Impact Factor ISSN: 1449-1907
- Issue 9; 2026
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Volume 23; 2026
- Past Issues
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2023; 20(5):572-580. doi:10.7150/ijms.79741 This issue Cite
Research Paper
Salt Restriction and Angiotensin-Converting Enzyme Inhibitor Improve the Responsiveness of the Small Artery in Salt-Sensitive Hypertension
Shi-Cheng Li1, Tong-Meng Jiang2,3, Jia-Hao Zhang1, Meng-Ying Zeng1, Yu-Xin Ma1, Shu-Yi Feng4, Qing-Hai Wang5, Xiao-Wei Yan1 ![]()
1. Department of Cardiology, Peking Union Medical College Hospital (PUMCH), Chinese Academy of Medical Sciences & Peking Union Medical College, Beijing 100730, China.
2. Laboratory of Hainan Trauma and Disaster Rescue, The First Affiliated Hospital of Hainan Medical University, Haikou 571199, China.
3. Key Laboratory of Emergency and Trauma, Ministry of Education, Engineering Research Center for Hainan Bio-Smart Materials and Bio-Medical Devices, Key Laboratory of Hainan Functional Materials and Molecular Imaging, College of Emergency and Trauma, Hainan Medical University, Haikou 571199, China.
4. Experimental Research Center, China Academy of Chinese Medical Sciences, Beijing 100700, China.
5. Department of Cardiology, the Second Hospital, Cheeloo College of Medicine, Shandong University, Jinan 250000, China.
Received 2022-10-10; Accepted 2023-2-8; Published 2023-3-5
Abstract

For salt-sensitive hypertension (SSH), salt restriction and angiotensin-converting enzyme (ACE) inhibitors are essential treatments, but their effect on the function of resistance arteries is unclear. Here, we present an intravital study to detect the effect of salt restriction and ACE inhibitors on the function of the mesenteric small artery (MSA) in SSH. Dahl salt-sensitive rats were randomized into the following groups: ACE inhibitor gavage, salt restriction, ACE inhibitor combined with salt restriction, and high-salt diet. After a 12-week intervention, the mesenteric vessels maintained their perfusion in vivo, and the changes in the diameter and blood perfusion of the MSAs to norepinephrine (NE) and acetylcholine (ACh) were detected. Switching from a high-salt diet to a low-salt diet (i.e., salt restriction) attenuated the vasoconstriction of the MSAs to NE and promoted the vasodilatation to ACh, while ACE inhibitor improved the vasodilatation more obviously. Pathologically, changes in local ACE, AT1R, and eNOS expression were involved in these processes induced by a high-salt diet. Our study suggests that salt restriction and ACE inhibitor treatment improve high salt intake-induced MSA dysfunction in SSH, and salt restriction is a feasible and effective treatment. Our findings may provide a scientific basis for the treatment of hypertension.
Keywords: Hypertension, salt intake, salt restriction, mesenteric small artery, vascular reactivity, angiotensin-converting enzyme (ACE) inhibitor
Introduction
More than 31.1% of the global adult population worldwide were affected by hypertension [1], and approximately 50-60% of hypertensives are salt sensitive [2]. Increased salt consumption is associated with increased BP and cardiovascular diseases [3], far more than < 5 grams per day, the WHO-recommended amount [4]. In addition, increased salt sensitivity usually occurs in obesity, metabolic syndrome, and aging people despite genetic polymorphisms [2, 5]. Therefore, the high salt intake of hypertensive individuals has become a problem worthy of attention and urgently needs to be solved. Although a low-salt diet is beneficial to improve the retention of water and sodium in hypertensive individuals, its effect on the function of resistance arteries and the cooperative effect with antihypertensive drugs have not been fully demonstrated.
In addition to regulating blood flow to organs, small resistance arteries contribute to total peripheral vascular resistance, which is a crucial factor affecting blood pressure [6]. The increases [7, 8], decreases [9, 10] and biphasic [11, 12] effects of the vascular response induced by vasoconstrictors and vasodilators in arterial tonus have been reported previously. In a study of salt and vasomotor function, a high-salt diet reduced endothelium-dependent relaxation of the thoracic aorta in response to ACh in vitro [13], and high salt intake induced increased alpha-1 receptor responsiveness in excised MSAs [14]. Angiotensin-angiotensin system (RAS) is one of the mechanisms that facilitate arterial perfusion [15], because it cannot be completely simulated by isolated blood vessels with Krebs buffer or saline solution [11], especially vascular reactions to various agents. In addition, in real life, excessive salt intake is a common problem in hypertensive individuals. High salt intake negatively influenced the response of resistant arteries to vasoactive agents, as confirmed by our previous study [16]. Therefore, we hope to further explore the relationship between resistance artery and BP from another perspective by limiting salt intake.
Currently, there is a global consensus to limit salt intake; in particular, the benefits of changing from a high to a low salt intake beyond lower BP are noteworthy. Therefore, in the present study, we assessed the impact of changing from high salt intake to low salt intake on the vascular reactivity of mesenteric small arteries (MSA) in vivo on the basis of benazepril, an angiotensin-converting enzyme (ACE) inhibitor, in the treatment of salt-sensitive hypertension.
Materials and Methods
Experimental animals
In all experiments, the US National Institutes of Health's Guide for the Care and Use of Laboratory Animals [17] and ARRIVE guidelines were followed [18]. Ethical approval for this study was granted by the Ethical Committee of PUMCH (XHDW-2017-011).
All male Dahl-SS rats were obtained from Vital River Laboratory Animal Technology Co. Ltd. (Beijing, China), weighed 200-230 grams, and were maintained on a 12-h light/dark cycle with unrestricted access to food and drinking water. After 7 days of adaptation, rats were randomly divided into 4 experimental groups and the feed was changed at the same time: HS (high-salt diet, 8% NaCl content feed[19], n = 8) for 12 weeks, HB (HS with Benazepril administered intragastrically, n = 9) for 12 weeks, HLS (high salt for the first 6 weeks, then low-salt diet for the next 6 weeks, 0.3% NaCl content feed, n = 8), and HLB (HLS with Benazepril administration, 10 mg/kg per day, n = 9) for 12 weeks. Feed quantities of 8% HS and 0.3% LS were adjusted according to Beijing Keao Xieli Feed CO., LTD. normal feed. A tail-cuff method (BP-2010A, Softron) was used to monitor blood pressure before randomization and at 6 and 12 weeks.
Biochemical analysis and vascular preparation
The inner canthal orbital vein blood and 24-hour urine samples were collected at the end of the 12th week, and analyzed by the Clinical Laboratory of PUMCH (Olympus AU5400 and SIEMENS Clinitek Atlas Analyzer), including for serum creatinine, potassium, 24 hours urinary sodium and urinary total protein. The plasma angiotensin II and aldosterone levels were measured by radioimmunoassay. The proximal small intestine and mesentery were removed and placed in a transparent chamber filled with a constant temperature physiological saline solution after pentobarbital sodium (30 mg/kg, Sigma-Aldrich) was administered intraperitoneally [16]. The mesenteric vessels maintained their perfusion and vascular tone in vivo and in situ.
MSA diameter changes in response to vasoactive agents
To evaluate the changes in the MSAs in vivo, the start time, a GigaView Suite was used to record the maximum changes, the time to maximum changes and the duration of vasoconstriction/vasodilation in the second branch of MSAs induced by vasoactive agents. These results for each rat were averaged from three segments of the MSAs. The optimal reaction concentrations [16] of both norepinephrine tartrate (NE) and acetylcholine chloride (ACh) were 10 μg/kg. The dynamic change in the MSA diameter was recorded immediately after NE and ACh injection, and the interval between injections was at least 15 minutes to ensure drug elution. The GigaView Suite includes a microscope (BX51W1, OLYMPUS), high-speed camera (GigaView, Southern Vision Systems Inc.) and supporting software (GigaView software, version 2.4.7). Image-Pro Plus 6.0 software was used to determine MSAs diameters. The MSA under a microscope are shown in Figure S1 and Video S1.
Measuring the effect of drug injection on MSA perfusion
To evaluate the changes in MSA blood perfusion in vivo, a full-field laser perfusion imaging system (LPI) (Moor Instruments Ltd., UK) was used to continuously record and analyze the small arteries of the proximal small intestine [20]. According to the measurement unit of the LPI suite software, blood perfusion was expressed as the perfusion unit (PU). An average blood perfusion of MSAs was determined between 10th and 40th seconds after NE or ACh injection, based on the baseline blood perfusion. In the 30 s interval, the maximum MSA perfusion changes relative to baseline were recorded. For each rat, the blood perfusion of three arteries was averaged. Analyzing the LPI measurements was carried out using moorFLPI reviewer version 3.0 (Moor Instruments Ltd.). The MSAs under LPI are shown in Figure S1.
Histological analysis
After measurement in vivo, the MSAs of each rat were fixed, embedded and sliced. Hematoxylin and eosin, Masson staining and immunohistochemistry were performed on the MSAs in sections. Immunohistochemistry detecting the expression of angiotensin II type 2 receptor (AT2R), angiotensin II type 1 receptor (AT1R), angiotensin-converting enzyme (ACE) and endothelial nitric oxide synthase (eNOS). All these antibodies were purchased from Abcam. To determine the rate of fibrosis or protein expression, Image-Pro Plus 6.0 (Media Cybemetics, USA) was used to calculate the ratio of positive expression areas in the media or endothelium.
Statistical analysis
The means and standard deviations of the data are given. Students' t-tests or one-way ANOVA were used to compare groups. GraphPad Prism 8.0.2 was used for all analyses and a significance level of p < 0.05 was used.
Results
An anesthetic accident during surgery caused the death of two rats in the HS group and two rats in the HLS group after a 12-week dietary intervention. The dead rats completed all tests except the MSAs under a microscope and LPI, and the remaining rats completed all the experimental procedures.
General characteristics
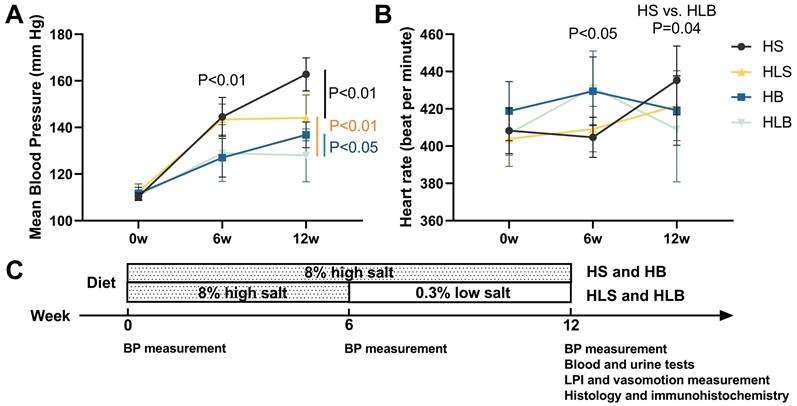
At baseline, neither group's mean blood pressure nor heart rate was significantly different. After 6 weeks of dietary and ACE inhibitor intervention, the mean BP of the high salt with Benazepril (HB, HLB) group was significantly lower than that of the HS without Benazepril (HS, HLS) group (HB vs. HS: P = 0.0006; HLB vs. HLS: P = 0.0093), and the heart rate of the HB and HLB groups was significantly higher than that of the HS and HLS groups (HB vs. HS: P = 0.0046; HLB vs. HLS: P = 0.0152). A significantly lower mean blood pressure was observed in the HB and HLS groups than HS group (P < 0.0001 and P = 0.0007, respectively) after the 12th week, but a significantly higher one than the HLB group (P = 0.0330 and P = 0.0072, respectively). The systolic and mean BP were not significantly different between the HB and HLS groups, but the diastolic BP of the HLS group was higher than that of the HB group (P = 0.0383). Table S1 and Figure 1 show that no difference in heart rate was observed among the four groups after the intervention of 12 weeks (all P > 0.05).
The mean blood pressure (A) and heart rate (B) of Dahl-SS rats at baseline and at the end of the 6-week and 12-week dietary interventions. (C) Timeline of the protocol. HS: the group with high salt intake for 12 weeks, n=8; HLS: the group with high salt for the first 6 weeks, then low salt intake for the next 6 weeks, n=8; HB: the group with high salt intake plus Benazepril administered intragastrically for 12 weeks, n=9; HLB: the group with high salt for the first 6 weeks, then low salt intake for the next 6 weeks, and with Benazepril administration throughout 12 weeks, n=9.
Biochemical analysis
After 12 weeks of intervention, high salt intake significantly inhibited plasma aldosterone and angiotensin II levels. As expected, low-salt diets significantly reduced 24-hour urinary sodium excretion in both HLS and HLB groups when salt consumption changed from high to low. The 24-hour urinary total protein excretion of the HB and HLS groups was significantly lower than that of the HS group (P = 0.0076 and P = 0.0396, respectively) (Table 1).
Blood and urine analysis at the end of 12th week.
| HS (n=8) | HLS (n=8) | HB (n=9) | HLB (n=9) | |
|---|---|---|---|---|
| AT II (pg/ml) | 135.75±8.86 | 170.37±12.34 ** | 154.36±31.52 | 178.80±47.61 |
| ALD (pg/ml) | 15.48±3.51 | 35.79±2.01 **, ##, @@ | 22.37±4.30 ** | 25.22±4.06 |
| Cr (μmol/L) | 31.76±8.09 | 27.35±1.68 # | 26.50±4.83 | 24.68±3.14 |
| K+ (mmol/L) | 4.33±0.66 | 4.68±0.25 | 4.32±0.47 | 4.58±0.25 |
| 24 hU-Na (mmol/24h) | 3.27±1.02 | 0.38±0.18 **, @@ | 2.21±1.53 ## | 0.34±0.08 |
| 24 hU-Pro (mg/24h) | 139.21±70.39 | 91.47±27.51 *, @ | 63.40±21.46 ** | 60.69±26.53 |
Notes: AT II: plasma angiotensin II; ALD: plasma aldosterone; Cr: serum creatinine; K+: serum potassium; 24 hU-Na: 24 hours urinary sodium; 24 hU-Pro: 24 hours urinary total protein. HS: high salt intake group; HB: high salt intake with Benazepril administered intragastrically; HLS: HS for the first 6 weeks, then low salt intake for the next 6 weeks; HLB: HLS with Benazepril administration. *: compared with HS group, P<0.05; **: compared with HS group, P<0.01; #: compared with HLB group, P<0.05; ##: compared with HLB group, P<0.01; @: compared with HB group, P<0.05; @@: compared with HB group, P<0.01.
The effects of NE and ACh on MSAs
After NE administration, the maximum percentage reduction in the MSA inner diameter was more significant in the HS group than in the HLS group (-50.48 ± 11.34 vs. -34.13 ± 10.27%, and P = 0.0257), was more significant in the HB group than in the HLB group (-42.25 ± 14.39 vs. -25.03 ± 12.08%, P = 0.0143), and was not different between the HB and HLS groups (Figure 2A). As compared to the HB and HLS groups, the HS group had significantly longer vasoconstriction duration (51.6 ± 17.5 vs. 33.1 ± 9.3 and 26.0 ± 5.5 seconds, P = 0.0191 and P = 0.0066, respectively), and there was a shorter duration in the HLB group compared to the HB group (24.9 ± 5.8 vs. 33.1 ± 9.3 seconds, P = 0.0383) (Figure 2B).
NE-induced vasoconstriction and ACh-induced vasodilatation of MSAs. (A) NE-induced maximum percentage change of the MSA inner vasoconstriction. (B) Duration of NE-induced vasoconstriction, from the beginning of vasoconstriction to the end of artery diameter recovery. (C) ACh-induced maximum percentage change in MSA inner vasodilatation. (D) Duration of ACh-induced vasodilatation, from the beginning of ACh injection to the end of artery diameter recovery. The percentage change in vasoconstriction/vasodilatation was calculated as the percentage of MSA inner diameter changes after NE/ACh injection divided by the baseline inner diameter. NE and ACh were injected through a femoral vein (10 μg/kg). The second order branch of the mesenteric arteries was recorded by a high-speed camera attached to a microscope. The measurements of three MSA segments were averaged. HS: the group with high salt intake for 12 weeks, n=8; HLS: the group with high salt for the first 6 weeks, then low salt intake for the next 6 weeks, n=8; HB: the group with high salt intake plus Benazepril administered intragastrically for 12 weeks, n=9; HLB: the group with high salt for the first 6 weeks, then low salt intake for the next 6-week, and with Benazepril administration throughout 12 weeks, n=9. MSA: mesenteric small artery; NE: norepinephrine; ACh: acetylcholine. *: compared with HS group, P<0.05; **: compared with the HS group, P<0.01; #: compared with the HLB group, P<0.05; ##: compared with the HLB group, P<0.01; @: compared with the HB group, P<0.05.
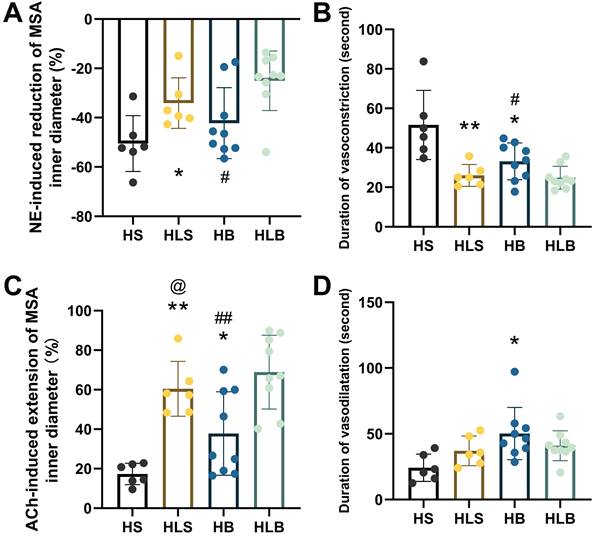
After ACh administration, the maximum percentage increase in the MSA inner diameter was significantly lower in the HS group than in the HB and HLS groups (17.27 ± 5.41 vs. 37.79 ± 21.18 and 60.43 ± 13.86%, P = 0.0389 and P < 0.0001, respectively), was significantly higher in the HLB group (68.87 ± 17.70%) than in the HB group (P = 0.0045) and was significantly lower in the HB group than in the HLS group (P = 0.0390) (Figure 2C). The duration of vasodilatation was significantly longer in the HB group than in the HS group (50.2 ± 19.9 vs. 24.2 ± 10.4 seconds, P = 0.0119) (Figure 2D and Table S2).
The effects of NE and ACh on blood perfusion of the MSAs
The baseline MSA blood perfusion did not significantly differ among the 4 groups before NE and ACh administration. After NE injection, the average percentage reduction in the MSA blood perfusion in the HLS group was significantly less than that in the HS group, and that in the HB group was also lower than that in the HLB group (HLS vs. HS: -39.25 ± 10.65 vs. -52.15 ± 7.13%, P = 0.0334; HLB vs. HB: -37.74 ± 13.07 vs. -48.83 ± 8.10, P = 0.0459, respectively); there was no difference between the HB and HLS groups (P = 0.0689) (Figure 3A). The maximum percentage reduction in blood perfusion after NE injection was significantly greater in the HB group than that in the HLB group (-62.35 ± 8.85% vs. -50.76 ± 13.28, P=0.0447), and there was no difference between the HB and HLS groups (P=0.1056) (Figure 3B).
Blood perfusion of the MSAs was recorded by full-field laser perfusion imaging. The MSAs of the proximal small intestine were recorded continuously for at least 10 minutes before and after injection of NE or ACh. (A) The average percentage changes in blood perfusion after NE injection. (B) The maximum percentage changes in blood perfusion after NE injection. (C) The average percentage increase in blood perfusion induced by ACh. (D) The maximum percentage increase in blood perfusion induced by ACh. The percentage changes within the MSAs were calculated as the average change in blood perfusion within a thirty-second interval after drug injection divided by baseline blood perfusion (i.e., thirty seconds of blood perfusion before NE/ACh administration). The maximum percentage change was calculated as the maximum blood perfusion induced by ACh (or minimum blood perfusion induced by NA) divided by the baseline blood perfusion. The measurements of three MSA segments were averaged. HS: the group with high salt intake for 12 weeks, n=8; HLS: the group with high salt for the first 6 weeks, then low salt intake for the next 6 weeks, n=8; HB: the group with high salt intake plus Benazepril administered intragastrically for 12 weeks, n=9; HLB: the group with high salt for the first 6 weeks, then low salt intake for the next 6 weeks, and with Benazepril administration throughout 12 weeks, n=9. MSA: mesenteric small artery; NE: norepinephrine; ACh: acetylcholine. *: compared with the HS group, P<0.05; **: compared with the HS group, P<0.01; #: compared with the HLB group, P<0.05.
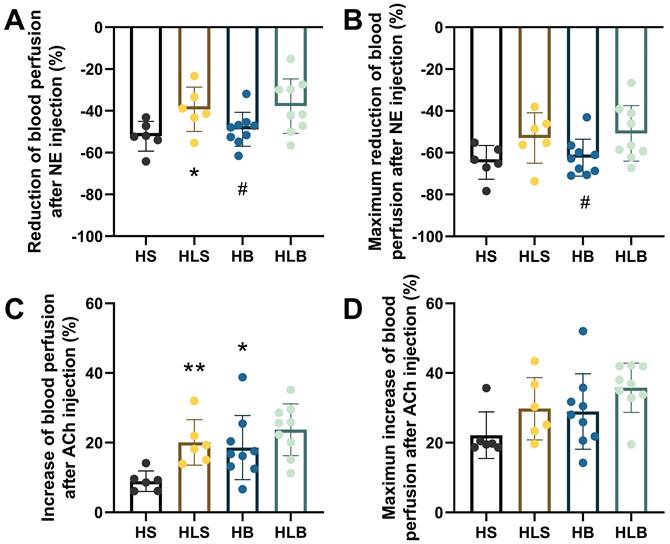
After ACh injection, ACh significantly increased MSA blood perfusion in the HB and HLS groups compared with the HS group (18.59 ± 9.20 and 20.05 ± 6.52 vs. 8.94 ± 2.96%, P = 0.0288 and P = 0.0035, respectively), however, HB and HLS did not differ (P = 0.7431) (Figure 3C). MSA blood perfusion, especially the maximum percentage change, was not significantly different among the 4 groups induced by ACh (Figure 3D).
Histological results
Figure 4 shows the histopathological changes in MSAs. After the 12-week intervention, according to the pathological sections, there was no significant difference in smooth muscle proliferation, intima-media thickness and internal lumen diameter among the 4 groups. In the HS group, collagen deposition rates in the intima-media of MSAs were significantly higher than in the HB group (P = 0.0004), according to Masson trichrome staining. Similar phenomenon was shown in HLS group than that in the HLB group (P < 0.0001).
Histological and immunohistochemical changes in the MSAs of Dahl-SS rats. The sectioned MSAs were stained with hematoxylin and eosin (HE), Masson's trichrome and immunohistochemistry (original magnification × 400, scale bar: 50 μm). For IHC, nuclei appeared brown in positive expression of MSA by immunohistochemistry assay. HS: the group with high salt intake for 12 weeks; HLS: the group with high salt for the first 6 weeks, then low salt intake for the next 6 weeks; HB: the group with high salt intake plus Benazepril administered intragastrically for 12 weeks; HLB: the group with high salt for the first 6 weeks, then low salt intake for the next 6 weeks, and with Benazepril administration throughout 12 weeks. MSA: mesenteric small artery; HE: hematoxylin and eosin; IHC: immunohistochemical staining; eNOS: endothelial nitric oxide synthase; ACE: angiotensin-converting enzyme; AT1R: angiotensin II type 1 receptor; AT2R: angiotensin II type 2 receptor. IMT/LD ratio: intima-media thickness/internal lumen diameter ratio; CD rate: collagen deposition rate. eNOS PE rate: positive expression rate of endothelial nitric oxide synthase; ACE PE rate: positive expression rate of angiotensin converting enzyme; AT1R PE rate: positive expression rate of angiotensin II type 1 receptor; AT2R PE rate: positive expression rate of angiotensin II type 2 receptor. *: compared with the HS group, P<0.05; **: compared with the HS group, P<0.01; #: compared with the HLB group, P<0.05; ##: compared with the HLB group, P<0.01; @: compared with the HB group, P<0.05; @@: compared with the HB group, P<0.01.
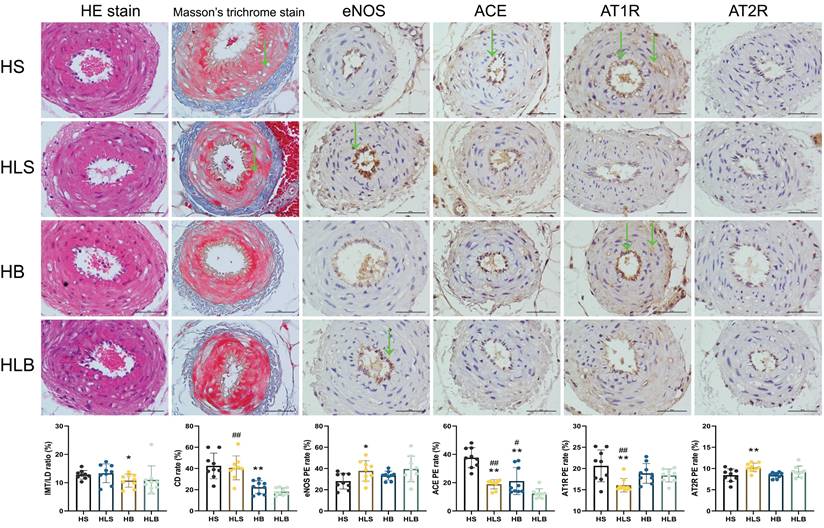
A significant difference existed between the HLS and HS groups in terms of eNOS expression in the MSAs by immunohistochemistry (P = 0.0308), and there was no difference between the HB and HLS groups (P = 0.2295). The ACE expression in the MSAs was significantly lower in the HB and HLS groups than in the HS group (P = 0.0007 and P < 0.0001, respectively) and higher than that in the HLB group (P = 0.0182 and P = 0.0007, respectively), but there was no difference between the HB and HLS groups (P = 0.5063). The AT1R expression in the MSAs were significantly lower in the HLS group than in the HS, HLB, and HB groups (P = 0.0041, P = 0.0064, and P = 0.0091, respectively). Compared to HS and HB, the HLS group showed significantly higher levels of AT2R in the MSAs (P = 0.0079 and P = 0.0004, respectively) (Figure 4 and Table S3).
Discussion
In this work, as shown in Dahl-SS rats, salt restriction improves resistance arteries' response to vasoactive agents in real-time when compared with benazepril. Six weeks of salt restriction had a comparable BP-lowering effect to benazepril in Dahl-SS rats. Both switching from high to low salt intake and ACE inhibitor treatment favorably affected NE-induced vasoconstriction and ACh-induced vasodilatation in the MSAs of rats, and the effect intensity of salt restriction was comparable to that of ACE inhibitors. The immunohistochemistry study showed that salt restriction was associated with an inhibition of the local RAS and an improvement in eNOS expression.
The hypertension and cardiovascular disease guidelines [1, 4, 21, 22] strongly recommend hypertensive patients, especially those with high salt intake and salt-sensitive individuals, to reduce salt intake. Most studies compared long-term intervention of high salt to low-salt diet, lacking a process of switching salt intake, which is a common lifestyle change of hypertensive people in real clinical world. Major gaps remain regarding the relationship of salt intake with human physiology and health. Although previous studies [23-25] have shown that high salt intake negatively impacts the response of resistant arteries to vasoactive agents, there was a lack of research focusing on the improvement effects of switching from a high- to low-salt diet in vivo. In a human study [26], found that salt reduction improved endothelium-dependent vasodilation [26] but did not improve arterial stiffness [27] and showed no impact on central and peripheral hemodynamics during exercise [28]. Perhaps it is quite impractical in human studies to design sophisticated procedures to observe the effects of salt restriction on vascular function. Therefore, our experiments used living animals through a sophisticated design to reveal the real-time effects of salt restriction on the function of MSAs, a phenomenon different from that of central and peripheral arteries and different from isolated arteries.
An animal study [29] showed that salt restriction can decrease arterial stiffness. The current study shows that salt restriction in salt-sensitive animals can not only reduce BP but also achieve significant improvement in vascular function in MSAs. As the purpose of lowering BP is to protect target organs, the improvement of vascular function may bring more benefits to individuals than the reduction of BP per se [30]. In this study, salt restriction for 6 weeks in the high-salt diet individuals did not completely reverse high BP and vascular dysfunction to the initial state. As this study showed that low salt elevated angiotensin II and aldosterone levels, the RAS was also activated by low salt in Sprague-Dawley rats [31]. Moreover, a previous study [32] showed that ong-term low salt diet-induced hypertension may be caused by activation of the RAS. Therefore, the effect of local RAS activated by a high-salt diet for a long time may not be completely eliminated [33], and the change to a low-salt diet activated circulating RAS, which may lead to some of the changes developed during salt loading that could not be reversed.
The function and structure of arteries can be improved by ACE inhibitors according to a large number of previous studies [34-36]. ACE inhibitors also reverse vascular dysfunction and hyperconstriction of peripheral arteries [37]. In this in vivo study, an ACE inhibitor improved RAS activation, both vasoconstriction and vasodilatation, in Dahl-SS rats, but the improvement in vasoconstriction was more obvious. Vasoconstriction is mainly driven by vascular smooth muscle, while ACE inhibition results in improved vascular function by reducing vascular smooth muscle activation and vessel wall remodeling [38]. The angiotensin receptors AT1R and AT2R play opposite roles in regulating sodium excretion, and the increased AT1R activity reduces sodium excretion in the proximal tubule and distal tubule and plays a stronger role [39]. Interestingly, in pathological conditions of sodium depletion, the expression of AT2R was enhanced compared with that in healthy tissue [40, 41]. In a study [42], chronic treatment with an AT2R agonist prevented salt-sensitive hypertension. However, ACE inhibitor treatment reduced the activation of both AT1 and AT2 receptors to improve sodium and fluid retention. The current study shows that switching from high to low salt intake almost achieved comparable effects on the local RAS as using ACE inhibitor in high salt intake animals after 6-week intervention, meanwhile, salt restriction slightly increased the AT2R expression without ACE inhibitor. Both excessively low- [43] and high-salt diets cause local RAS activation. Our study showed that moderately low salt levels can improve RAS activation, including downregulating the expression of ACE and AT1R and upregulating the expression of AT2R.
A dysfunctional activated circulatory RAS maintains high blood pressure [44], while activated tissue RAS damages and malfunctions target organs [45]. The chronic activation of RAS can participate in structural and functional alteration of the arterial wall [46, 47]. In the current study, salt restriction was associated with an upregulation of eNOS expression and an inhibition of local RAS activity, including downregulation of ACE and AT1R in the MSAs. These results were similar to those of the ACE inhibitors, and may be related to the reversal of unfavorable response of the MSAs to vasoactive agents. Moreover, ACE inhibitor treatment plus salt restriction could inhibit local RAS and promote eNOS expression synergistically in animals with salt-sensitive hypertension. The results showed that salt restriction was almost equivalent to the same inhibitory effect on the local RAS of ACE inhibitors, which may be a change in a short time. In a study [48] of high fructose-induced hypertension in Dahl-SS rats, an ACE inhibitor attenuated hypertension and renal damage after 12 weeks of treatment. According to the inhibition of local RAS based on salt restriction, ACE inhibitors may have a more obvious inhibition effect in the long-range that may be longer than 12 weeks, but further exploration is needed.
We attempted to elucidate the critical role of salt restriction and ACE inhibitors in salt-sensitive hypertension, but this study had some limitations. First, we have described a remarkable phenomenon with immunohistochemistry but lack further mechanistic exploration. Second, we have shown in animals that salt restriction was almost as effective as ACE inhibitors in MSAs of Dahl-SS rats, but the implication in clinical practice remains to be studied. Third, it is difficult to switch from a high- to low-salt diet in real clinical practice due to poor patient compliance. Therefore, we enthusiastically explored and emphasized the favorable impacts of salt restriction on vascular function. Fourth, salt restriction and ACE inhibitors did not return BP values to baseline but only prevented further increases in the 6th to 12th week period. It would have been interesting to have weekly BP measurements to better appreciate the BP trend. Finally, we are actually interested in the impact of different levels of salt intake on BP and vascular function; unfortunately, we only set two grades of salt intake in the present study.
In conclusion, we found that switching from high-salt to low-salt diets and ACE inhibitor treatment improved the responsiveness of resistant arteries to vasoactive agents synergistically. The expression of ACE, AT1R, and eNOS at the local level may aid these processes. Moreover, the improvements in BP and vascular function highlight the importance of salt restriction for hypertension and shed light on reducing salt-sensitive hypertension.
Supplementary Material
Supplementary figure and tables, video legend.
Supplementary video 1.
Acknowledgements
We thank the linguistic editing service by AJE. We also thank the help of Home for Researchers (www.home-for-researchers.com) and the graphical abstract by Figdraw (www.figdraw.com).
Funding
This work was supported by grants from the National Natural Science Foundation of China (No. 81970360).
Author Contributions
SCL, TMJ, JHZ, and SYF performed the experiments. SCL and XWY designed the experiments, performed the analyses, and wrote the manuscript. MYZ, QHW, and YXM contributed to manuscript preparation.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Unger T, Borghi C, Charchar F, Khan NA, Poulter NR, Prabhakaran D. et al. 2020 International Society of Hypertension global hypertension practice guidelines. Journal of hypertension. 2020;38:982-1004
2. Rust P, Ekmekcioglu C. Impact of Salt Intake on the Pathogenesis and Treatment of Hypertension. In: Islam MS, editor. Hypertension: from basic research to clinical practice. Cham: Springer International Publishing. 2017 p. 61-84
3. He FJ, Tan M, Ma Y, Macgregor GA. Salt Reduction to Prevent Hypertension and Cardiovascular Disease. Journal of the American College of Cardiology. 2020;75:632-47
4. Al-Makki A, DiPette D, Whelton PK, Murad MH, Mustafa RA, Acharya S. et al. Hypertension Pharmacological Treatment in Adults: A World Health Organization Guideline Executive Summary. Hypertension. 2022;79:293-301
5. Faulkner JL, Belin de Chantemèle EJ. Female Sex, a Major Risk Factor for Salt-Sensitive Hypertension. Current hypertension reports. 2020;22:99
6. Kario K, Kanegae H, Oikawa T, Suzuki K. Hypertension Is Predicted by Both Large and Small Artery Disease. Hypertension. 2019;73:75-83
7. Wang T, Liao L, Tang X, Li B, Huang S. Effects of different vasopressors on the contraction of the superior mesenteric artery and uterine artery in rats during late pregnancy. BMC Anesthesiology. 2021;21:185
8. Christie MJ, Romano T, Murphy RM, Posterino GS. Effects of intrauterine growth restriction on Ca2+-activated force and contractile protein expression in the mesenteric artery of 1-year-old Wistar-Kyoto rats. Journal of Physiology and Biochemistry. 2020;76:111-21
9. Matsumoto T, Takayanagi K, Katome T, Kojima M, Taguchi K, Kobayashi T. Reduced Relaxant Response to Adenine in the Superior Mesenteric Artery of Spontaneously Hypertensive Rats. Biological and Pharmaceutical Bulletin. 2021;44:1530-5
10. Isidoro-García L, Villalpando DM, Ferrer M. Vasomotor action of androgens in the mesenteric artery of hypertensive rats. Role of perivascular innervation. PloS one. 2021;16:e0246254-e
11. Nyvad J, Mazur A, Postnov DD, Straarup MS, Soendergaard AM, Staehr C. et al. Intravital investigation of rat mesenteric small artery tone and blood flow. The Journal of physiology. 2017;595:5037-53
12. Liu H, Li XZ, Peng M, Ji W, Zhao L, Li L. et al. Role of gap junctions in the contractile response to agonists in the mesenteric resistance artery of rats with acute hypoxia. Molecular medicine reports. 2017;15:1823-31
13. Yoshida M, Nakamura K, Miyoshi T, Yoshida M, Kondo M, Akazawa K. et al. Combination therapy with pemafibrate (K-877) and pitavastatin improves vascular endothelial dysfunction in dahl/salt-sensitive rats fed a high-salt and high-fat diet. Cardiovascular diabetology. 2020;19:149
14. Mokotedi L, Michel FS, Woodiwiss AJ, Norton GR, Millen AME. Effects of High Salt-Low Potassium Diet on Blood Pressure and Vascular Reactivity in Male Sprague Dawley Rats. Journal of cardiovascular pharmacology. 2018 71
15. Emdin M, Fatini C, Mirizzi G, Poletti R, Borrelli C, Prontera C. et al. Biomarkers of activation of renin-angiotensin-aldosterone system in heart failure: how useful, how feasible? Clinica Chimica Acta. 2015;443:85-93
16. Li SC, Wang QH, Chen LF, Feng SY, Wu YX, Yan XW. High Sodium Intake Impairs Small Artery Vasoreactivity in vivo in Dahl Salt-Sensitive Rats. Journal of vascular research. 2019;56:65-76
17. National Research Council Committee for the Update of the Guide for the C, Use of Laboratory A. The National Academies Collection: Reports funded by National Institutes of Health. In: th, editor. Guide for the Care and Use of Laboratory Animals. Washington (DC): National Academies Press (US) National Academy of Sciences. 2011
18. Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. Animal research: reporting in vivo experiments: the ARRIVE guidelines. British journal of pharmacology. 2010;160:1577-9
19. Yang P, Zhao X, Zhou L, Jin Y, Zheng X, Ouyang Y. et al. Protective effect of oral histidine on hypertension in Dahl salt-sensitive rats induced by high-salt diet. Life sciences. 2021;270:119134
20. Aubdool AA, Graepel R, Kodji X, Alawi KM, Bodkin JV, Srivastava S. et al. TRPA1 is essential for the vascular response to environmental cold exposure. Nature communications. 2014;5:5732
21. Whelton PK, Carey RM, Aronow WS, Casey DE Jr, Collins KJ, Dennison Himmelfarb C. et al. 2017 ACC/AHA/AAPA/ABC/ACPM/AGS/APhA/ASH/ASPC/NMA/PCNA Guideline for the Prevention, Detection, Evaluation, and Management of High Blood Pressure in Adults: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Journal of the American College of Cardiology. 2018;71:e127-e248
22. Bruce A, Diane L. 2013 ACC/AHA guideline on the assessment of cardiovascular risk. Journal of the American College of Cardiology. 2014;63:2886 -
23. Sofola O, Knill A, Myers D, Hainsworth R, Drinkhill M. High-salt diet and responses of the pressurized mesenteric artery of the dog to noradrenaline and acetylcholine. Clinical and experimental pharmacology & physiology. 2004;31:696-9
24. Ling WC, Mustafa MR, Vanhoutte PM, Murugan DD. Chronic administration of sodium nitrite prevents hypertension and protects arterial endothelial function by reducing oxidative stress in angiotensin II-infused mice. Vascular Pharmacology. 2018;102:11-20
25. Bridges LE, Williams CL, Awumey EM. High Salt Upregulates Ca2+-Sensing Receptor Expression and Ca2+-Induced Relaxation of Contracted Mesenteric Arteries from Dahl Salt-Sensitive Rats. Journal of Pharmacology and Experimental Therapeutics. 2022;381:120
26. Dickinson KM, Keogh JB, Clifton PM. Effects of a low-salt diet on flow-mediated dilatation in humans. The American journal of clinical nutrition. 2009;89:485-90
27. Ishida A, Isotani A, Fujisawa M, Del Saz EG, Okumiya K, Kimura Y. et al. Effects of a Low-Salt and High-Potassium Diet on Arterial Stiffness and Left Ventricular Function in Indigenous Papuans. Journal of the American Heart Association. 2021;10:e021789
28. Ratchford SM, Broxterman RM, La Salle DT, Kwon OS, Park SY, Hopkins PN. et al. Salt restriction lowers blood pressure at rest and during exercise without altering peripheral hemodynamics in hypertensive individuals. American journal of physiology Heart and circulatory physiology. 2019;317:H1194-h202
29. Grigorova YN, Wei W, Petrashevskaya N, Zernetkina V, Juhasz O, Fenner R. et al. Dietary Sodium Restriction Reduces Arterial Stiffness, Vascular TGF-β-Dependent Fibrosis and Marinobufagenin in Young Normotensive Rats. International journal of molecular sciences. 2018 19
30. Grillo A, Salvi L, Coruzzi P, Salvi P, Parati G. Sodium Intake and Hypertension. Nutrients. 2019 11
31. Shao W, Seth DM, Prieto MC, Kobori H, Navar LG. Activation of the renin-angiotensin system by a low-salt diet does not augment intratubular angiotensinogen and angiotensin II in rats. Am J Physiol Renal Physiol. 2013;304:F505-14
32. Wang J, Deng Y, Zou X, Luo H, Jose PA, Fu C. et al. Long-term low salt diet increases blood pressure by activation of the renin-angiotensin and sympathetic nervous systems. Clinical and experimental hypertension (New York, NY: 1993). 2019;41:739-46
33. Allen TJ, Waldron MJ, Casley D, Jerums G, Cooper ME. Salt restriction reduces hyperfiltration, renal enlargement, and albuminuria in experimental diabetes. Diabetes. 1997;46:19-24
34. Ding H, Liu S, Zhao K-X, Pu J, Xie Y-F, Zhang X-W. Comparative Efficacy of Antihypertensive Agents in Flow-Mediated Vasodilation of Patients with Hypertension: Network Meta-Analysis of Randomized Controlled Trial. International journal of hypertension. 2022;2022:2432567
35. Raffai G, Lombard JH. Angiotensin-(1-7) Selectively Induces Relaxation and Modulates Endothelium-Dependent Dilation in Mesenteric Arteries of Salt-Fed Rats. Journal of vascular research. 2016;53:105-18
36. Lezama-Martinez D, Elena Hernandez-Campos M, Flores-Monroy J, Valencia-Hernandez I, Martinez-Aguilar L. Time-Dependent Effects of Individual and Combined Treatments with Nebivolol, Lisinopril, and Valsartan on Blood Pressure and Vascular Reactivity to Angiotensin II and Norepinephrine. Journal of Cardiovascular Pharmacology and Therapeutics. 2021;26:490-9
37. Li Z, Lindner DP, Bishop NM, Cipolla MJ. ACE (Angiotensin-Converting Enzyme) Inhibition Reverses Vasoconstriction and Impaired Dilation of Pial Collaterals in Chronic Hypertension. Hypertension. 2020;76:226-35
38. Te Riet L, van Esch JH, Roks AJ, van den Meiracker AH, Danser AH. Hypertension: renin-angiotensin-aldosterone system alterations. Circulation research. 2015;116:960-75
39. Giani JF, Eriguchi M, Bernstein EA, Katsumata M, Shen XZ, Li L. et al. Renal tubular angiotensin converting enzyme is responsible for nitro-L-arginine methyl ester (L-NAME)-induced salt sensitivity. Kidney international. 2017;91:856-67
40. Peluso AA, Santos RA, Unger T, Steckelings UM. The angiotensin type 2 receptor and the kidney. Current opinion in nephrology and hypertension. 2017;26:36-42
41. Ozono R, Wang ZQ, Moore AF, Inagami T, Siragy HM, Carey RM. Expression of the subtype 2 angiotensin (AT2) receptor protein in rat kidney. Hypertension. 1997;30:1238-46
42. Ali Q, Patel S, Hussain T. Angiotensin AT2 receptor agonist prevents salt-sensitive hypertension in obese Zucker rats. Am J Physiol Renal Physiol. 2015;308:F1379-85
43. Okamoto C, Hayakawa Y, Aoyama T, Komaki H, Minatoguchi S, Iwasa M. et al. Excessively low salt diet damages the heart through activation of cardiac (pro) renin receptor, renin-angiotensin-aldosterone, and sympatho-adrenal systems in spontaneously hypertensive rats. PloS one. 2017;12:e0189099
44. Bayorh MA, Ganafa AA, Emmett N, Socci RR, Eatman D, Fridie IL. Alterations in Aldosterone and Angiotensin II Levels in Salt-Induced Hypertension. Clinical & Experimental Hypertension. 2005;27:355-67
45. Muñoz-Durango N, Fuentes CA, Castillo AE, González-Gómez LM, Vecchiola A, Fardella CE. et al. Role of the Renin-Angiotensin-Aldosterone System beyond Blood Pressure Regulation: Molecular and Cellular Mechanisms Involved in End-Organ Damage during Arterial Hypertension. 2016; 17: 797.
46. Laurent S, Boutouyrie P. The structural factor of hypertension: large and small artery alterations. Circulation research. 2015;116:1007-21
47. De Silva TM, Modrick ML, Grobe JL, Faraci FM. Activation of the Central Renin-Angiotensin System Causes Local Cerebrovascular Dysfunction. Stroke; a journal of cerebral circulation. 2021;52:2404-13
48. Xu L, Hu G, Qiu J, Fan Y, Ma Y, Miura T. et al. High Fructose-Induced Hypertension and Renal Damage Are Exaggerated in Dahl Salt-Sensitive Rats via Renal Renin-Angiotensin System Activation. Journal of the American Heart Association. 2021;10:e016543
Author contact
![]() Corresponding author: Xiao-Wei Yan, Professor of Department of Cardiology. Permanent address: Peking Union Medical College Hospital, No.1 Shuai Fu Yuan, Beijing 100730, China; Telephone: +86-13701178433; Email: yanxw_pumchcom
Corresponding author: Xiao-Wei Yan, Professor of Department of Cardiology. Permanent address: Peking Union Medical College Hospital, No.1 Shuai Fu Yuan, Beijing 100730, China; Telephone: +86-13701178433; Email: yanxw_pumchcom