Impact Factor ISSN: 1449-1907
- Issue 9; 2026
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Volume 23; 2026
- Past Issues
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and methods
Results
Discussion
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2023; 20(6):725-736. doi:10.7150/ijms.82667 This issue Cite
Research Paper
Role of the gut microbiota and their metabolites in hemodialysis patients
Ying Ting Chao1#, Ying-Kuang Lin1,2#, Liang-Kun Chen1, Poyin Huang3,4,5,6, Yi-Chiung Hsu1 ![]()
1. Department of Biomedical Sciences and Engineering, National Central University, Taoyuan 320317, Taiwan, R.O.C.
2. Division of Nephrology, Department of Medicine, Landseed International Hospital, Taoyuan City 324609, Taiwan, R.O.C.
3. Department of Neurology, Kaohsiung Medical University Hospital, Kaohsiung Medical University, Kaohsiung City, 807, Taiwan, R.O.C.
4. Department of Neurology, Kaohsiung Municipal Siaogang Hospital, Kaohsiung Medical University, Kaohsiung City, 807, Taiwan R.O.C.
5. Neuroscience Research Center, Kaohsiung Medical University, Kaohsiung City, 807, Taiwan R.O.C.
6. Department of Neurology, Faculty of Medicine, College of Medicine, Kaohsiung Medical University, Kaohsiung City, 807, Taiwan R.O.C.
# means equal contribution.
Received 2023-1-15; Accepted 2023-3-25; Published 2023-4-17
Abstract

High serum phosphate levels in chronic kidney disease (CKD) are linked to adverse health outcomes, including cardiovascular disease, kidney disease progression, and all-cause mortality. This study is aimed to find out which microorganisms or microbial functions have a significant impact on higher calcium-phosphorus product (Ca x P) after they undergo hemodialysis (HD) treatment. Feces samples from 30 healthy controls, 15 dialysis patients with controlled Ca xP (HD), and 16 dialysis patients with higher Ca xP (HDHCP) were collected to perform in 16S amplicon sequencing.
We found gut microbial composition was significantly different between hemodialysis patients and healthy controls. Three phyla including Firmicutes, Actinobacteria, and Proteobacteria were significantly enriched in hemodialysis patients. Although only one genus, Lachnospiraceae_FCS020_group, was significantly increased in higher Ca xP group, there were four metabolic pathways predicted by PICRUSt significantly increased in higher Ca xP group and associated with causing VC, including the pentose phosphate pathway, steroid biosynthesis, terpenoid backbone biosynthesis, and fatty acid elongation pathway. Characterizing dysbiosis of gut microbiome played the important role in hemodialysis patients.
Keywords: calcium-phosphorus product, chronic kidney disease, gut microbiota, dysbiosis
Introduction
Chronic kidney disease (CKD) is one of the leading causes of death globally in the 21st century and affects 10% of the general population worldwide, amounting to more than 800 million individuals [1]. Cardiovascular disease (CVD) is the main cause of morbidity and mortality among patients with CKD [2, 3], and the mortality of CVD patients with chronic renal disease was higher. The mortality rate of the patients who undergo hemodialysis (HD), and peritoneal dialysis (PD) are 10 to 20 times than the general population [4]. Furthermore, vascular calcification (VC) is a risk factor associated with major adverse cardiovascular events [5]. Vascular calcification is linked to elevated calcium-phosphorus product (Ca × P) [6.7]. End-stage renal failure, also known as end-stage renal disease (ESRD), occurs when chronic kidney disease. The kidney of ESRD patients lost filtering abilities can no longer function on their own.
A ESRD patient must receive HD, PD or kidney transplantation in order to survive. Kraus et al. demonstrated that 100% of patients withESRD on the treatment with hemodialysis for 3 months suffer from calcification in the aortic valve, mitral valve, or mitral annulus by echocardiography, and 77.8% of X-rayed patients suffer from abdominal aortic calcification [8]. In those CKD patients who haven't applied dialysis, the incidence rate of coronary artery calcification is 25.5% [9].
VC was also induced by high phosphorus caused by CKD, parathyroid hormone (PTH), inflammatory cytokines, oxidative stress, and uremic toxins [10]. Moreover, the gut microbiota-derived metabolites like p-cresol (pCS), indoxyl sulfate (IS), Trimethylamine N-oxide (TMAO), bile acid, and phenylacetylglutamine affected vascular smooth muscle cells or vascular endothelial cells which directly or indirectly induce VC [10, 11]. The human gut microbiota collectively made up to 100 trillion archaeal and bacterial cells and contained over 99% of bacteria distributed over more than 1,000 species [12]. The relative abundance of major bacterial populations in fecal samples from patients with CKD and patients with ESRD versus healthy controls was shifted [11]. Much direct experimental evidence supported the contribution of fecal microbial community changes in CVD [13]. Therefore, we mainly focus on microorganisms or microbial functions which may be contributing to the increased risk of higher calcium-phosphorus product (Ca × P) by discovering gut microbial profiles between healthy controls, and hemodialysis patients with or without higher Ca × P.
Materials and methods
Patients and Controls
The hemodialysis or with higher Ca × P patients who signed informed consent and would be compliant for follow-up were recruited to the study. The study conformed to the ethical guidelines of the 1975 Declaration of Helsinki and was approved by the Institutional Review Board of the [IRB-20-046-B1]. The recruited patients will be collected their basic demographics, hospital courses, use of antibiotics, type of feedings, and other treatments in the Landseed International Hospital. The patients we collected in the study have been excluded if they use either antibiotics or probiotics. We used calcium-phosphate product (Ca x P) as an indicator which is a clinically relevant tool to estimate the risk of patients with renal failure. The calcium phosphorus product must be kept <55 mg2 / dL2 [6]. CKD patients are controlled their metabolism by monitoring the plasma levels of phosphorus within the range of target values. When the score of calcium-phosphate product is greater than 60 mg2 / dL2, we employed these cut-off values to identify patients with higher Ca x P levels.
Both patients on hemodialysis and control subjects were asked to bring a fecal sample to the laboratory within 12 h of defecation. After collection of the fecal sample, an aliquot was immediately frozen and stored at -20°C until further analysis.
Stool DNA extraction and 16S rRNA gene amplification
DNA was extracted with QIAamp Fast DNA Stool Mini Kit (Qiagen, Hilden, Germany) all according to respective manufacturers' instructions.
The 16S rRNA sequencing libraries are followed according to the manufacturer's instructions provided by Illumina (Illumina, CA, USA). Briefly, 12.5 ng of DNA is used for PCR amplification of the V3 and V4 regions of 16S rRNA gene. The PCR products are purified with AMPure XP beads (Beckman Coulter, USA) and subjected to a secondary PCR reaction with primers from Nextera XT Index kit (Illumina, CA, USA) by adding dual indices and Illumina sequencing adapters. After PCR reaction, the final libraries (~630 bp) are purified with AMPure XP beads and ready for next-generation sequencing.
16S rRNA Gene Amplicon Sequencing
The concentrations of 16S rRNA sequencing libraries are determined by real-time quantitative PCR with Illumina adapter-specific primers provided by KAPA library quantification kit (KAPA Biosystems, USA). Libraries are denatured and sequenced by Illumina MiSeq platform with reagent v3 for pair-end sequencing (2*250 bp/ 2*300 bp). Instrument control, cluster generation, image capture and base calling are processed by Real-Time Analysis software (RTA), MiSeq Control software (MCS) and MiSeq Report software (MSR) on MiSeq platform. FASTQ files generated by MiSeq Report are used for further analysis.
Bioinformatics and statistical analysis
Raw reads were processed using the mothur [14] V1.35.0 and were based upon a protocol developed for MiSeq by the mothur creators [15] with default parameters. The read pairs were assembled into contigs by aligning the reads and any sequences which failed to align were discarded. The sequence was then aligned to SILVA reference alignment [16] and any reads aligned outside of the V3-V4 region were removed from the dataset. Chimeras were identified and removes using UCHIME [17]. The sequences were classified using mothur's Bayesian classifier against the SILVA database (release 128) [16]. Sequences identified as chloroplast, mitochondria or not originating from bacteria were removed from further analysis. The sequences were then clustered into Operational Taxonomic Units (OTUs) based on 97% sequence similarity. Taxa summaries, OTU heatmap and alpha rarefaction were performed in QIIME [18]. Principal coordinate analyses (PCoA) were based on unweighted and weighted UniFrac distances and calculated in QIIME. Non-metric multidimensional scaling (NMDS) and canonical correspondence analysis (CCA) were analyzed using vegan (R package: v2.4 1) [19] package and heatmap were analyzed using gplots [20] package in R. High-dimensional biomarker discovery and explanation that identifies genomics taxonomy characterizing the differences between two or more biological conditions were performed in LEfSe v1.0 [21]. Differential analyses of the count data at the OTU level were applied to use negative binomial generalized linear model implemented in DESeq2 [22, 23] R package. Functional contents of the 16S rRNA were predicted using the PICRUSt software v2.2.0 b [24]. Differential analyses of the functional contents were also using negative binomial generalized linear model implemented in DESeq2 R package. The statistical significance related to the relative abundance was using one‐way ANOVA and post‐hoc Tukey's test, GraphPad Prism® version 9 (GraphPad Software, USA).
Results
Clinical characteristics of the Participants
A total of 61 stool samples were collected, including 30 healthy controls (Ctrl), 15 hemodialysis patients without higher Ca x P (HD), and 16 hemodialysis patients with higher Ca x P (HDHCP).
Clinical characteristics were described in Table 1. In the validation cohort, the proportions of women and men were similar in each group. The average age of Ctrl, HD and HDHCP patients were at age 58.3, 63.5 and 55.3. Hemodialysis time was not significantly different between HD and HDHCP. However, in HD group, serum levels of creatinine (Cr), uric acid (UA), albumin (Alb), sodium (Na), calcium-phosphate product (Ca×P), calcium (Ca) and Intact Parathyroid hormone (iPTH) were significantly increased in HDHCP (p < 0.05).
Clinical characteristics in this study were exhibited by patients with healthy controls (Ctrl), hemodialysis dialysis patients without higher Ca x P (HD), and hemodialysis patients with higher Ca x P (HDHCP). Categorical variables were compared using one-way ANOVA or Student's t-test.
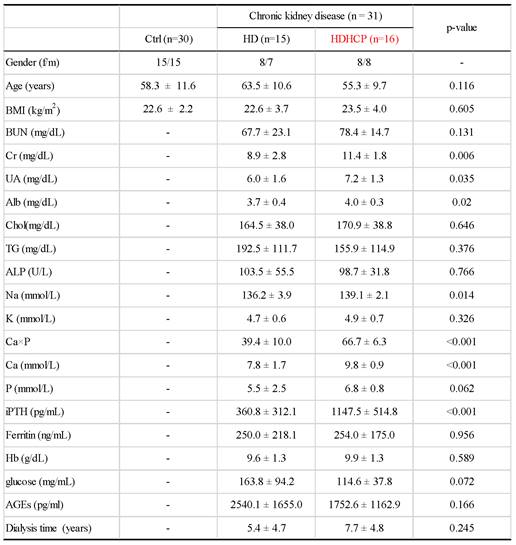
Gut microbial diversity of healthy controls and hemodialysis patients
A total of 19,647,778 sequencing sequences were generated from stool samples collected from 61 participants (range, 166,892- 772,186). After quality and size filtered, and host sequence removed, 15,326,912 reads were clustered into OTUs (range, 123,032- 715,042). A total of 100,8282 OTUs with more than 1 read were generated for downstream analysis. Rarefaction analysis showed that the number of OTUs was significantly increased in hemodialysis patients (n = 31) compared to that in healthy controls (n = 30) (p < 0.0001) (Supplementary data S1 A). As measured by the Chao index (Supplementary data S1 B), the Ace index (Supplementary data S1 C), and the Shannon index (Supplementary data S1 D), the gut microbial diversity was significantly increased in HD and HDHCP compared to Ctrl (p ≦ 0.0001).
In addition, the principal coordinate analysis (PCoA), non-metric multidimensional scaling (NMDS) and the statistical analysis of Adonis based on OTUs distribution were executed to illustrate the diversity. The gut microbial composition was significantly different between hemodialysis patients and healthy controls (Figure 1A and 1B). While diversity in hemodialysis patients with higher Ca x P was not different significantly from hemodialysis patients without higher Ca x P.
Gut microbial diversity based on OTUs distribution in healthy controls and hemodialysis patients (A-B).
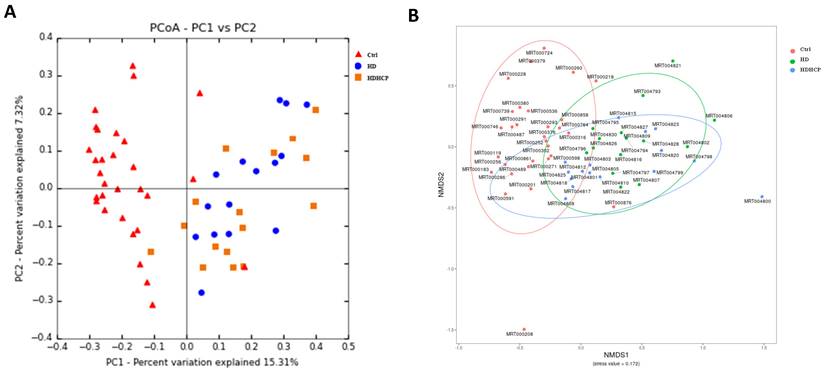
Gut microbiome in healthy controls and hemodialysis patients
Further analysis is to do taxonomic analysis. Average compositions and relative abundance of the bacterial community in three groups at the phylum and the genus levels have been shown in Figure 2A and 2B. Three phyla including Firmicutes, Actinobacteria and Proteobacteria were significantly enriched, while Bacteroidetes and Verrucomicrobia were significantly decreased in hemodialysis patients.
Gut microbial composition of healthy controls and hemodialysis patients. Average compositions and relative abundance of the bacterial community in three groups at levels of the phylum (A) and the genus (B). (C) The difference in gut microbial composition was shown in Ctrl, HD and HDHCP.
At the genus level, Blautia, Streptococcus, Lachnospiraceae_unclassified, Ruminococcus_gnavus_group, Collinsella and Burkholderia-Paraburkholderia in hemodialysis patients are the most abundant and more than that in the healthy controls. The difference in gut microbial composition was shown in Ctrl, HD and HDHCP (Figure 2C).
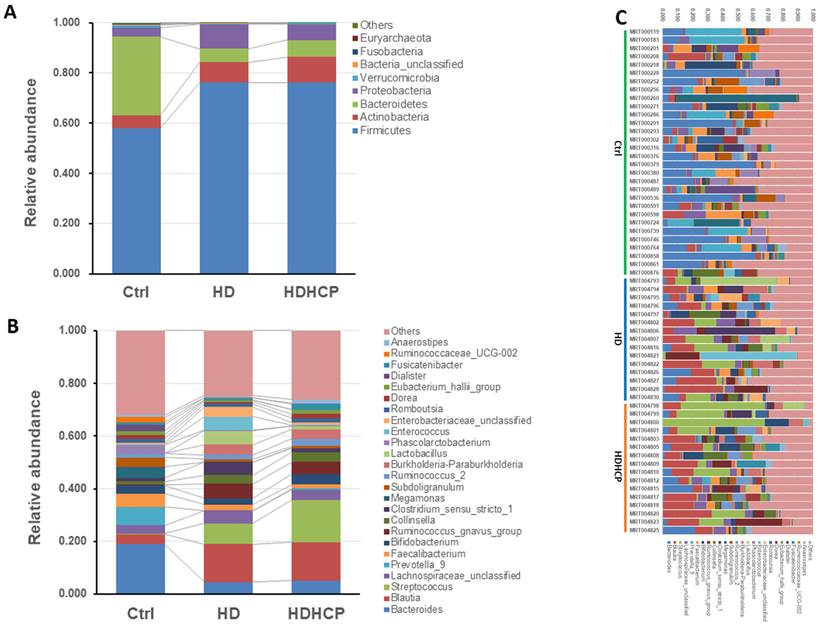
Potential bacteria related to higher Ca x P in hemodialysis patients
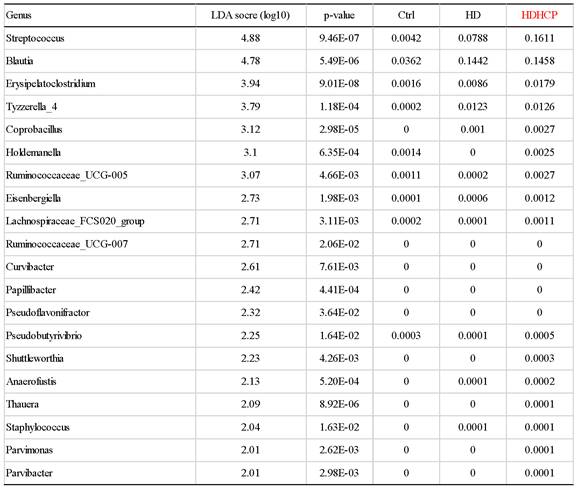
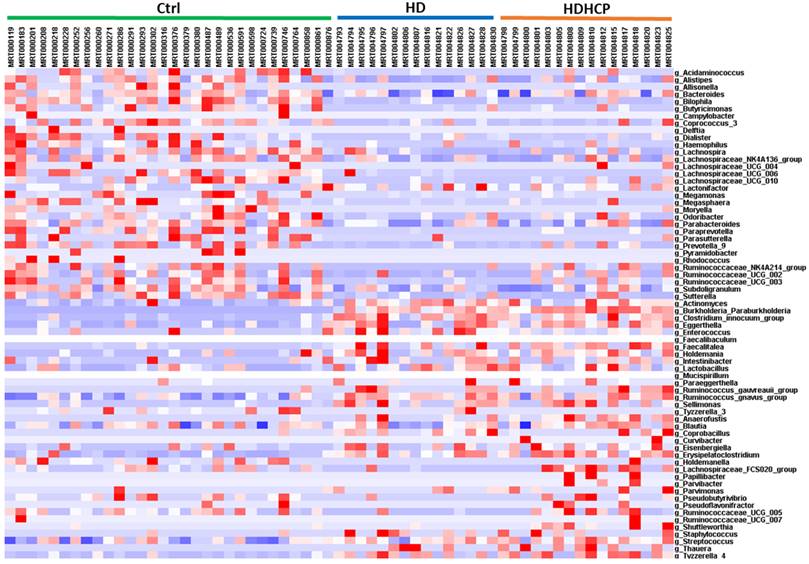
Linear discriminant analysis effect size (LEfSe) was used to compare the presence and impact of region-specific OTUs in three groups, to determine the specific bacterial taxa and predominant. The threshold of the linear discriminant analysis (LDA) was >2.0 and p<0.05. The relative abundances of the family Streptococcaceae, Coriobacteriaceae, Erysipelotrichaceae, Dermabacteraceae, Dermatophilaceae, Micrococcaceae, Family_XI and Rhodocyclaceae were found to be higher in HDHCP. The taxonomic clusters based on LDA selection at the genus level were shown in Supplementary data S2. According to the LDA selection, 32 genera were significantly enriched in Ctrl, 16 genera in HD and 20 genera in HDHCP. Heatmap analysis of the relative abundance of the 68 genera was demonstrated the LEfSe result (Figure 3). In particular, the relative abundances of the genera Streptococcus, Blautia, Erysipelatoclostridium, Tyzzerella_4, Coprobacillus, Holdemanella, Ruminococcaceae_UCG-005, Eisenbergiella Lachnospiraceae_FCS020_group, Ruminococcaceae_UCG-007, Curvibacter, Papillibacter, Pseudoflavonifractor, Pseudobutyrivibrio, Shuttleworthia, Anaerofustis, Thauera, Staphylococcus, Parvimonas and Parvibacter were higher in HDHCP (Table 2).
According to LEfSe results, 20 genera were significantly enriched in HDHCP.
Heatmap showing the relative abundance of the 68 genera that were significantly enriched between Ctrl, HD, and HDHCP.
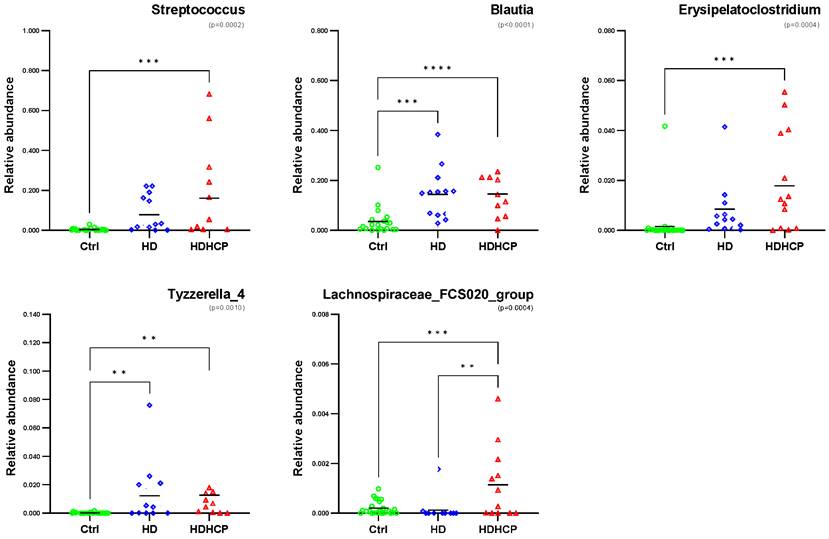
One-way ANOVA was used to analyze the statistical significance of 20 genera significantly enriched in HDHCP. As the result showed in Figure 4, 5 genera including Streptococcus, Blautia, Erysipelatoclostridium, Tyzzerella_4 and Lachnospiraceae_FCS020_group were significantobserved in HDHCP. Besides, only the Lachnospiraceae_FCS020_group was significantly enriched in HDHCP compared to HD.
Relative abundance of 5 genera with significantly enriched in HDHCP and average abundance more than 0.001 in Ctrl, HD and HDHCP.
Correlation between the Gut Microbiome and Clinical Indicators of hemodialysis patients
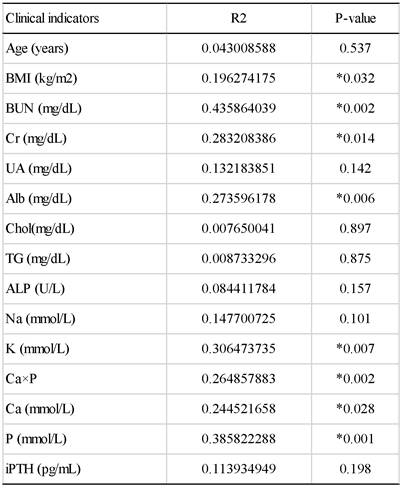
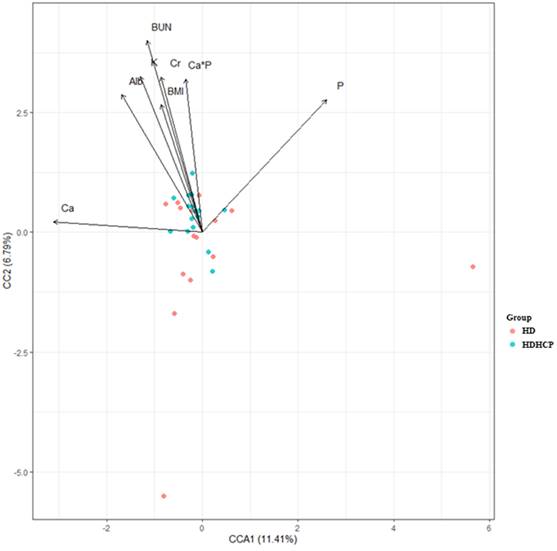
To further understand the impact of clinical indicators of hemodialysis patients on the gut microbiome in the HD and HDHCP, canonical correspondence analysis (CCA) was performed at the OTU level. Fifteen clinical indicators of hemodialysis patients (Age, BMI, BUN, Cr, UA, Alb, Chol, TG, ALP, Na, K, (Ca×P), Ca, P and iPTH) were analyzed. CCA of the gut microbiome and these clinical indicators were shown in Table 3. Eight clinical indicators (BMI, BUN, Cr, Alb, K, Ca*P, Ca and P) were closely related to the gut microbiome in hemodialysis patients (Figure 5).
The canonical correspondence analysis of the associations between the gut microbiome and clinical indicators for hemodialysis patients.
The CCA analysis of the associations between the gut microbiome and clinical indicators for hemodialysis patients in the HD and HDHCP.
Crucial microbial functions related to higher Ca x P in dialysis patients
PICRUSt software was used to predict the relative abundance of functions according to the obtained 16S rRNA sequences to determine the abundance and enrichment of functional genes at different levels in the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways. The predicted functions classified as metabolism at the first level of the KEGG were conducted to identify the predominant microbial functions by LEfSe. According to the LDA selection, 67 predicted microbial functions were shown in Figure 6. The result exhibited 17 functions including tryptophan metabolism, tyrosine metabolism and secondary bile acid biosynthesis were increased in HD. In addition, 17 functions including pentose phosphate pathway (PPP), steroid biosynthesis, terpenoid backbone biosynthesis, and fatty acid elongation pathway were increased in HDHCP.
Functional alterations of the gut microbiome in healthy controls and hemodialysis patients. LEfSe results showed a statistically significant increase in the abundance metabolism of KEGG pathways in the Ctrl, HD, and HDHCP.
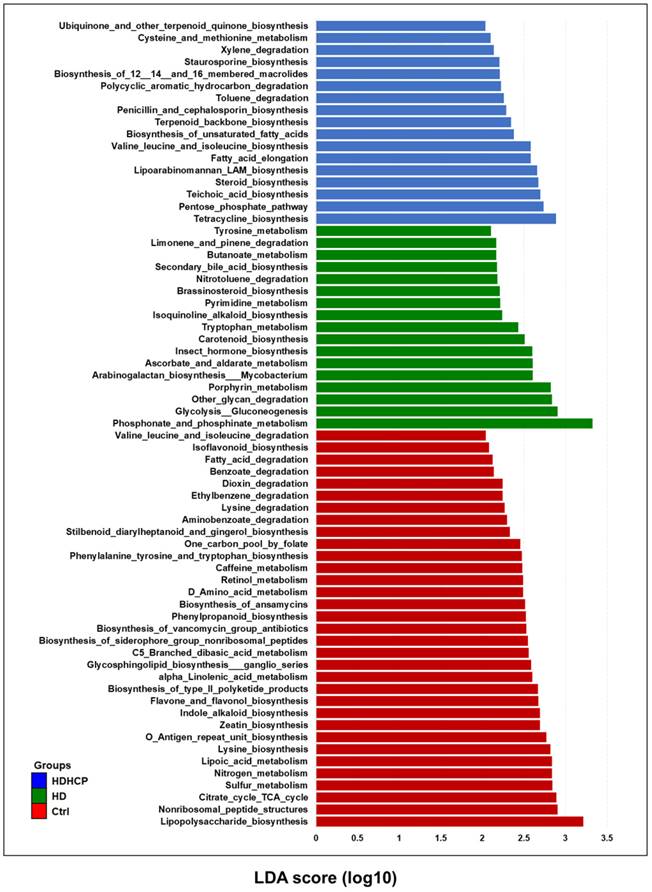
Discussion
Two dominant bacterial phyla, Bacteroidetes and Firmicutes, were found in more than 90% of the gut microbiome of healthy people [26]. The total percentage of Bacteroidetes and Firmicutes of the gut microbiome was almost 90% in the healthy controls, but only 81.6% and 82.9% in HD and HDHCP, respectively. The proportion changed was affected by the decrease of Bacteroidetes in the gut microbiome of hemodialysis patients (Ctrl:31.2%, HD:5.5% and HDHCP: 6,7%) (Supplementary data S3). According to Bao et al. study, their study purpose is similar with us [27]. They indicated the majority of phyla in the gut microbiome of hemodialysis (HD) patients were Firmicutes, Actinobacteriota, Proteobacteria, and Bacteroidota which had same trend compared to our results. In terms of abundant phyla, our result was consistent with theirs. The percentages in our result of these two phyla Firmicutes and Actinobacteriota were over 70% and 8-10% respectively in HD patients, which the proportion is 70% and 10% in their study. In Ana Merino-Ribas' study, they included 44 CKD patients undergoing peritoneal dialysis [28]. The results of the gut microbiome at the phylum level enriched in Firmicutes and Bacteroidota which were consistent with us. However, the abundance composition ratio in PD patients was not in consonance with our study.
The decrease of protein digestion and absorption capacity occurring in CKD patients when the decrease of fiber intake and the increase of protein intake in the dietary treatment, leads to the decrease of short-chain fatty acids and the increase of protein in the gut, which in turn causes their dysbiosis [29]. Shin et al proposed that an increased prevalence of Proteobacteria would be a potential diagnostic signature of dysbiosis and risk of disease due to an imbalanced gut microbiota often arose from a sustained increase in abundance of Proteobacteria [30]. The content of Proteobacteria in the gut microbiome of healthy people was less than 1% [26]. While the relative abundance of Proteobacteria was increased more significantly in hemodialysis patients than healthy controls. This result was concordant with other studies [31, 32] which is a characterization of dysbiosis in CKD patients.
The diversity of the gut microbiome in heathy controls was higher than mild CKD and moderate CKD patients, but lower than advanced CKD patients [32]. Our result also demonstrated that the diversity involved both taxon richness and evenness in hemodialysis patients was significantly higher than healthy controls. Nevertheless, the diversity involved both taxon richness and evenness were similar as estimated by observed species, Chao1, Ace, Shannon diversity indexes between HD and HDHCP. Similarly, very few bacteria were showed significant differences between HD and HDHCP. Among the 65 genera selected by LDA, only Lachnospiraceae_FCS020_group was significantly different between HD and HDHCP.
Lachnospiraceae_FCS020_group was a genus of Lachnospiraceae family producing short chain fatty acids (SCFA). TMAO induced inflammatory gene expression on vascular smooth muscle cells to contribute to atherosclerosis and is associated with coronary artery disease risk [33]. While serum TMAO levels were positively associated with decreased abundance of Lachnospiraceae_FCS020_group [34]. In addition, Lachnospiraceae_FCS020_group was found to be negatively associated with triglycerides in VLDL particles of various sizes, small HDL particles, and medium HDL particles [35]. According to these research data, Lachnospiraceae_FCS020_group seemed to be negatively associated with VC, but it was probably due to the insufficient published data.
pCS, IS, TMAO, bile acid and phenylacetylglutamine induced VC were derived from gut microbiome [10, 11]. pCS, IS and bile acid originated from tyrosine metabolism, tryptophan metabolism and secondary bile acid biosynthesis, respectively. All three metabolic pathways were significantly increased in HD, but not in HDHCP. Although the blood vessels did not show calcification in HD, the three metabolic pathways were significantly increased. This indicated that the production of pCS, IS and bile acid probably caused the blood vascular abnormalities and raised the risk of VC. In HDHCP group, the metabolic pathways which significantly increased and potentially causing VC were PPP, steroid biosynthesis, terpenoid backbone biosynthesis, and fatty acid elongation pathway.
The PPP was related to the regulation of the intracellular Ca2+. The inhibition of PPP decreased intracellular Ca2+ to oxidize NADPH and GSH and appears to activate a novel coordination of redox-controlled relaxing mechanisms in bovine coronary arteries [36]. Peiró et al indicated that activation of the PPP by pro-inflammatory cytokines caused a pro-oxidant environment by creating a situation in which free radical formation exceeds the capacity of the cell to generate GSH to increased vascular inflammation and induced the vascular damage [37]. In addition, the PPP was inhibited by AGEs, but AGEs accelerated VC in VSMC [38]. Although the HDHCP group had a lower mean value of AGEs compared to the HD group and slightly influenced VC, the activation of the PPP was contributed to developing higher calcium-phosphorus product.
The steroid biosynthesis pathway and the terpenoid backbone biosynthesis pathway were two increased lipid metabolic pathways in HDHCP. The terpenoid backbone biosynthesis produces steroid, and steroid and was converted to form cholesterol by the steroid biosynthesis pathway. Cholesterol produced cholesteryl esters by the esterification of cholesterol with long-chain fatty acids. Cholesterol ester will interact with triacylglycerol to produce very low-density lipoprotein (VLDL). In nondiabetic subjects, higher VLDL size was significantly associated with CAC [39]. In a high-risk type-2 diabetic population, very low-density lipoprotein cholesterol (VLDL-C) levels were positively associated with increasing CAC [40]. The VLDL-C level is an independent risk factor for all-cause and cardiovascular mortality in peritoneal dialysis patients [41]. The elevated VLDL-C was significantly associated with elevated coronary heart disease risk in a large Chinese cohort study [42].
In addition, the fatty acid elongation pathway also enriched in HDHCP. The fatty acid elongation pathway produced palmitic acid (PA) which was the most abundant long-chain saturated fatty acid (LCFA) in plasma. PA was quantitatively the major fatty acid produced during hepatic lipogenesis to generate VLDL [43]. PA induced osteoblastic differentiation and calcium deposition in VSMC through increasing the expression of the genes for bone-related proteins in human aortic smooth muscle cells and then inducing the activation of NF-kB [44]. Long-chain fatty acids activated calcium channels and increased intracellular calcium in ventricular myocytes [45]. The accumulation of LCFAs in mature erythrocytes was sensitive to single HD treatment which increased the cardiovascular risk in ESRD patients [46].
Our study demonstrated that Lachnospiraceae_FCS020_group was significantly increased in hemodialysis patients with VC compared to hemodialysis patients without VC. Perhaps Lachnospiraceae_FCS020_group was a potential biomarker for VC diagnosis. Besides, we found the changes of metabolic functions in hemodialysis patients with VC and the changes were related to causing VC. The clinical characteristics showed that Ca, Ca×P and iPTH of blood were significantly increased in hemodialysis patients with VC. Ca, P and iPTH are crucial to the simultaneous control of the mortality risk and cardiovascular hospitalization in dialysis patients [47-49]. Differences of the gut microbiome, microbial metabolism and several clinical characteristics between two hemodialysis groups were probably factors to cause VC, including the gut microbiome, microbial metabolism and several clinical characteristics.
Overall, the gut microbiota appears to play a significant role in the development of VC in patients with CKD. Although the prediction of the functional profile using PICRUSt is sufficiently linked to the phylogeny and provides useful insights, it is not a direct evidence [24]. Further research is needed to characterize the specific mechanisms through which the gut microbiota contributes to VC and to identify potential therapeutic strategies that may target these mechanisms to prevent or reverse VC in patients with CKD.
Supplementary Material
Supplementary data.
Acknowledgements
Funding
This work was supported by the National Science and Technology Council (NSTC- 111-2221-E-008 -066 -), NCU & Landseed Chronic disease Research Center (109A1510-1) and the National Central University-Landseed Hospital United Research Center (NCU-LSH-109-B-00).
Author contributions
YK L. and YC H. designed the study; YT C. and LK C. performed the experiments and analyzed the data; YT C. YC H. and LK C. wrote the manuscript; YK L. and P H. participated in discussion, language editing and review of the manuscript. All authors have read and agreed to the published version of the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Kovesdy CP. Epidemiology of chronic kidney disease: an update 2022. Kidney International Supplements. 2022;12:7-11
2. Haarhaus M, Brandenburg V, Kalantar-Zadeh K, Stenvinkel P, Magnusson P. Alkaline phosphatase: a novel treatment target for cardiovascular disease in CKD. Nature Reviews Nephrology. 2017;13:429-42
3. Sarnak MJ, Amann K, Bangalore S, Cavalcante JL, Charytan DM, Craig JC. et al. Chronic Kidney Disease and Coronary Artery Disease: JACC State-of-the-Art Review. J Am Coll Cardiol. 2019;74:1823-38
4. Foley RN, Parfrey PS, Sarnak MJ. Clinical epidemiology of cardiovascular disease in chronic renal disease. Am J Kidney Dis. 1998;32:S112-9
5. Strauss HW, Nakahara T, Narula N, Narula J. Vascular calcification: the evolving relationship of vascular calcification to major acute coronary events. Journal of Nuclear Medicine. 2019;60:1207-12
6. Hioki H, Miyashita Y, Shiraki T, Iida O, Uematsu M, Miura T. et al. Impact of deteriorated calcium-phosphate homeostasis on amputation-free survival after endovascular revascularization in patients with critical limb ischemia on hemodialysis. Vascular Medicine. 2016 2
7. London GM, Guerin AP, Marchais SJ, Metivier F, Pannier B, Adda H. Arterial media calcification in end-stage renal disease: impact on all-cause and cardiovascular mortality. Nephrol Dial Transplant. 2003;18:1731-40
8. Kraus MA, Kalra PA, Hunter J, Menoyo J, Stankus N. The prevalence of vascular calcification in patients with end-stage renal disease on hemodialysis: a cross-sectional observational study. Therapeutic advances in chronic disease. 2015;6:84-96
9. Zhu H, Yin C, Schoepf UJ, Wang D, Zhou C, Lu GM. et al. Machine Learning for the Prevalence and Severity of Coronary Artery Calcification in Nondialysis Chronic Kidney Disease Patients: A Chinese Large Cohort Study. Journal of Thoracic Imaging. 2022 10.1097
10. Yin L, Li X, Ghosh S, Xie C, Chen J, Huang H. Role of gut microbiota-derived metabolites on vascular calcification in CKD. J Cell Mol Med. 2021;25:1332-41
11. Jovanovich A, Isakova T, Stubbs J. Microbiome and Cardiovascular Disease in CKD. Clin J Am Soc Nephrol. 2018;13:1598-604
12. Qin J, Li R, Raes J, Arumugam M, Burgdorf KS, Manichanh C. et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature. 2010;464:59-65
13. Witkowski M, Weeks TL, Hazen SL. Gut Microbiota and Cardiovascular Disease. Circ Res. 2020;127:553-70
14. Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB. et al. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Applied and environmental microbiology. 2009;75:7537-41
15. Kozich JJ, Westcott SL, Baxter NT, Highlander SK, Schloss PD. Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the MiSeq Illumina sequencing platform. Applied and environmental microbiology. 2013;79:5112-20
16. Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P. et al. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic acids research. 2012;41:D590-D6
17. Edgar RC, Haas BJ, Clemente JC, Quince C, Knight R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics. 2011;27:2194-200
18. Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK. et al. QIIME allows analysis of high-throughput community sequencing data. Nature methods. 2010;7:335-6
19. Oksanen J, Blanchet F, Kindt R, Legendre P, Minchin P, O'Hara R, et al. Vegan: community ecology package http://CRAN.R-projectorg/package=vegan. 2013
20. Warnes GR, Bolker B, Bonebakker L, Gentleman R, Huber W, Liaw A. et al. gplots: Various R programming tools for plotting data. R package version. 2009;2:1
21. Segata N, Izard J, Waldron L, Gevers D, Miropolsky L, Garrett WS. et al. Metagenomic biomarker discovery and explanation. Genome biology. 2011;12:1-18
22. Jonsson V, Österlund T, Nerman O, Kristiansson E. Statistical evaluation of methods for identification of differentially abundant genes in comparative metagenomics. BMC genomics. 2016;17:1-14
23. Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome biology. 2014;15:1-21
24. Langille MG, Zaneveld J, Caporaso JG, McDonald D, Knights D, Reyes JA. et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nature biotechnology. 2013;31:814-21
25. Hioki H, Miyashita Y, Shiraki T, Iida O, Uematsu M, Miura T. et al. Impact of deteriorated calcium-phosphate homeostasis on amputation-free survival after endovascular revascularization in patients with critical limb ischemia on hemodialysis. Vascular Medicine. 2016 21
26. Eckburg PB, Bik EM, Bernstein CN, Purdom E, Dethlefsen L, Sargent M. et al. Diversity of the human intestinal microbial flora. Science. 2005;308:1635-8
27. Bao WH, Yang WL, Su CY, Lu XH, He L, Zhang AH. Relationship between gut microbiota and vascular calcification in hemodialysis patients. Renal Failure. 2023 45
28. Merino-Ribas A, Araujo R, Pereira L, Campos J, Barreiros L, Segundo MA. et al. Vascular Calcification and the Gut and Blood Microbiome in Chronic Kidney Disease Patients on Peritoneal Dialysis: A Pilot Study. Biomolecules. 2022 21
29. Chung S, Barnes JL, Astroth KS. Gastrointestinal Microbiota in Patients with Chronic Kidney Disease: A Systematic Review. Adv Nutr. 2019;10:888-901
30. Shin NR, Whon TW, Bae JW. Proteobacteria: microbial signature of dysbiosis in gut microbiota. Trends Biotechnol. 2015;33:496-503
31. Ren Z, Fan Y, Li A, Shen Q, Wu J, Ren L. et al. Alterations of the Human Gut Microbiome in Chronic Kidney Disease. Adv Sci (Weinh). 2020;7:2001936
32. Wu I-W, Lin C-Y, Chang L-C, Lee C-C, Chiu C-Y, Hsu H-J. et al. Gut microbiota as diagnostic tools for mirroring disease progression and circulating nephrotoxin levels in chronic kidney disease: discovery and validation study. International journal of biological sciences. 2020;16:420
33. Seldin MM, Meng Y, Qi H, Zhu W, Wang Z, Hazen SL. et al. Trimethylamine N-Oxide Promotes Vascular Inflammation Through Signaling of Mitogen-Activated Protein Kinase and Nuclear Factor-κB. J Am Heart Assoc. 2016 5
34. Gao J, Yan K-T, Wang J-X, Dou J, Wang J, Ren M. et al. Gut microbial taxa as potential predictive biomarkers for acute coronary syndrome and post-STEMI cardiovascular events. Scientific Reports. 2020;10:2639
35. Vojinovic D, Radjabzadeh D, Kurilshikov A, Amin N, Wijmenga C, Franke L. et al. Relationship between gut microbiota and circulating metabolites in population-based cohorts. Nat Commun. 2019;10:5813 -
36. Gupte SA, Arshad M, Viola S, Kaminski PM, Ungvari Z, Rabbani G. et al. Vascular Signaling by Free Radicals Pentose phosphate pathway coordinates multiple redox-controlled relaxing mechanisms in bovine coronary arteries. Am J Physiol Heart Circ Physiol. 2003;285:H2316-H26
37. Peiró C, Romacho T, Azcutia V, Villalobos L, Fernández E, Bolaños JP. et al. Inflammation, glucose, and vascular cell damage: the role of the pentose phosphate pathway. Cardiovascular diabetology. 2016;15:1-15
38. Zhu Y, Ma W-Q, Han X-Q, Wang Y, Wang X, Liu N-F. Advanced glycation end products accelerate calcification in VSMCs through HIF-1α/PDK4 activation and suppress glucose metabolism. Scientific reports. 2018;8:1-12
39. Colhoun HM, Otvos JD, Rubens MB, Taskinen M, Underwood SR, Fuller JH. Lipoprotein subclasses and particle sizes and their relationship with coronary artery calcification in men and women with and without type 1 diabetes. Diabetes. 2002;51:1949-56
40. Prenner SB, Mulvey CK, Ferguson JF, Rickels MR, Bhatt AB, Reilly MP. Very low-density lipoprotein cholesterol associates with coronary artery calcification in type 2 diabetes beyond circulating levels of triglycerides. Atherosclerosis. 2014;236:244-50
41. Xie X, Zhang X, Xiang S, Yan X, Huang H, Tian Y. et al. Association of very low-density lipoprotein cholesterol with all-cause and cardiovascular mortality in peritoneal dialysis. Kidney and Blood Pressure Research. 2017;42:52-61
42. Ren J, Grundy SM, Liu J, Wang W, Wang M, Sun J. et al. Long-term coronary heart disease risk associated with very-low-density lipoprotein cholesterol in Chinese: the results of a 15-Year Chinese Multi-Provincial Cohort Study (CMCS). Atherosclerosis. 2010;211:327-32
43. Aarsland A, Wolfe RR. Hepatic secretion of VLDL fatty acids during stimulated lipogenesis in men. J Lipid Res. 1998;39:1280-6
44. Kageyama A, Matsui H, Ohta M, Sambuichi K, Kawano H, Notsu T. et al. Palmitic acid induces osteoblastic differentiation in vascular smooth muscle cells through ACSL3 and NF-κB, novel targets of eicosapentaenoic acid. PLoS One. 2013;8:e68197
45. Huang JM, Xian H, Bacaner M. Long-chain fatty acids activate calcium channels in ventricular myocytes. Proceedings of the National Academy of Sciences of the United States of America. 1992;89:6452-6
46. Liu T, Dogan I, Rothe M, Reichardt J, Knauf F, Gollasch M. et al. Bioaccumulation of blood long-chain fatty acids during hemodialysis. Metabolites. 2022;12:269
47. Block GA, Kilpatrick RD, Lowe KA, Wang W, Danese MD. CKD-mineral and bone disorder and risk of death and cardiovascular hospitalization in patients on hemodialysis. Clin J Am Soc Nephrol. 2013;8:2132-40
48. Danese MD, Belozeroff V, Smirnakis K, Rothman KJ. Consistent control of mineral and bone disorder in incident hemodialysis patients. Clin J Am Soc Nephrol. 2008;3:1423-9
49. Viegas C, Araújo N, Marreiros C, Simes D. The interplay between mineral metabolism, vascular calcification and inflammation in Chronic Kidney Disease (CKD): challenging old concepts with new facts. Aging (Albany NY). 2019;11:4274
Author contact
![]() Corresponding author: Yi-Chiung Hsu, PhD, Department of Biomedical Sciences and Engineering, National Central University, Taoyuan, Taiwan. E-mail address: syicncuncu.edu.tw
Corresponding author: Yi-Chiung Hsu, PhD, Department of Biomedical Sciences and Engineering, National Central University, Taoyuan, Taiwan. E-mail address: syicncuncu.edu.tw