 2021;31:980-97
75. Liang J, Cao R, Wang X, Zhang Y, Wang P, Gao H. et al. Mitochondrial PKM2 regulates oxidative stress-induced apoptosis by stabilizing Bcl2. Cell Res. 2017;27:329-51
76. Khan MR, Xiang S, Song Z, Wu M. The p53-inducible long noncoding RNA TRINGS protects cancer cells from necrosis under glucose starvation. EMBO J. 2017;36:3483-500
77. Liang M, Li JW, Luo H, Lulu S, Calbay O, Shenoy A. et al. Epithelial-mesenchymal transition suppresses AMPK and sensitizes cancer cells to pyroptosis under energy stress. Cells. 2022;11:2208
78. Zhang T, Xu D, Trefts E, Lv M, Inuzuka H, Song G. et al. Metabolic orchestration of cell death by AMPK-mediated phosphorylation of RIPK1. Science (New York, NY). 2023;380:1372-80
79. Hsieh PF, Jiang WP, Basavaraj P, Huang SY, Ruangsai P, Wu JB. et al. Cell suspension culture extract of Eriobotrya japonica attenuates growth and induces apoptosis in prostate cancer cells via targeting SREBP-1/FASN-driven metabolism and AR. Phytomedicine. 2021;93:153806
80. Huang Y, Yong P, Dickey D, Vora SM, Wu H, Bernlohr DA. Inflammasome activation and pyroptosis via a lipid-regulated SIRT1-p53-ASC axis in macrophages from male mice and humans. Endocrinology. 2022;163:bqac014
81. Li D, Li Y. The interaction between ferroptosis and lipid metabolism in cancer. Sig Transduct Target Ther. 2020;5:108
82. Liu X, Lieberman J. Knocking 'em dead: pore-forming proteins in immune defense. Annu Rev Immunol. 2020;38:455-85
83. Hu B, Jin C, Li HB, Tong J, Ouyang X, Cetinbas NM. et al. The DNA-sensing AIM2 inflammasome controls radiation-induced cell death and tissue injury. Science (New York, NY). 2016;354:765-8
84. Yaron JR, Gangaraju S, Rao MY, Kong X, Zhang L, Su F. et al. K(+) regulates Ca(2+) to drive inflammasome signaling: dynamic visualization of ion flux in live cells. Cell Death Dis. 2015;6:e1954
85. Xiao J, Wang C, Yao JC, Alippe Y, Yang T, Kress D. et al. Radiation causes tissue damage by dysregulating inflammasome-gasdermin D signaling in both host and transplanted cells. PLoS Biol. 2020;18:e3000807
86. Smith AO, Ju W, Adzraku SY, Wenyi L, Yuting C, Qiao J. et al. Gamma radiation induce inflammasome signaling and pyroptosis in microvascular endothelial Cells. J Inflamm Res. 2021;14:3277-88
87. Han C, Godfrey V, Liu Z, Han Y, Liu L, Peng H. et al. The AIM2 and NLRP3 inflammasomes trigger IL-1-mediated antitumor effects during radiation. Sci Immunol. 2021;6:eabc6998
88. Yu J, Li S, Qi J, Chen Z, Wu Y, Guo J. et al. Cleavage of GSDME by caspase-3 determines lobaplatin-induced pyroptosis in colon cancer cells. Cell Death Dis. 2019;10:193
89. Zhou B, Zhang JY, Liu XS, Chen HZ, Ai YL, Cheng K. et al. Tom20 senses iron-activated ROS signaling to promote melanoma cell pyroptosis. Cell Res. 2018;28:1171-85
90. Cao W, Chen G, Wu L, Yu KN, Sun M, Yang M. et al. Ionizing radiation triggers the antitumor immunity by inducing Gasdermin E-mediated pyroptosis in tumor cells. Int J Radiat Oncol Biol Phys. 2023;115:440-52
91. Annibaldi A, Wicky John S, Vanden Berghe T, Swatek KN, Ruan J, Liccardi G. et al. Ubiquitin-Mediated Regulation of RIPK1 Kinase Activity Independent of IKK and MK2. Mol Cell. 2018;69:566-80.e5
92. Fritsch M, Günther SD, Schwarzer R, Albert MC, Schorn F, Werthenbach JP. et al. Caspase-8 is the molecular switch for apoptosis, necroptosis and pyroptosis. Nature. 2019;575:683-7
93. Liu Z, Dagley LF, Shield-Artin K, Young SN, Bankovacki A, Wang X. et al. Oligomerization-driven MLKL ubiquitylation antagonizes necroptosis. Embo J. 2021;40:e103718
94. Yatim N, Jusforgues-Saklani H, Orozco S, Schulz O, Barreira da Silva R, Reis e Sousa C. et al. RIPK1 and NF-κB signaling in dying cells determines cross-priming of CD8⁺ T cells. Science. 2015;350:328-34
95. Jiao H, Wachsmuth L, Kumari S, Schwarzer R, Lin J, Eren RO. et al. Z-nucleic-acid sensing triggers ZBP1-dependent necroptosis and inflammation. Nature. 2020;580:391-5
96. Yang Y, Wu M, Cao D, Yang C, Jin J, Wu L. et al. ZBP1-MLKL necroptotic signaling potentiates radiation-induced antitumor immunity via intratumoral STING pathway activation. Sci Adv. 2021;7:eabf6290
97. Jia Y, Wang F, Guo Q, Li M, Wang L, Zhang Z. et al. Curcumol induces RIPK1/RIPK3 complex-dependent necroptosis via JNK1/2-ROS signaling in hepatic stellate cells. Redox Biol. 2018;19:375-87
98. Stockwell BR, Friedmann Angeli JP, Bayir H, Bush AI, Conrad M, Dixon SJ. et al. Ferroptosis: a regulated cell death nexus linking metabolism, redox biology, and disease. Cell. 2017;171:273-85
99. Lei G, Mao C, Yan Y, Zhuang L, Gan B. Ferroptosis, radiotherapy, and combination therapeutic strategies. Protein Cell. 2021;12:836-57
100. Yan B, Ai Y, Sun Q, Ma Y, Cao Y, Wang J. et al. Membrane damage during ferroptosis is caused by oxidation of phospholipids catalyzed by the oxidoreductases POR and CYB5R1. Mol Cell. 2021;81:355-69.e10
101. Doll S, Proneth B, Tyurina YY, Panzilius E, Kobayashi S, Ingold I. et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat Chem Biol. 2017;13:91-8
102. Zou Y, Li H, Graham ET, Deik AA, Eaton JK, Wang W. et al. Cytochrome P450 oxidoreductase contributes to phospholipid peroxidation in ferroptosis. Nat Chem Biol. 2020;16:302-9
103. Conrad M, Sato H. The oxidative stress-inducible cystine/glutamate antiporter, system x (c) (-): cystine supplier and beyond. Amino Acids. 2012;42:231-46
104. Wiernicki B, Maschalidi S, Pinney J, Adjemian S, Vanden Berghe T, Ravichandran KS. et al. Cancer cells dying from ferroptosis impede dendritic cell-mediated anti-tumor immunity. Nat Commun. 2022;13:3676
105. Han L, Bai L, Qu C, Dai E, Liu J, Kang R. et al. PPARG-mediated ferroptosis in dendritic cells limits antitumor immunity. Biochem Biophys Res Commun. 2021;576:33-9
106. Ugolini A, Tyurin VA, Tyurina YY, Tcyganov EN, Donthireddy L, Kagan VE. et al. Polymorphonuclear myeloid-derived suppressor cells limit antigen cross-presentation by dendritic cells in cancer. JCI Insight. 2020;5:e138581
107. Kim R, Hashimoto A, Markosyan N, Tyurin VA, Tyurina YY, Kar G. et al. Ferroptosis of tumour neutrophils causes immune suppression in cancer. Nature. 2022;612:338-46
108. Wang W, Green M, Choi JE, Gijón M, Kennedy PD, Johnson JK. et al. CD8(+) T cells regulate tumour ferroptosis during cancer immunotherapy. Nature. 2019;569:270-4
109. Guo S, Yao Y, Tang Y, Xin Z, Wu D, Ni C. et al. Radiation-induced tumor immune microenvironments and potential targets for combination therapy. Sig Transduct Target Ther. 2023;8:205
110. Chen J, Levy A, Tian AL, Huang X, Cai G, Fidelle M. et al. Low-dose irradiation of the gut improves the efficacy of PD-L1 blockade in metastatic cancer patients. Cancer Cell. 2025;43:361-79.e10
111. Formenti SC, Demaria S. Combining radiotherapy and cancer immunotherapy: a paradigm shift. J Natl Cancer Inst. 2013;105:256-65
112. Yu WD, Sun G, Li J, Xu J, Wang X. Mechanisms and therapeutic potentials of cancer immunotherapy in combination with radiotherapy and/or chemotherapy. Cancer Lett. 2019;452:66-70
113. Szklener K, Mazurek M, Wieteska M, Wacławska M, Bilski M, Mańdziuk S. New Directions in the Therapy of Glioblastoma. Cancers (Basel). 2022;14:5377
114. Szklener K, Bilski M, Nieoczym K, Mańdziuk D, Mańdziuk S. Enhancing glioblastoma treatment through the integration of tumor-treating fields. Front Oncol. 2023;13:1274587
115. Verginadis II, Citrin DE Ky B, Feigenberg SJ Georgakilas AG, Hill-Kayser CE et al. Radiotherapy toxicities: mechanisms, management, and future directions. Lancet. 2025;405:338-52
116. De Ruysscher D, Niedermann G, Burnet NG, Siva S, Lee AWM, Hegi-Johnson F. Radiotherapy toxicity. Nat Rev Dis Primers. 2019;5:13
117. Kuncman Ł, Pajdziński M, Smółka K, Bilski M, Socha J, Stando R. et al. Early lymphocyte levels and low doses radiation exposure of lung predict lymphopenia in radiotherapy for lung cancer. Front Immunol. 2024;15:1426635
118. Dai D, Tian Q, Shui Y, Li J, Wei Q. The impact of radiation induced lymphopenia in the prognosis of head and neck cancer: A systematic review and meta-analysis. Radiother Oncol. 2022;168:28-36
119. Kuncman Ł, Stawiski K, Bilski M, Nowak-Potemska J, Bilewicz M, Fijuth J. Radiation induced lymphopenia depends on lymph node irradiation in prostate cancer radiotherapy. Int J Radiat Oncol Biol Phys. 2024;120:e549-e50
120. Pike LRG, Bang A, Mahal BA, Taylor A, Krishnan M, Spektor A. et al. The impact of radiation therapy on lymphocyte count and survival in metastatic cancer patients receiving PD-1 immune checkpoint inhibitors. Int J Radiat Oncol Biol Phys. 2019;103:142-51
121. McCullum L, Shin J, Xing S, Beekman C, Schuemann J, Hong T. et al. Predicting severity of radiation induced lymphopenia in individual proton therapy patients for varying dose rate and fractionation using dynamic 4-dimensional blood flow simulations. Int J Radiat Oncol Biol Phys. 2023;116:1226-33
122. Martin OA, Yin X, Forrester HB, Sprung CN, Martin RF. Potential strategies to ameliorate risk of radiotherapy-induced second malignant neoplasms. Semin Cancer Biol. 2016;37-38:65-76
123. Kouvaris JR, Kouloulias VE, Vlahos LJ. Amifostine: the first selective-target and broad-spectrum radioprotector. Oncologist. 2007;12:738-47
124. Epperly MW, Defilippi S, Sikora C, Gretton J, Kalend A, Greenberger JS. Intratracheal injection of manganese superoxide dismutase (MnSOD) plasmid/liposomes protects normal lung but not orthotopic tumors from irradiation. Gene Ther. 2000;7:1011-8
125. Mitchell JB, Anver MR, Sowers AL, Rosenberg PS, Figueroa M, Thetford A. et al. The antioxidant tempol reduces carcinogenesis and enhances survival in mice when administered after nonlethal total body radiation. Cancer Res. 2012;72:4846-55
126. Choi YJ, Kim MJ, Lee YJ, Choi M, Shim WS, Park M. et al. Prevention of radiotherapy-induced pro-tumorigenic microenvironment by SFK inhibitors. Theranostics. 2025;15:875-93
Author contact
![]() Corresponding authors: An Song, Zhi-Jun Sun; ansongedu.cn, sunzjedu.cn.
Corresponding authors: An Song, Zhi-Jun Sun; ansongedu.cn, sunzjedu.cn.