Impact Factor ISSN: 1449-1907

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2023; 20(2):262-266. doi:10.7150/ijms.81381 This issue Cite
Review
Mechanism of CD38 via NAD+ in the Development of Non-alcoholic Fatty Liver Disease
Min Dong1, Shuo Wang2, Zuowei Pei3,4 ![]()
1. Department of Cardiology, Beijing Hospital, National Center of Gerontology, Institute of Geriatric Medicine, Chinese Academy of Medical Sciences, Beijing, China.
2. Department of Internal Medicine, The Affiliated Zhong Shan Hospital of Dalian University, Dalian, China.
3. Department of Cardiology, Central Hospital of Dalian University of Technology, Dalian, China.
4. Faculty of Medicine, Dalian University of Technology, Dalian, China.
Received 2022-11-30; Accepted 2023-1-12; Published 2023-1-22
Abstract

Non-alcoholic fatty liver disease (NAFLD) is the most common chronic liver disease globally, and it can proceed to cirrhosis and hepatocellular carcinoma, as well as cardiovascular disease, chronic renal disease, and other complications, resulting in a massive economic burden. At the moment, nicotinamide adenine dinucleotide (NAD+) is thought to be a possible treatment target for NAFLD, besides Cluster of differentiation 38(CD38) is the primary NAD+ degrading enzyme in mammals and may play a role in the pathophysiology of NAFLD. For example, CD38 regulates Sirtuin 1 activity and hence affects inflammatory responses. CD38 inhibitors enhance glucose intolerance and insulin resistance in mice and lipid accumulation in the liver is greatly decreased in CD38-deficient mice. This review describes the role of CD38 in the development of NAFLD in terms of Macrophage-1, insulin resistance, and abnormal lipid accumulation in order to offer recommendations for future NAFLD pharmacological trials.
Keywords: Non-alcoholic fatty liver disease, CD38, NAD+, Macrophage-1 Insulin resistance, Lipid accumulation.
Introduction
Non-alcoholic fatty liver disease (NAFLD) is the most prevalent kind of chronic liver disease in the world, accounting for 25.24% of all cases, and is rapidly becoming the primary cause of liver-related morbidity and mortality [1]. NAFLD encompasses a range of pathological changes including non-alcoholic fatty liver (NAFL) and nonalcoholic steatohepatitis (NASH), and can progress to cirrhosis and hepatocellular carcinoma (HCC) [2]. NAFLD can also cause extrahepatic symptoms such as cardiovascular disease chronic renal disease, and respiratory illness [3]. However, the pathogenesis of NAFLD is unknown. Nicotinamide adenine dinucleotide (NAD+) has been identified as a potential target for NAFLD prevention and reversal. NAD+ is a critical cofactor in redox processes and a key regulator of several mammalian metabolisms. It is involved in a variety of biological processes and is essential for energy production, fatty acid and cholesterol synthesis, oxidation reactions, adenosine triphosphate (ATP) production, gluconeogenesis and ketogenesis [1, 4]. The cluster of differentiation 38(CD38) is a single-chain type II transmembrane glycoprotein with a large extracellular domain at the C-terminus and a small cytoplasmic domain at the N-terminus [5]. It was first identified as a lymphocyte-specific antigen in lymphocytes, but it was soon discovered to also be present intracellularly, where it is responsible for metabolizing intracellular NAD+ and generating second messengers like cyclic adenosine dinucleotide phosphate ribose (cADPR) in mammalian cells, as well as in pathological conditions that promote the development of a variety of diseases like cardiovascular disease, aging, obesity, diabetes, and inflammation. [5, 6]. CD38 is one of the major NAD glycohydrolase (NADase) in mammals, and it is currently believed that NAD+ levels can be effectively increased by inhibiting CD38, several inhibitors of CD38 exist or are under development [7-9]. As of now, dietary and lifestyle modifications are the cornerstone of treatment for NAFLD and NASH despite the fact that there is no Food or Drug Administration-approved drug for these conditions. However, the sustainability of these treatments over the long term is poor. This review covers the role of CD38 on NAD+ and explores the effect of CD38 on the onset of NAFLD to clarify the pathophysiology of NAFLD further and offer ideas for clinical therapeutic research.
Typing and function of CD38
A cADPR synthetase is called CD38. The cADPR synthase indirectly influences a number of Ca2+-dependent processes, such as cell proliferation, muscular contraction, immunological response, and glucose-stimulated insulin production by pancreatic β-cells [10]. It has been demonstrated that flavonoids, such as apigenin and quercetin, decrease CD38 activity. These flavonoids raise the level of NAD+ in hepatocytes and may attenuate NAFLD [11]. According to studies, CD38 is a membrane protein with an extracellular type II and an intracellular type III orientation [12]. Extracellular NAD+ is hydrolyzed to form nicotinamide and ADP-ribose at the catalytic site, which is directed toward the extracellular space (type II CD38). The extracellular enzymatic properties of type II CD38 raised long ago the question about the topological paradox of the entry of intracellular NAD+ substrates into the extracellular active site and the entry of extracellular cADPR products into its intracellular receptor ryanodine (RyR) channel, besides, according to the topological paradox theory, intracellular cADPR can be generated [13, 14]. The salvage pathways then utilize nicotinamide for the production of NAD+ [5]. It was shown that cellular levels of NAD+ were differentially increased in the tissues of CD38-deficient mice and that CD38-deficient protected mice from high fat diet (HFD)-induced obesity[15].It has been demonstrated that CD38 (type IIICD38) can be introduced into the endoplasmic reticulum (ER) membrane with its catalytic site facing the cytoplasm[16].However, the type III CD38 catalytic structural domain is cytoplasm-oriented with low abundance, but can effectively cyclize cytoplasmic NAD+ to generate cADPR[17]. The type of membrane insertion may be determined by protein kinases. Protein kinases may diminish the charge of positive amino acids close to the transmembrane domain [16]. Nicotinamide adenine dinucleotide phosphate (NADPH) oxidases have the ability to activate Type III CD38, which first appears to be dormant [18]. In endolysosomal membranes, type II CD38 is thought to play a role in the production of the calcium-mobilizing second messenger nicotinic acid adenine dinucleotide phosphate (NAADP), While type III CD38 is thought to contribute to the ER membranes' production of the calcium-mobilizing cADPR[16]. There is still much to learn about the regulation, localization, and function of CD38.
NAFLD is a metabolic disease with a complex etiology involving inflammation, excessive fatty acid accumulation and insulin resistance.[2]. NAD+ is currently thought to be a promising target for the therapy of NAFLD. CD38 is the major depleting enzyme of NAD+ in mammalian tissues. Mice deficient in CD38 have increased NAD+ in the brain and liver, suggesting that CD38 plays a critical role in maintaining NAD+ homeostasis [19].As a kind of NAD+-consuming enzymes, CD38 may play a specific role in the development of NAFLD.
Roles of CD38 in NAFLD
1. The activation of M1 macrophages
Macrophages in the liver, also known as Kupffer cells, accounts for 15% of total hepatocytes and 80-90% of all tissue-resident macrophages. Activation of Kupffer cells is closely associated with the development of NAFLD [20, 21]. It has been demonstrated that CD38 is a Macrophage-1(M1) specific gene [22]. Obesity increases adipose tissue and the liver must absorb excess lipids stored in lipid droplets, leading to ongoing inflammation and activation of resident and infiltrating macrophages [11]. Studies have shown that CD38 cleaves NAD+ inside and outside the cell and that a large fraction of CD38 ecto-NADase activity in macrophages is intracellular [23]. Besides, enhanced NADase activity of pro-inflammatory M1 macrophages is mediated by CD38[23]. Sirtuin 1 (SIRT1) activation attenuated hepatic steatosis and inflammation in HFD-induced NAFLD by inhibiting CD38 expression and NF-κB signaling pathway [24-26]. Moreover, CD38 deficiency has been demonstrated to shield the heart from HFD-induced oxidative stress by activating the Sirtuin3 (SIRT3)/FOXO3-mediated antioxidant stress pathway [15]. However, there are no studies of this pathway in the liver. It is possible that pro- and anti-inflammatory responses seem to be related to cell type and cellular environment. Inhibition of the ectoenzymatic activity of CD38 in vivo with antibody 68(Ab68) leads to a decrease in the level of the NAD+ degradation product adenosine diphosphate ribose (ADPR) [27]. It was found that inflammation can reduce NAD+ by increasing CD38 and that blocking the exoenzyme activity of CD38 can increase NAD+ through a nicotinamide mononucleotide (NMN)-dependent process [27].
Recent findings reveal a major role for CD38 in inflammation, suggesting that the age-related decline in NAD+ is mediated in part by aging/ the senescence-associated secretory phenotype (SASP)-induced accumulation of CD38 inflammatory cells in tissues [23]. Besides, chronic inflammation affects energy metabolism by interfering with the AMPK-NAD+-PGC1α-SIRT1 pathway via CD38[28]. This may provide new clues for further studies to explore the exact mechanism of CD38 in the progression of NAFLD to cirrhosis [23, 27]. Hepatocytes with NAFLD have considerably increased glycolysis, which raises the amounts of pyruvate in the blood and liver. The precise mechanism by which increased glycolysis can result in hepatic inflammation is unknown [29]. Clinical research on the interactions between various organs in patients will ultimately be important. Future research must take into account novel players in NAFLD and lipid homeostasis, such as lipid droplet dysregulation. NAFLD is linked to mutations in the lipid droplet lipase PNPLA3 (patatin-like phospholipase A3) [1, 30]. It was discovered that PTIP, also known as Paxip1 (interaction of Pax with transcriptional activation domain protein-1), controls the expression of CD38 in macrophages by creating an H3K27ac-enriched enhancer of CD38 activity in conjunction with acetyltransferase p300.Additionally, by comparing histone changes with gene expression profiles for NAD+ metabolism, PTIP was discovered to be a crucial regulator of CD38 expression, an enzyme that predominantly uses NAD+ in macrophages [31].
2. Insulin resistance
Type 2 diabetes (T2DM) and insulin resistance are both directly related to the evolution of NAFLD [32]. Besides, improved NASH histopathology and fibrosis regression were both strongly correlated with enhanced insulin sensitivity [33]. The largest independent risk factor for the onset of cirrhosis and HCC in NASH patients was a baseline diagnosis of T2DM [33, 34]. A recent study has demonstrated that the decline of NAD+ levels with aging are primarily dependent on CD38, besides, CD38 deficiency activates the NAD+/SIRT1 pathway [35, 36]. Activation of SIRT1 improves glucose tolerance, lowers hyperinsulinemia, and increases systemic insulin sensitivity by significantly repressing the jun-n-terminal kinase (JNK) and inhibitor of NF-κB kinase (IKK) inflammatory pathways [37]. CD38-deficient mice exhibited improved glucose tolerance compared to wild-type mice [35]. 78c is a specific inhibitor of CD38 and does not directly affect the activity or expression of other enzymes involved in NAD+ metabolism, it could improve glucose intolerance and insulin resistance in mice [8]. Insulin resistance promotes adipose tissue lipolysis, resulting in elevated levels of non-esterified fatty acids (NEFA) in the blood, which are taken up by the liver. Serum insulin levels were greater in CD38-transgenic mice than in control mice in a glucose tolerance test performed on live animals [38]. When anti-CD38 antibodies are present in patients, glucose-induced insulin secretion may be impaired and lead to type 2 diabetes [38]. The researchers demonstrated that peroxisome proliferators-activated receptor γ(PPARγ)-mediated insulin sensitization enhances NAADP production by upregulating CD38 in adipocytes and that CD38-/ -completely blocked this effect [36]. Cyclic ADP-ribose acts as a second messenger for intracellular Ca2+ mobilization in glucose-induced insulin secretion [39]. This suggests that CD38 plays a key role in enhancing insulin sensitivity. However, more experiments are needed to explore the mechanism of CD38's role in the relationship between NAFLD and insulin resistance.
3. Lipid accumulation
One of the critical pathogenic characteristics of NAFLD is excessive lipid accumulation [2]. It has been demonstrated that CD38-deficient animals have much less lipid accumulation in their livers [40]. It is possible that the energy depletion is caused by increased NAD+ due to CD38 deficiency and the data suggest that levels of NAD+ and the NAD+ precursor nicotinamide mononucleotide (NMN) are significantly increased [40]. CD38 inhibition in a mouse model of HFD-induced obesity treated with the flavonoid apigenin showed a reduction in lipid accumulation in the liver through increased lipid oxidation [41]. Additionally, elevated CD38 expression in cells causes mitochondrial metabolic dysfunction [35]. Part of the mechanism may be increased lipid oxidation, NAD+ reduction and SIRT1 activation [36, 41]. The exact mechanism requires further research. In the HFD model, CD38-deficient mice displayed considerably less hepatic fat infiltration than wild-type controls [42]. However, more data are needed to demonstrate that CD38-deficient mice can prevent the abnormal accumulation of hepatic lipids in NAFLD. CD38 is also associated with the browning of white fat and the development of "classic" brown fat in mice. NAD+ and NADP(H) levels in thermogenic adipose tissues are increased when CD38 is downregulated [43]. Ablation of SIRT3 in CD38-deficient mice eliminates the protective role of CD38 inhibition in HFD-induced obesity [35]. Thus, the role of CD38 as a regulator of obesity and energy expenditure may also be related to thermogenesis and mediated by a NAD+-SIRT-dependent mechanism. Increased glutathione/oxidative glutathione (GSH/ GSSG) ratio in CD38-deficient hearts was found to be one of the mechanisms that reduce HFD-induced oxidative stress. Progress studies are needed regarding the mechanism of oxidative stress in the liver by CD38[15]. In pre-diabetic patients, CD38 cells were negatively correlated with cholesterol and low-density lipoprotein, and CD38 cells were negatively correlated with high-density lipoprotein values in patients with T2DM [44]. The study demonstrated that CD38-deficient mice were substantially more resistant to the obesity-inducing effects of HFD, and that WT mice fed HFD had significantly higher CD38 expression [36]. This protective mechanism may be related to the inhibition of PPARγ and CCAAT/enhancer-binding proteinα (C/EBPα) expression in adipose tissue [36]. The expression and activity of sterol regulatory element-binding protein-1(SREBP1) and its target gene fatty acid synthase (FASN) were found to be significantly diminished in CD38-/ - mouse embryonic fibroblasts (MEFs) [36]. This suggests that CD38 deficiency not only inhibits adipocyte differentiation, but also suppresses adipogenesis.
CD38 can influence the course of NAFLD through lipid accumulation, macrophage-1 and insulin secretion.
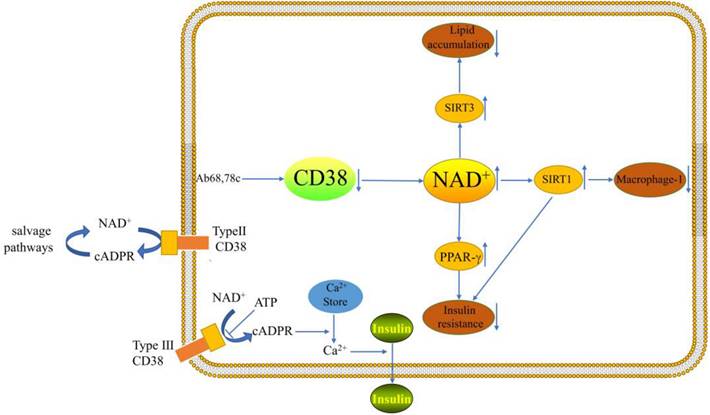
Conclusion
NAFLD is the most common form of liver disease, but there are no licensed medications available for its treatment. The progression of NAFLD is closely related to chronic inflammation, insulin resistance, and lipid accumulation. NAD+ is currently considered as a potential therapeutic target for NAFLD. As the critical NAD+ degrading enzyme in mammals, inhibiting of CD38 can significantly increase NAD+ levels and prevent NAFLD progression. These ideas have been confirmed in animal studies, but clinical trials still need to be verified.
Outlook
In summary, there is still a long way to go in the pathogenesis and clinical studies of CD38 in NAFLD. First, the pathogenesis of CD38 in NAFLD has not been adequately studied, and more studies are needed to demonstrate the correlation between CD38 and inflammation, abnormal accumulation of fatty acids, and insulin resistance. The interaction of inflammation and abnormal lipid accumulation promotes the pathological development of NAFLD, while abnormal lipid accumulation can be caused by insulin resistance. Abnormal accumulation of lipids in the liver leads to chronic inflammation and activation of Macrophage-1, and the development of inflammation in turn acts on hepatocytes, leading to abnormal accumulation of lipids. This interaction complicates the study of the pathogenesis of NAFLD, so more experimental investigations are needed in the future.
Acknowledgements
Funding
This study was funded by grant from the Beijing Hospital Research Project (No. BJ-2021-188).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Conze D, Brenner C, Kruger CL. Safety and Metabolism of Long-term Administration of NIAGEN (Nicotinamide Riboside Chloride) in a Randomized, Double-Blind, Placebo-controlled Clinical Trial of Healthy Overweight Adults. Sci Rep. 2019;9:9772
2. Friedman SL, Neuschwander-Tetri BA, Rinella M, Sanyal AJ. Mechanisms of NAFLD development and therapeutic strategies. Nat Med. 2018;24:908-22
3. Dall M, Hassing AS, Treebak JT. NAD(+) and NAFLD - caution, causality and careful optimism. J Physiol. 2022;600:1135-54
4. Bogan KL, Brenner C. Nicotinic acid, nicotinamide, and nicotinamide riboside: a molecular evaluation of NAD+ precursor vitamins in human nutrition. Annu Rev Nutr. 2008;28:115-30
5. Malavasi F, Deaglio S, Funaro A, Ferrero E, Horenstein AL, Ortolan E. et al. Evolution and function of the ADP ribosyl cyclase/CD38 gene family in physiology and pathology. Physiol Rev. 2008;88:841-86
6. Chini EN, Chini CCS, Espindola Netto JM, de Oliveira GC, van Schooten W. The Pharmacology of CD38/NADase: An Emerging Target in Cancer and Diseases of Aging. Trends Pharmacol Sci. 2018;39:424-36
7. Covarrubias AJ, Perrone R, Grozio A, Verdin E. NAD(+) metabolism and its roles in cellular processes during ageing. Nat Rev Mol Cell Biol. 2021;22:119-41
8. Tarrago MG, Chini CCS, Kanamori KS, Warner GM, Caride A, de Oliveira GC. et al. A Potent and Specific CD38 Inhibitor Ameliorates Age-Related Metabolic Dysfunction by Reversing Tissue NAD(+) Decline. Cell Metab. 2018;27:1081-95 e10
9. Boslett J, Reddy N, Alzarie YA, Zweier JL. Inhibition of CD38 with the Thiazoloquin(az)olin(on)e 78c Protects the Heart against Postischemic Injury. J Pharmacol Exp Ther. 2019;369:55-64
10. Quarona V, Zaccarello G, Chillemi A, Brunetti E, Singh VK, Ferrero E. et al. CD38 and CD157: a long journey from activation markers to multifunctional molecules. Cytometry B Clin Cytom. 2013;84:207-17
11. Bock KW. Aryl hydrocarbon receptor (AHR) functions: Balancing opposing processes including inflammatory reactions. Biochem Pharmacol. 2020;178:114093
12. Bock KW. Modulation of aryl hydrocarbon receptor (AHR) and the NAD(+)-consuming enzyme CD38: Searches of therapeutic options for nonalcoholic fatty liver disease (NAFLD). Biochem Pharmacol. 2020;175:113905
13. Beyer K. Age at Onset: An Essential Variable for the Definition of Genetic Risk Factors for Sporadic Alzheimer's Disease. Annals of the New York Academy of Sciences. 2005;1057:260-78
14. Astigiano C, Benzi A, Laugieri ME, Piacente F, Sturla L, Guida L. et al. Paracrine ADP Ribosyl Cyclase-Mediated Regulation of Biological Processes. Cells. 2022 11
15. Wang LF, Huang CC, Xiao YF, Guan XH, Wang XN, Cao Q. et al. CD38 Deficiency Protects Heart from High Fat Diet-Induced Oxidative Stress Via Activating Sirt3/FOXO3 Pathway. Cell Physiol Biochem. 2018;48:2350-63
16. Lee HC, Zhao YJ. Resolving the topological enigma in Ca(2+) signaling by cyclic ADP-ribose and NAADP. J Biol Chem. 2019;294:19831-43
17. Deng QW, Zhang J, Li T, He WM, Fang L, Lee HC. et al. The transferrin receptor CD71 regulates type II CD38, revealing tight topological compartmentalization of intracellular cyclic ADP-ribose production. J Biol Chem. 2019;294:15293-303
18. Park DR, Nam TS, Kim YW, Bae YS, Kim UH. Oxidative activation of type III CD38 by NADPH oxidase-derived hydrogen peroxide in Ca(2+) signaling. FASEB J. 2019;33:3404-19
19. Chini CCS, Tarrago MG, Chini EN. NAD and the aging process: Role in life, death and everything in between. Mol Cell Endocrinol. 2017;455:62-74
20. Jager J, Aparicio-Vergara M, Aouadi M. Liver innate immune cells and insulin resistance: the multiple facets of Kupffer cells. J Intern Med. 2016;280:209-20
21. Stahl EC, Haschak MJ, Popovic B, Brown BN. Macrophages in the Aging Liver and Age-Related Liver Disease. Front Immunol. 2018;9:2795
22. Jablonski KA, Amici SA, Webb LM, Ruiz-Rosado Jde D, Popovich PG, Partida-Sanchez S. et al. Novel Markers to Delineate Murine M1 and M2 Macrophages. PLoS One. 2015;10:e0145342
23. Covarrubias AJ, Kale A, Perrone R, Lopez-Dominguez JA, Pisco AO, Kasler HG. et al. Senescent cells promote tissue NAD(+) decline during ageing via the activation of CD38(+) macrophages. Nat Metab. 2020;2:1265-83
24. Niu B, He K, Li P, Gong J, Zhu X, Ye S. et al. SIRT1 upregulation protects against liver injury induced by a HFD through inhibiting CD36 and the NFkappaB pathway in mouse kupffer cells. Mol Med Rep. 2018;18:1609-15
25. Braidy N, Berg J, Clement J, Khorshidi F, Poljak A, Jayasena T. et al. Role of Nicotinamide Adenine Dinucleotide and Related Precursors as Therapeutic Targets for Age-Related Degenerative Diseases: Rationale, Biochemistry, Pharmacokinetics, and Outcomes. Antioxid Redox Signal. 2019;30:251-94
26. Kitada M, Ogura Y, Monno I, Koya D. Sirtuins and Type 2 Diabetes: Role in Inflammation, Oxidative Stress, and Mitochondrial Function. Front Endocrinol (Lausanne). 2019;10:187
27. Chini CCS, Peclat TR, Warner GM, Kashyap S, Espindola-Netto JM, de Oliveira GC. et al. CD38 ecto-enzyme in immune cells is induced during aging and regulates NAD(+) and NMN levels. Nat Metab. 2020;2:1284-304
28. Guimera AM, Clark P, Wordsworth J, Anugula S, Rasmussen LJ, Shanley DP. Systems modelling predicts chronic inflammation and genomic instability prevent effective mitochondrial regulation during biological ageing. Exp Gerontol. 2022;166:111889
29. Lu Q, Tian X, Wu H, Huang J, Li M, Mei Z. et al. Metabolic Changes of Hepatocytes in NAFLD. Front Physiol. 2021;12:710420
30. Elhassan YS, Kluckova K, Fletcher RS, Schmidt MS, Garten A, Doig CL. et al. Nicotinamide Riboside Augments the Aged Human Skeletal Muscle NAD(+) Metabolome and Induces Transcriptomic and Anti-inflammatory Signatures. Cell Rep. 2019;28:1717-28 e6
31. Wang Q, Hu J, Han G, Wang P, Li S, Chang J. et al. PTIP governs NAD(+) metabolism by regulating CD38 expression to drive macrophage inflammation. Cell Rep. 2022;38:110603
32. Cotter TG, Rinella M. Nonalcoholic Fatty Liver Disease 2020: The State of the Disease. Gastroenterology. 2020;158:1851-64
33. Kleiner DE, Brunt EM, Wilson LA, Behling C, Guy C, Contos M. et al. Association of Histologic Disease Activity With Progression of Nonalcoholic Fatty Liver Disease. JAMA Netw Open. 2019;2:e1912565
34. Alexander M, Loomis AK, van der Lei J, Duarte-Salles T, Prieto-Alhambra D, Ansell D. et al. Risks and clinical predictors of cirrhosis and hepatocellular carcinoma diagnoses in adults with diagnosed NAFLD: real-world study of 18 million patients in four European cohorts. BMC Med. 2019;17:95
35. Camacho-Pereira J, Tarrago MG, Chini CCS, Nin V, Escande C, Warner GM. et al. CD38 Dictates Age-Related NAD Decline and Mitochondrial Dysfunction through an SIRT3-Dependent Mechanism. Cell Metab. 2016;23:1127-39
36. Wang LF, Miao LJ, Wang XN, Huang CC, Qian YS, Huang X. et al. CD38 deficiency suppresses adipogenesis and lipogenesis in adipose tissues through activating Sirt1/PPARgamma signaling pathway. J Cell Mol Med. 2018;22:101-10
37. Yoshizaki T, Schenk S, Imamura T, Babendure JL, Sonoda N, Bae EJ. et al. SIRT1 inhibits inflammatory pathways in macrophages and modulates insulin sensitivity. Am J Physiol Endocrinol Metab. 2010;298:E419-28
38. Takasawa S. CD38-Cyclic ADP-Ribose Signal System in Physiology, Biochemistry, and Pathophysiology. Int J Mol Sci. 2022 23
39. Okamoto H, Takasawa S. Okamoto model for necrosis and its expansions, CD38-cyclic ADP-ribose signal system for intracellular Ca(2+) mobilization and Reg (Regenerating gene protein)-Reg receptor system for cell regeneration. Proc Jpn Acad Ser B Phys Biol Sci. 2021;97:423-61
40. Xie L, Wen K, Li Q, Huang CC, Zhao JL, Zhao QH. et al. CD38 Deficiency Protects Mice from High Fat Diet-Induced Nonalcoholic Fatty Liver Disease through Activating NAD(+)/Sirtuins Signaling Pathways-Mediated Inhibition of Lipid Accumulation and Oxidative Stress in Hepatocytes. Int J Biol Sci. 2021;17:4305-15
41. Escande C, Nin V, Price NL, Capellini V, Gomes AP, Barbosa MT. et al. Flavonoid apigenin is an inhibitor of the NAD+ ase CD38: implications for cellular NAD+ metabolism, protein acetylation, and treatment of metabolic syndrome. Diabetes. 2013;62:1084-93
42. Barbosa MT, Soares SM, Novak CM, Sinclair D, Levine JA, Aksoy P. et al. The enzyme CD38 (a NAD glycohydrolase, EC 3.2.2.5) is necessary for the development of diet-induced obesity. FASEB J. 2007;21:3629-39
43. Benzi A, Sturla L, Heine M, Fischer AW, Spinelli S, Magnone M. et al. CD38 downregulation modulates NAD(+) and NADP(H) levels in thermogenic adipose tissues. Biochim Biophys Acta Mol Cell Biol Lipids. 2021;1866:158819
44. Mendez-Frausto G, Romero-Aguilera G, Sanchez-Gutierrez R, Garcia-Jacobo RE, Lara-Ramirez EE, Uresti-Rivera EE. et al. B regulatory cells associated with changes in biochemical and inflammatory parameters in normal-glycemic individuals, pre-diabetes and T2DM patients. Diabetes Res Clin Pract. 2021;173:108692
Author contact
![]() Corresponding author: Zuowei Pei, Department of Cardiology, Central Hospital of Dalian University of Technology, No.826 Xinan Road, 116033, Dalian, Telephone number: +86-0411-84412001, E-mail: pzw_dlcom.
Corresponding author: Zuowei Pei, Department of Cardiology, Central Hospital of Dalian University of Technology, No.826 Xinan Road, 116033, Dalian, Telephone number: +86-0411-84412001, E-mail: pzw_dlcom.