Impact Factor ISSN: 1449-1907
- Issue 9; 2026
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Volume 23; 2026
- Past Issues
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
T lymphocytes and granulocytes
Natural Killer Cells and...
Dendritic cells
Macrophages
Conclusions
References

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2022; 19(12):1787-1795. doi:10.7150/ijms.73134 This issue Cite
Review
SARS-CoV-2 and HIV-1: So Different yet so Alike. Immune Response at the Cellular and Molecular Level
Catherine Demoliou, Christos Papaneophytou, Vicky Nicolaidou ![]()
Department of Life and Health Sciences, School of Sciences and Engineering, University of Nicosia, 46 Makedonitissas Avenue, 2417, Nicosia, Cyprus
Received 2022-3-22; Accepted 2022-9-7; Published 2022-10-3
Abstract

In the past half century, humanity has experienced two devastating pandemics; the HIV-1 pandemic and the recent pandemic caused by SARS-CoV-2. Both emerged as zoonotic pathogens. Interestingly, SARS-CoV-2 has rapidly migrated all over the world in less than two years, much as HIV-1 did almost 40 years ago. Despite these two RNA viruses being different in their mode of transmission as well as the symptoms they generate, recent evidence suggests that they cause similar immune responses. In this mini review, we compare the molecular basis for CD4+ T cell lymphopenia and other effects on the immune system induced by SARS-CoV-2 and HIV-1 infections. We considered features of the host immune response that are shared with HIV-1 and could account for the lymphopenia and other immune effects observed in COVID-19. The information provided herein, may cast the virus-induced lymphopenia and cytokine storm associated with the acute SARS-CoV-2 infection and pathogenesis in a different light for further research on host immune responses. It can also provide opportunities for the identification of novel therapeutic targets for COVID-19. Furthermore, we provide some basic information to enable a comparative framework for considering the overlapping sets of immune responses caused by HIV-1 and SARS-CoV-2.
Keywords: COVID-19, SARS-CoV-2, HIV-1, lymphopenia, immune response
Introduction
In the past half century, two distinct, novel RNA viruses have caused global pandemics, both having emerged as zoonotic pathogens: human immunodeficiency virus type 1 (HIV-1) and severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) (1). HIV-1, a member of the Retroviridae virus family, is most closely related to immunodeficiency viruses found in wild chimpanzees and most likely made the transfer to humans early in the 20th century (2). HIV-1 is transmitted sexually or through body fluids, such as blood and breast milk, and leads to a chronic infection that eventually results in immunodeficiency and death by opportunistic infections.
Coronaviruses (CoVs), like the SARS-CoV-1 (3), the Middle East Respiratory Syndrome Coronavirus (MERS-CoV) (4), and their recently isolated relative SARS-CoV-2 (5), are all enveloped viruses with a closely related positive RNA genome, and zoonotic origin. The most recent member, SARS-CoV-2, is most closely related to a virus isolated from bats, it is transmitted via the respiratory route and has spread worldwide causing a pandemic (6). The WHO reports that to date there are more than 583 million confirmed cases of infection, including more than 6 million deaths of the related disease, Coronavirus disease 2019 (COVID-19) (7).
CoVs contain four major structural proteins, of which the Spike (S) protein is used for host receptor attachment and host cell entry, facilitated via membrane fusion(8) and via priming by the Transmembrane serine protease 2 (TMPRSS2) (9). The host receptor is the cell membrane angiotensin-converting enzyme 2 (ACE2) receptor, which converts angiotensin II into angiotensin. The ACE2 receptor is an interferon (IFN)-stimulated gene, co-expressed with the serine protease TMPRSS2 in the upper airway human respiratory epithelial cells, human absorptive enterocytes, nasal goblet secretory cells (10) and vascular cells of many tissues, including the lungs, heart and kidney, and it contributes to the SARS-CoV-2 organotropism especially for patients with pre-existing conditions (11-14).
COVID-19 symptoms can range from asymptomatic to acute and may result even in death. In mild cases, viral infiltration of lung parenchymal tissue triggers an innate immune response. This is accompanied by the secretion of cytokines and chemokines (15), triggered by the viral RNA (16) due to the activation of pattern recognition receptors (PRRS), RIG-I-like receptors (RLRs) and the Toll-like receptors (TLRs) of Natural Killer (NK) cells and monocytes/macrophages of the host. If the innate response is compromised at the alveolar cell level, the virus has the chance to replicate, causing extensive tissue injury that dictates the severity of infection. In acute infection cases, the overproduction of pro-inflammatory cytokines, known as “cytokine storm”, driven by IL-6, CXCL10 and infiltrating macrophages (17-20), results in the suppression/deregulation of the immune response by T cells and in lymphopenia (21,22), as well as the acute respiratory distress syndrome (ARDS), which may lead to organ dysfunction, and death (23-25).
CoVs and HIV-1 belong to different viral families, differ significantly in their ecology, mode of infection, and genome. Another important difference is the fact that HIV-1 infection cannot be cleared, which constitutes a major challenge in eliminating the virus, something which is not an issue in SARS-CoV-2 infection. The two viruses do however, share important similarities; recent reviews have compared them in terms of their evolution (1). Similarities also exist regarding the immune responses caused by the two viruses.
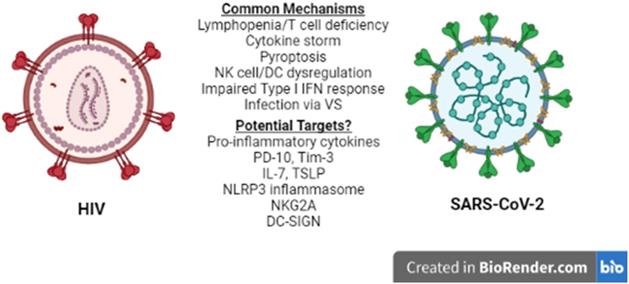
Lymphopenia is associated with several other viral infections including HIV-1 (26), and is considered a prognostic value parameter for individuals that are infected by SARS-CoV-2 or HIV-1. Although the manifestations of HIV-1 infection on the immune system become apparent in the chronically ill patients (26), they are driven by the cytokine profile set early in the disease and, therefore, several mechanisms and molecules proposed to play a role in acute HIV-1 lymphopenia may also be shared in the SARS-CoV-2-induced pathogenesis. A recent comparison of the pathologies and side effects of infection by these two viruses has highlighted the similarities in the pro-inflammatory cytokine response, the modification of intestinal microbiota and the formation of Neutrophil Extracellular Traps (NETs) (27). Understanding COVID-19-associated lymphopenia in the framework of HIV-1 infection may identify potential molecular targets for further research and/or development of treatment(s) for COVID-19 (Figure 1).
HIV-1 vs SARS-CoV-2. Similarities between HIV-1 and SARS-CoV-2 immune response to infection at the cellular level reveal common mechanisms and may lead to the identification of potential therapeutic targets for COVID-19. NK: Natural Killer, DC: Dendritic cell, VS: Virological Synapse.
T lymphocytes and granulocytes
HIV-1 and SARS-CoV-2 are different viruses. However, T lymphocyte deficiency or lymphocyte ineffectiveness and a “cytokine storm” due to the hyperinflammatory response of cells involved in the innate response, are characteristics of both (26-30). In the case of acute HIV-1 subtype B and C infections, the pro-inflammatory “cytokine storm” activation contributes to CD4+ T cell lymphopenia and prevention of IL-2 producing memory CD4+ and CD8+ T cells, which lead to the development of the acquired immune deficiency syndrome (AIDS). The majority of T cells die by pyroptosis, linking the HIV-1 infection and lymphopenia with dying CD4+ T cells releasing signals that cause more cells to die thus, preventing T cell homeostasis and renewal (31-33).
There are distinct differences in the numbers of blood cells like macrophages, CD8+ T cells, Th17 cells and naive T cells between HIV-1 and severe COVID-19 patients. However, in both types of infections, the common feature is that cells like NK cells, B cells, CD4+ T cells, regulatory T cells, memory T and B cells are reduced significantly in numbers (26). There is a reduction in CD8+ T cell observed in COVID-19, which is attributed to cell exhaustion due to continuous exposure to cytokines like IL-10, IL-6, and Tumor Necrosis Factor-alpha (TNF-α), to damage-associated molecular patterns and to virus-derived antigens (34). The cell cytokine profile resembles that of terminally exhausted CD8+ T cells with compromised cytotoxic activity, seen in patients chronically infected with HIV-1 (35-37). Increased COVID-19 severity and chronic HIV-1 infection are accompanied by high expression of T cell inhibitory receptors including programmed cell death-1 (PD-10) and T cell immunoglobulin mucin 3 (Tim-3) proteins, which contribute to the low CD4+ T cell counts and reduction of IFN-γ production. This immunopathogenic late CD4+ T cell response is considered to provide potential targets for treating T cell exhaustion via inhibition of expression of these inhibitory receptors (36-39).
Recent studies in T cells infected with HIV-1 have indicated the role of restriction factors like APOBEC3G (apolipoprotein B mRNA editing enzyme, catalytic polypeptide-like 3G), Tetherin (CD317), SAMHD1 (SAM domain and HD domain-containing protein 1), and TRIM5α (Tripartite motif-containing protein 5,) in the front-line defense against HIV-1 infection. These factors are associated with the intracellular protein degradation pathways involved in viral antigens production and the activation of HIV-1-specific T cell immunity. HIV-1 has evolved the ability to counteract these by inhibiting their expression using several accessory viral proteins or evading through protein variability, and thus replicate efficiently in the hostile environment of the host cell contributing to T cell death (39,40). Functional proteomic atlases of HIV-1 and SARS-CoV-2 cell infections in vitro (41,42), have identified a large number of cellular host restriction factors against HIV-1 and SARS-CoV-2. Comparisons of these along with the atlas of phosphorylated proteins in SARS-CoV-2 infection (43) could provide valuable clues in the understanding of the mechanisms underlying viral inactivation at the transcriptional level, and help identify molecular targets and therapeutic or curative strategies for COVID-19 treatment.
Little is known about the “cytokine storm” contribution in HIV-1 or SARS-CoV-2 immunopathology of eosinophils, basophils and neutrophils. A positive correlation between neutrophil cell counts and cytokines, like IP-10 and IL-8, has been reported in acute HIV-1 infection (29). In COVID-19 cases, neutrophil and granulocyte pathophysiology has been linked to the adverse effects of SARS-CoV-2 on tissues, as well as the vascular and coagulation systems in severely affected patients (44). The increase in mature and immature neutrophil numbers and decrease in the numbers of eosinophils and basophils in severe COVID-19 infections, have been associated with altered expression of several receptors involved in activation, adhesion and migration of granulocytes (e.g. CD62L, CD11a/b, CD69, CD63, CXCR4) (45). As in the case of acute HIV-1 infection (29) and HIV-1 dissemination to CD4+ T cells (46), identifying SARS-CoV-2 role in the expression of the chemokines/chemokine receptors responsible for mononuclear cell trafficking and tissue infiltration observed in post-mortem analysis of COVID-19 patients (47), will be important. These routes of infection (i.e. blood monocytes) could explain the persistent tissue infectivity of the SARS-CoVs and contribute to the persistent inflammation caused by the antibody-dependent enhancement (ADE) mechanism, a viral dependent phenomenon observed in patients who remain virus positive although they have early, suboptimal antibody activity (21).
Alternatively, identifying upstream regulators of the inflammatory pathway, as for example IL-17 studied in mice (48), or regulators of lymphopoiesis, as for example IL-7 and thymic stromal lymphopoietin (TSLP), may be needed to understand COVID-19 cytokine pathogenesis. IL-7 is produced by intestinal epithelial and epithelial goblet cells and plays an important role in the development, proliferation, and survival of innate and adaptive lymphoid cells as well as in the development of the multiple effector functions during infections (49,50). IL-7 was used in HIV-1 infections to increase de novo T cell formation but led to rapid proliferation of the latent HIV-1 reservoir in resting memory CD4+ T cells, and any further treatment was abandoned (28). IL-7, however, may be of use in the treatment of severely ill COVID-19 patients to counterbalance lymphopenia and enhance antiviral response of the immune system (51).
TSLP is produced by epithelial cells of the skin, gut and lungs and it is considered to play a critical role in driving Th2 mediated inflammation, and to contribute to asthma and allergic inflammation (52). TSLP expression is activated by cytokines including IL-4, IL-1 and TNF-α (53), and high TSLP expression appears to play a role in platelet activation and thrombosis in Kawasaki disease (54), as well as in the metabolic syndrome and high blood pressure of severely obese individuals (52-55). HIV-1-induced expression of TSLP in epithelial cells triggers dendritic cell (DCs)-mediated amplification of HIV-1 infection in activated CD4+ T cells (56). TSLP, therefore, could be a good candidate for further investigations in relation to SARS-CoV-2 infection and related side effects. TSLP and its isoforms may contribute to the Kawasaki Syndrome seen in SARS-CoV-2 infected children, or to increased thrombosis in COVID-19 severe cases, through activation of vascular endothelial cells and/or platelet activation via TSLP-dependent PI3K/Akt signaling (57-59).
It has been well documented that the lymphopenia in acute SARS-CoV-2 infection is the result of tissue damage inflammation and a dysregulated innate and adaptive immune system that involves a dramatic loss of CD4+ and an even greater loss of CD8+ T cells (60, 61). Since T cell infection by SARS-CoV-2 is abortive (62), tissue destruction and cell death have been attributed to cytokine induced tissue necrosis and apoptosis and to T cell driven pyroptosis via inflammasomes activation, rather than viral replication (63, 64). Pyroptosis-driven CD4+ T cell death following SARS-CoV-2 infection (65, 66) is supported by the increased serum levels of IL-1β and IL-18 in COVID-19 patients. Pyroptosis occurs faster than apoptosis and the release of cell contents results in the recruitment of increasing numbers of effector immune cells, thus promoting further the inflammatory cascade and T cell exhaustion (63, 66-69). In HIV-1 acute infection, studies have shown that more than 95% of CD4+ T cells depleted from lymphoid tissue die by pyroptosis due to abortive HIV-1 infection and inefficient reverse transcription (31, 68). Pyroptosis is also involved in CD4+ T cell loss in chronically HIV-1-infected patients via the NLR family pyrin domain containing 3 (NLRP3) sensor in both peripheral blood and lymphoid tissues in a bystander manner (70). HIV-1 induced CD4+ T cell death by pyroptosis is not inhibited by antibodies to IFN α/β, suggesting that pyroptosis is not normally part of the innate immune responses that promote cell death in acute infections. Additionally, caspase-3 expressing cells in HIV-1 productively infected cells, appear to be anatomically separated from cells with abortive infections (63, 68). Targeting pyroptosis could potentially be a beneficial approach to address lymphopenia in COVID-19 (62, 71), and implicate pyroptosis signaling as a target for anti-HIV-1 treatment.
Natural Killer Cells and CD8+ Cytotoxic T Cells
NK cells along with CD8+ cytotoxic T cells are central to the host defense against viral infection and tumour cells. NK cells recognize and kill virally-infected and neoplastic cells directly or via antibody dependent cellular cytotoxicity (ADCC), using the granzyme-perforin system that leads to apoptosis. NK cells are also able to kill via cytokine release (i.e. TNF) and expression of ligands such as FasL and the Tumor necrosis factor-related apoptosis-inducing ligand (TRAIL). NK cells have additionally an immunoregulatory role via secretion of and response to various cytokines (IFNs, IL-12, -15 and -18), which prime them to respond to viral infection via IFN-γ production and/or NK cell proliferation (72, 73). NK cells are also activated by stress induced ligands via their cell surface lectin receptors like CD94/NKG2C, and express inhibitory receptors, like CD94/NKG2A, to repress NK cell activation and cytotoxicity. NKG2A positive NK cells, therefore, play an important role in limiting excessive activation, preventing apoptosis, and preserving CD8+ T cell responses during viral infections (73-75).
Upon chronic cytokine stimulation there is exhaustion of adaptive NK cells, associated with down modulation of CD94/NKG2C receptor expression via epigenetic reprograming, which is prevented through signaling involving the NKG2A inhibitory receptors (76). NK cells from HIV-1 infected individuals have increased- whereas seronegative individuals have decreased-NKG2A receptors due to changes in cell phenotype and exhaustion. High levels of HIV-1 replication alters the NK cell phenotype and promotes the expansion of an anergic subset, unable to perform ADCC and to kill virus infected CD4+ T cells. The reduced numbers of cytotoxic NK cells and higher NKG2A expression in patients with late-stage HIV-1 infection were related to the escape of HIV-1-infected CD4+ T cells from NK cells cytotoxicity (77, 78).
A similar dysregulation of NK cell expression of the NKG2A inhibitory receptor due to SARS-CoV-2 induced hyperinflammation could also lead to NK cells exhaustion with diminished capacity to produce TNF and IFN-γ cytokines, as in the case of HIV-1 infected cells (76, 79). The decreased numbers of NK and CD8+ T cells have been positively related to the severity of COVID-19, to the increased expression of NKG2A inhibitory receptors, and to the reduced expression of lysosomal-associated membrane protein-1, IFN-γ, IL-2, granzyme B, and TNF-α (normally present in chronic viral infections). In contrast, NKG2A expression was reduced in patients recovering from SARS-CoV-2 infection, suggesting an early functional exhaustion of cytotoxic lymphocytes (30, 80, 81). Furthermore, SP1 expression in lung epithelial cells was shown to contribute to the exhaustion of NK cells via HLA-E/NKG2A interactions (80). It is of interest to note that patients who recover from COVID-19 have SARS-CoV-2 specific CD8+ and CD4+ T cells (82), supporting the fact that, the SARS-CoV-2 virus may have mechanisms that evade the efficient innate immune response (83). Taken together, these findings highlight the importance of improving the early immune response of NK cells in addition to cytotoxic T cells to prevent their exhaustion, and that the NKG2A receptor in NK cells could be a potential target to enable virus elimination in the early stage of SARS-CoV-2 infection.
Dendritic cells
Antigen presentation to T cells is a key event for their activation, which requires the contribution of mature DCs, the immune cells that are considered as the link between the innate and adaptive immune systems. HIV-1 infection of DCs prevents DC maturation, which results in their continuous stimulation and elimination of infected T cells, enhancement of central memory CD4+ T cell proliferation and differentiation, leading to exhaustion of the CD4+ T cell pool, while at the same time generating increased target cell numbers for HIV-1 infection. The secreted type I IFNs also induce the expression of immunosuppressive molecules like PD-L1 (a suppressor of the adaptive immune response as discussed earlier) at the surface of DCs, leading to diminished anti-viral responses (84).
Studies on DCs infected with SARS-CoV-1, concluded that although infection could be abortive, the virus negatively modulated the DC expression of antiviral cytokines and promoted death receptor ligands and chemokine receptors. This facilitated the migration of cytotoxic DCs from the infection site to lymph nodes of SARS-CoV-1 infected patients for further depletion of lymphoid cell through TRAIL induced apoptosis (85-87).
At lymph nodes and peripheral tissue, infected DCs may promote SARS-CoV-2 infection of T cells through virological synapses (VS), in a manner similar to that of HIV-1 infected DCs (88, 89). VS, (i.e. cell to cell transfer), is used by a number of intracellular pathogens and viruses in order to spread through their host (90). More interestingly, studies on HIV-1 transmission have shown DCs participation in CD4+ T cell trans-infection, despite the low infectivity of DCs. This involves the virus attachment to C-type lectins, such as the DC‐specific intercellular adhesion molecule‐3‐grabbing nonintegrin (DC-SIGN). Attachment is followed by endocytosis, storage and viral dissemination through the exosome pathway, or through an “infectious synapse” between uninfected mature DCs harboring HIV-1 and uninfected target cells with retention of the virus on the DC surface and transmission to target cell through adhesion protein interactions. DCs were reported to take up SARS-CoV-1 via the DC‐SIGN-receptor and transfer the virus to uninfected target cells through direct cell-to-cell transmission, suggesting that the CoVs family of viruses may also infect T cells “in trans” via VS (91, 92). DC-SING and DC-SING receptor (DC-SINR) enhance SARS-CoV-1 and SARS-CoV-2 S protein-driven transinfection (93, 94), and there is evidence that DC-SINGR expression is induced in alveolar macrophages by IL-4 and IL-13 upon infection by other viruses (95). Since the levels of these cytokines increase in the early phases of SARS-CoV-2 infection, they could also be involved in macrophage infectivity at lymph node sites. Lymphopenia, in COVID-19, in addition to innate response (60) may also occur via transmission via DC-T cell VS, T cell-T cell VS and even macrophage-T cell VS infection with SARS-CoV-2, in a manner similar to that of HIV-1 transmission (92, 94). It is of interest to note that HIV-1 taken up via DC-SIGN remains infectious for prolonged periods of time, protected possibly by trafficking through non-lysosomal endosomes and infecting T cells through DC-mediated viral transmission (89). DCs' role in the transmission of SARS-CoV-2 virus, T cell dysregulation and functional exhaustion could provide targets for development of therapeutic approaches.
Macrophages
The acute onset of lung inflammation is key to ARDS present in severe COVID-19 cases and mortality. Alveolar and lung interstitial macrophages have a central role in SARS-CoVs infection and ARDS since they contribute to the “cytokine storm” through massive production of cytokines, monocyte chemoattractant proteins, nitric oxide (NO) and reactive oxygen species (ROS) (17, 58, 96, 97). This cytokine storm may also be followed by a secondary haemophagocytic lymphohistiocytosis (sHLH) response, a hyperinflammatory syndrome, and more likely result in fatal acute lung injury and multi-organ failure (98).
Early timing of type I IFN production by activated macrophages, appears to prevent the lethal inflammatory cytokine storm (99, 100). The sera from critically ill patients who died of SARS promoted the accumulation of pulmonary proinflammatory macrophages with enhanced SARS-CoV-1-induced MCP1 and IL-8 production, and this was counteracted by the blockade of FcγR (101). It has therefore been considered, that macrophage activation by SARS-CoV-1 could be mediated by antibody (anti-S-IgG) dependent enhancement (ADE) via the engagement of Fc receptors (FcR) (102, 103). It has also been suggested that ADE may facilitate the spread of SARS-CoV-2 in infected hosts, although in vivo evidence from human studies is still required for further support regarding this path of infection (104). However, internalization of virus-antibody immune complexes through the FcRs can upregulate pro-inflammatory cytokines, downregulate anti-inflammatory cytokines and induce cell death of recruited immune cells infected with SARS-CoVs either through ADE or the ACE2 receptor and/or through VS (69, 105).
In the case of HIV-1, non-dividing, metabolically active macrophages have been shown to contain large quantities of biologically active HIV-1 DNA, which is unintegrated and can remain stable and contribute to HIV-1 pathogenesis (106). HIV-1-infected macrophages can engage motile T cells in stable contacts through binding of virus- and host-derived adhesive molecules and contribute to high viral spread and HIV-1 progression (107). Using a similar path of dissemination, SARS-CoV-2 may also persist in resident macrophages of many tissues after recovery from COVID-19, since they are differentiated cells and have a long life span. GU-rich RNA derived from SARS-CoV-2, SARS-CoV-1, and HIV-1 have been shown to induce cytokine release (IL-6, TNF, and IL-1b) from human primary macrophages in the absence of viral infection, associating SARS-CoV-2 and HIV-1 to chronic inflammation (69). The presence of infected macrophages at lymph nodes and their tissue redistribution may prevent the elimination of the virus and explain how a small percentage of patients recovering from SARS-CoV-2 may become positive again for SARS-CoV-2 RNA, and/or be characterized as virus carriers (108, 109).
Conclusions
COVID-19 is multiphasic in its pathogenesis, and includes a cytokine storm, lymphopenia and ARDS, which indicate poor prognosis. COVID-19 progression and severity are associated with a systemic dysregulation of the immune system and impaired type I IFN early responses. Tissue damage and signatures of early innate antiviral responses by lymphocyte and monocyte infiltration and macrophage accumulation in the infected alveolar lumen, are all markers of ARDS. The death of infected monocytes/macrophages, NK cell activation, NK cell-mediated ADCC, NK cell and T cell dysregulation and exhaustion are considered to be the contributors of proinflammatory cytokines and to drive the cytokine storm and lymphopenia. Although lymphopenia is observed in many viral infections, it is poorly understood and more so in the case of COVID-19, where the immune cell dynamics and those of cytokine secretion remain unclear. Furthermore, the evidence for T cell death due to direct infection by SARS-CoV-2 is insufficient and needs to be confirmed.
Comparative studies on immune cell dysfunction in SARS-CoV-1, MERS-CoV and HIV-1 infection, may help in understanding the pathogenesis of SARS-CoV-2 and identifying important components for further research. Common mechanisms highlighted in this review are summarized in Table 1. Infection of T lymphocytes by SARS-CoV-2 via VS in a manner similar to that used by HIV-1 for its dissemination in the host, although abortive, could contribute to lymphopenia due to virus-induced pyroptosis. Furthermore, SARS-CoV-2 infection and the systemic hyperinflammatory response may cause a state of T cell exhaustion with effector functions that may contribute via cytokine secretion and cytotoxicity to COVID-19 lymphopenia and pathogenesis. Such processes would justify strategies aiming the blockade of cytokine activity and cytokine receptor signaling at the early stages of COVID-19 using anti-inflammatory medication. Effective drug design and targeting of specific cytokines and their receptors, however, may first require a better understanding of the crosstalk and interactions of the pathways activated by these cytokines, as well as the stage specific factors that contribute to the pathology of SARS-CoV-2 infection.
Common mechanisms affecting immunity in HIV-1 and SARS-CoV-2 infection.
| Common molecular/cellular mechanisms | Associated immunological/pathological effect | Reference |
|---|---|---|
| Cytokine storm/ inflammasome activation | T cell lymphopenia due to death by pyroptosis T cell exhaustion Immunodeficiency | (26-34) |
| Elevated T cell inhibitory receptors | T cell lymphopenia Reduced I IFN-γ production | (36-39) |
| Altered host restriction factors | Inability to control viral infection | (41,42) |
| NK cell dysregulation | Reduced cytokine/inhibitory receptor expression NK cell exhaustion, inability to kill virally infected cells | (76-79) |
| Dysregulated and infected DCs | Reduced T cell activation Viral spreading via VS | (84-89) (91-94) |
| Impaired Type I IFN response | Inability to control viral infection | (76,79) |
| Viral persistence in macrophages | Production of pro-inflammatory cytokines Viral spreading | (107-109) |
Competing Interests
The authors have declared that no competing interest exists.
References
1. Fischer W, Giorgi EE, Chakraborty S, Nguyen K, Bhattacharya T, Theiler J. et al. HIV-1 and SARS-CoV-2: Patterns in the evolution of two pandemic pathogens. Cell Host & Microbe. 2021;29(7):1093-110
2. Sharp PM, Hahn BH. Origins of HIV and the AIDS Pandemic. Available from: http://www.unaids.org/.
3. Drosten C, Gunther S, Preiser W, van der Werf S, Brodt HR, Becker S. et al. Identification of a novel coronavirus in patients with severe acute respiratory syndrome. N Engl J Med. 2003;348(20):1967-76 Available from: https://www.ncbi.nlm.nih.gov/pubmed/12690091
4. Zaki AM, van Boheemen S, Bestebroer TM, Osterhaus AD, Fouchier RA. Isolation of a novel coronavirus from a man with pneumonia in Saudi Arabia. N Engl J Med. 2012;367(19):1814-20 Available from: https://www.ncbi.nlm.nih.gov/pubmed/23075143
5. Huang C, Wang Y, Li X, Ren L, Zhao J, Hu Y. et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet. 2020;395(10223):497-506 Available from: https://www.ncbi.nlm.nih.gov/pubmed/31986264
6. Zhou P, Yang XL, Wang XG, Hu B, Zhang L, Zhang W. et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. 2020;579(7798):270-3 Available from: https://www.ncbi.nlm.nih.gov/pubmed/32015507
7. WHO COVID-19 Dashboard. Geneva: World Health Organization, 2020. Available from: https://covid19.who.int/.
8. Wu F, Zhao S, Yu B, Chen YM, Wang W, Song ZG. et al. A new coronavirus associated with human respiratory disease in China. Nature. 2020;579(7798):265-9 Available from: https://www.ncbi.nlm.nih.gov/pubmed/32015508
9. Hoffmann M, Kleine-Weber H, Schroeder S, Krüger N, Herrler T, Erichsen S. et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell. 2020 Available from: http://www.sciencedirect.com/science/article/pii/S0092867420302294
10. Ziegler CGK, Allon SJ, Nyquist SK, Mbano IM, Miao VN, Tzouanas CN. et al. SARS-CoV-2 Receptor ACE2 Is an Interferon-Stimulated Gene in Human Airway Epithelial Cells and Is Detected in Specific Cell Subsets across Tissues. Cell. 2020;181(5):1016-1035 e19. Available from: https://www.ncbi.nlm.nih.gov/pubmed/32413319
11. Akhmerov A, Marban E. COVID-19 and the Heart. Circ Res. 2020;126(10):1443-55 Available from: https://www.ncbi.nlm.nih.gov/pubmed/32252591
12. Hamming I, Timens W, Bulthuis ML, Lely AT, Navis G, van Goor H. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J Pathol. 2004;203(2):631-7 Available from: https://www.ncbi.nlm.nih.gov/pubmed/15141377
13. Puelles VG, Lutgehetmann M, Lindenmeyer MT, Sperhake JP, Wong MN, Allweiss L. et al. Multiorgan and Renal Tropism of SARS-CoV-2. N Engl J Med. 2020/05/14. 2020 Available from: https://www.ncbi.nlm.nih.gov/pubmed/32402155
14. Walls AC, Park YJ, Tortorici MA, Wall A, McGuire AT, Veesler D. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell. 2020;181(2):281-292.e6 Available from: /pmc/articles/PMC7102599/
15. Khalil BA, Elemam NM, Maghazachi AA. Chemokines and chemokine receptors during COVID-19 infection. Computational and Structural Biotechnology Journal, Elsevier B.V. 2021:976-88 Available from: https://pubmed.ncbi.nlm.nih.gov/33558827/
16. Lee HC, Chathuranga K, Lee JS. Intracellular sensing of viral genomes and viral evasion. Exp Mol Med. 2019;51(12):1-13 Available from: https://www.ncbi.nlm.nih.gov/pubmed/31827068
17. Coperchini F, Chiovato L, Rotondi M. Interleukin-6, CXCL10 and Infiltrating Macrophages in COVID-19-Related Cytokine Storm: Not One for All But All for One!. Frontiers in Immunology, Frontiers Media S.A. 2021 Available from: https://pubmed.ncbi.nlm.nih.gov/33981314/
18. Vardhana SA, Wolchok JD. The many faces of the anti-COVID immune response. J Exp Med. 2020 217(6). Available from: https://www.ncbi.nlm.nih.gov/pubmed/32353870
19. Tisoncik JR, Korth MJ, Simmons CP, Farrar J, Martin TR, Katze MG. Into the eye of the cytokine storm. Microbiol Mol Biol Rev. 2012;76(1):16-32 Available from: https://www.ncbi.nlm.nih.gov/pubmed/22390970
20. Fajgenbaum DC, June CH. Cytokine Storm. New England Journal of Medicine. 2020];383(23):2255-73. Available from: https://www.nejm.org/doi/full/10.1056/NEJMra2026131.
21. Fu Y, Cheng Y, Wu Y. Understanding SARS-CoV-2-Mediated Inflammatory Responses: From Mechanisms to Potential Therapeutic Tools. Virol Sin. 2020 Available from: https://www.ncbi.nlm.nih.gov/pubmed/32125642
22. Lotfi R, Kalmarzi RN, Roghani SA. A review on the immune responses against novel emerging coronavirus (SARS-CoV-2). Immunologic Research. Springer. 2021 Available from: https://pubmed.ncbi.nlm.nih.gov/33928531/
23. Mehta P, McAuley DF, Brown M, Sanchez E, Tattersall RS, Manson JJ. et al. COVID-19: consider cytokine storm syndromes and immunosuppression. Lancet. 2020;395(10229):1033-4 Available from: https://www.ncbi.nlm.nih.gov/pubmed/32192578
24. Sarzi-Puttini P, Giorgi V, Sirotti S, Marotto D, Ardizzone S, Rizzardini G. et al. COVID-19, cytokines and immunosuppression: what can we learn from severe acute respiratory syndrome? Clin Exp Rheumatol. 2020;38(2):337-42 Available from: https://www.ncbi.nlm.nih.gov/pubmed/32202240
25. Haque SKM, Ashwaq O, Sarief A, Azad John Mohamed AK. A comprehensive review about SARS-CoV-2. Future Virology, Future Medicine Ltd. 2020 p. 625-48. Available from: https://pubmed.ncbi.nlm.nih.gov/33224265/
26. Peng X, Ouyang J, Isnard S, Lin J, Fombuena B, Zhu B. et al. Sharing CD4+ T Cell Loss: When COVID-19 and HIV Collide on Immune System. Vol. 11, Frontiers in Immunology. Frontiers Media S.A. 2020 p. 3307. Available from: www.frontiersin.org
27. Illanes-álvarez F, Márquez-Ruiz D, Márquez-Coello M, Cuesta-Sancho S, Girón-González JA. Similarities and differences between hiv and sars-cov-2. International Journal of Medical Sciences. 2021;18(3):846-51 Available from: /pmc/articles/PMC7797543/
28. Hokello J, Sharma AL, Dimri M, Tyagi M. Insights into the HIV Latency and the Role of Cytokines. Pathogens. 2019;8(3):10.3390 /pathogens8030137
29. Muema DM, Akilimali NA, Ndumnego OC, Rasehlo SS, Durgiah R, Ojwach DBA. et al. Association between the cytokine storm, immune cell dynamics, and viral replicative capacity in hyperacute HIV infection. BMC Medicine. 2020;18(1):1-17 Available from: https://doi.org/10.1186/s12916-020-01529-6
30. Zheng M, Gao Y, Wang G, Song G, Liu S, Sun D. et al. Functional exhaustion of antiviral lymphocytes in COVID-19 patients. Cell Mol Immunol. 2020;17(5):533-5 Available from: https://www.ncbi.nlm.nih.gov/pubmed/32203188
31. Doitsh G, Galloway NLK, Geng X, Yang Z, Monroe KM, Zepeda O. et al. Cell death by pyroptosis drives CD4 T-cell depletion in HIV-1 infection. Nature. 2014;505(7484):509-14 Available from: https://pubmed.ncbi.nlm.nih.gov/24356306/
32. Douek DC, Picker LJ, Koup RA. T cell dynamics in HIV-1 infection. Annu Rev Immunol. 2003;21:265-304 Available from: https://www.ncbi.nlm.nih.gov/pubmed/12524385
33. Vidya Vijayan KK, Karthigeyan KP, Tripathi SP, Hanna LE. Pathophysiology of CD4+ T-Cell Depletion in HIV-1 and HIV-2 Infections. Front Immunol. 2017;8:580. Available from: https://www.ncbi.nlm.nih.gov/pubmed/28588579
34. Diao B, Wang C, Tan Y, Chen X, Liu Y, Ning L. et al. Reduction and Functional Exhaustion of T Cells in Patients With Coronavirus Disease 2019 (COVID-19). Front Immunol. 2020;11:827. Available from: https://www.ncbi.nlm.nih.gov/pubmed/32425950
35. Perdomo-Celis F, Taborda NA, Rugeles MT. CD8+ T-cell response to HIV infection in the era of antiretroviral therapy. Frontiers in Immunology, Frontiers Media S.A. 2019 p. 1896. Available from: /pmc/articles/PMC6697065/
36. Varchetta S, Mele D, Oliviero B, Mantovani S, Ludovisi S, Cerino A. et al. Unique immunological profile in patients with COVID-19. Cellular and Molecular Immunology. 2021;18(3):604-12 Available from: https://doi.org/10.1038/s41423-020-00557-9
37. Okoye IS, Houghton M, Tyrrell L, Barakat K, Elahi S. Coinhibitory receptor expression and immune checkpoint blockade: Maintaining a balance in CD8+ T cell responses to chronic viral infections and cancer. Frontiers in Immunology, Frontiers Media S.A. 2017 Available from: https://pubmed.ncbi.nlm.nih.gov/29033936/
38. Aghbash PS, Eslami N, Shamekh A, Entezari-Maleki T, Baghi HB. SARS-CoV-2 infection: The role of PD-1/PD-L1 and CTLA-4 axis. Life Sciences, Elsevier Inc. 2021 p. 119124. Available from: /pmc/articles/PMC7838580/
39. Colomer-Lluch M, Ruiz A, Moris A, Prado JG. Restriction Factors: From Intrinsic Viral Restriction to Shaping Cellular Immunity Against HIV-1. Front Immunol. 2018;9:2876. Available from: https://www.ncbi.nlm.nih.gov/pubmed/30574147
40. Shukla E, Chauhan R. Host-HIV-1 Interactome: A Quest for Novel Therapeutic Intervention. Cells, NLM (Medline). 2019 Available from: /pmc/articles/PMC6830350/
41. Naamati A, Williamson JC, Greenwood EJD, Marelli S, Lehner PJ, Matheson NJ. Functional proteomic atlas of HIV infection in primary human CD4+ T cells. Elife. 2019 Mar 1;8
42. Martin-Sancho L, Lewinski MK, Pache L, Stoneham CA, Yin X, Becker ME. et al. Functional landscape of SARS-CoV-2 cellular restriction. Molecular Cell. 2021;81(12):2656-2668.e8
43. Bouhaddou M, Memon D, Meyer B, White KM, Rezelj V v, Correa Marrero M. et al. The Global Phosphorylation Landscape of SARS-CoV-2 Infection. Cell. 2020;182(3):685-712.e19 Available from: https://pubmed.ncbi.nlm.nih.gov/32645325/
44. Barnes BJ, Adrover JM, Baxter-Stoltzfus A, Borczuk A, Cools-Lartigue J, Crawford JM. et al. Targeting potential drivers of COVID-19: Neutrophil extracellular traps. Journal of Experimental Medicine, Rockefeller University Press. 2020 Available from: https://pubmed.ncbi.nlm.nih.gov/32302401/
45. Lourda M, Dzidic M, Hertwig L, Bergsten H, Palma LM, Kvedaraite E. et al. Title: High-dimensional profiling reveals phenotypic heterogeneity and disease-specific. medRxiv. 2021. 2021 01.27.21250591. Available from: https://doi.org/10.1101/2021.01.27.21250591
46. Jiang AP, Jiang JF, Guo MG, Jin YM, Li YY, Wang JH. Human Blood-Circulating Basophils Capture HIV-1 and Mediate Viral trans-Infection of CD4+ T Cells. J Virol. 2015;89(15):8050-62 Available from: https://www.ncbi.nlm.nih.gov/pubmed/26018157
47. Merad M, Martin JC. Pathological inflammation in patients with COVID-19: a key role for monocytes and macrophages. Nat Rev Immunol. 2020;20(6):355-62 Available from: https://www.ncbi.nlm.nih.gov/pubmed/32376901
48. Pacha O, Sallman MA, Evans SE. COVID-19: a case for inhibiting IL-17? Nat Rev Immunol. 2020;20(6):345-6 Available from: https://www.ncbi.nlm.nih.gov/pubmed/32358580
49. Lin J, Zhu Z, Xiao H, Wakefield MR, Ding VA, Bai Q. et al. The role of IL-7 in Immunity and Cancer. Anticancer Res. 2017;37(3):963-7 Available from: https://www.ncbi.nlm.nih.gov/pubmed/28314253
50. Sheikh A, Abraham N. Interleukin-7 Receptor Alpha in Innate Lymphoid Cells: More Than a Marker. Frontiers in Immunology, Frontiers Media S.A. 2019 p. 2897. Available from: www.frontiersin.org
51. Toor SM, Saleh R, Sasidharan Nair V, Taha RZ, Elkord E. T-cell responses and therapies against SARS-CoV-2 infection. Immunology, Blackwell Publishing Ltd. 2021 p. 30-43. Available from: https://pubmed.ncbi.nlm.nih.gov/32935333/
52. He R, Geha RS. Thymic stromal lymphopoietin. Ann N Y Acad Sci. 2010;1183:13-24 Available from: https://www.ncbi.nlm.nih.gov/pubmed/20146705
53. Varricchi G, Pecoraro A, Marone G, Criscuolo G, Spadaro G, Genovese A. et al. Thymic Stromal Lymphopoietin Isoforms, Inflammatory Disorders, and Cancer. Front Immunol. 2018;9:1595. Available from: https://www.ncbi.nlm.nih.gov/pubmed/30057581
54. Gu X. High Thymic Stromal Lymphopoietin (TSLP) Expression Promotes.. ISTH Academy. 2019; 273821. Available from: https://academy.isth.org/isth/2019/melbourne/273821/xiaoqiong.gu.high.thymic.stromal.lymphopoietin.%28tslp%29.expression.promotes.html
55. Turcot V, Bouchard L, Faucher G, Garneau V, Tchernof A, Deshaies Y. et al. Thymic stromal lymphopoietin: an immune cytokine gene associated with the metabolic syndrome and blood pressure in severe obesity. Clin Sci (Lond). 2012;123(2):99-109 Available from: https://www.ncbi.nlm.nih.gov/pubmed/22304237
56. Fontenot D, He H, Hanabuchi S, Nehete PN, Zhang M, Chang M. et al. TSLP production by epithelial cells exposed to immunodeficiency virus triggers DC-mediated mucosal infection of CD4+ T cells. Proc Natl Acad Sci U S A. 2009;106(39):16776-81 Available from: https://www.ncbi.nlm.nih.gov/pubmed/19805372
57. Klok FA, Kruip M, van der Meer NJM, Arbous MS, Gommers D, Kant KM. et al. Incidence of thrombotic complications in critically ill ICU patients with COVID-19. Thromb Res. 2020;191:145-7 Available from: https://www.ncbi.nlm.nih.gov/pubmed/32291094
58. Zhang L, Yan X, Fan Q, Liu H, Liu X, Liu Z. et al. D-dimer levels on admission to predict in-hospital mortality in patients with Covid-19. J Thromb Haemost. 2020;18(6):1324-9 Available from: https://www.ncbi.nlm.nih.gov/pubmed/32306492
59. Dong J, Lin J, Wang B, He S, Wu C, Kushwaha KK. et al. Inflammatory cytokine TSLP stimulates platelet secretion and potentiates platelet aggregation via a TSLPR-dependent PI3K/Akt signaling pathway. Cell Physiol Biochem. 2015;35(1):160-74 Available from: https://www.ncbi.nlm.nih.gov/pubmed/25591759
60. Ortega MA, Fraile-Martínez O, García-Montero C, García-Gallego S, Sánchez-Trujillo L, Torres-Carranza D. et al. An integrative look at SARS-CoV-2 (Review). International Journal of Molecular Medicine. 2021;47(2):415-34 Available from: http://www.spandidos-publications.com/10.3892/ijmm.2020.4828/abstract
61. Perico L, Benigni A, Casiraghi F, Ng LFP, Renia L, Remuzzi G. Immunity, endothelial injury and complement-induced coagulopathy in COVID-19. Nature Reviews Nephrology, Nature Research. 2021 p. 46-64. Available from: www.nature.com/nrneph
62. Wang X, Xu W, Hu G, Xia S, Sun Z, Liu Z. et al. SARS-CoV-2 infects T lymphocytes through its spike protein-mediated membrane fusion. Cell Mol Immunol. 2020 Available from: https://www.ncbi.nlm.nih.gov/pubmed/32265513
63. Schroder K, Tschopp J. The inflammasomes. Cell. 2010;140(6):821-32 Available from: https://www.ncbi.nlm.nih.gov/pubmed/20303873
64. Hachim MY, Khalil BA, Elemam NM, Maghazachi AA. Pyroptosis: The missing puzzle among innate and adaptive immunity crosstalk. J Leukoc Biol. 2020 Available from: https://www.ncbi.nlm.nih.gov/pubmed/32083338
65. Chen IY, Moriyama M, Chang MF, Ichinohe T. Severe Acute Respiratory Syndrome Coronavirus Viroporin 3a Activates the NLRP3 Inflammasome. Front Microbiol. 2019;10:50. Available from: https://www.ncbi.nlm.nih.gov/pubmed/30761102
66. Rodrigues TS, de Sá KSG, Ishimoto AY, Becerra A, Oliveira S, Almeida L. et al. Inflammasomes are activated in response to SARS-cov-2 infection and are associated with COVID-19 severity in patients. Journal of Experimental Medicine. 2020 218(3). Available from: https://pubmed.ncbi.nlm.nih.gov/33231615/
67. Ratajczak MZ, Kucia M. SARS-CoV-2 infection and overactivation of Nlrp3 inflammasome as a trigger of cytokine “storm” and risk factor for damage of hematopoietic stem cells. Leukemia. 2020;34(7):1726-9 Available from: https://doi.org/10.1038/s41375-020-0887-9
68. Choudhury SM, Ma X, Abdullah SW, Zheng H. Activation and inhibition of the nlrp3 inflammasome by rna viruses. Journal of Inflammation Research. 2021;14:1145-63 Available from: http://doi.org/10.2147/JIR.S295706
69. Campbell GR, To RK, Hanna J, Spector SA. SARS-CoV-2, SARS-CoV-1, and HIV-1 derived ssRNA sequences activate the NLRP3 inflammasome in human macrophages through a non-classical pathway. iScience. 2021;24(4):102295
70. Zhang C, Song JW, Huang HH, Fan X, Huang L, Deng JN. et al. NLRP3 inflammasome induces CD4+ T cell loss in chronically HIV-1-infected patients. Journal of Clinical Investigation. 2021 131(6). Available from: https://doi.org/10.1172/JCI138861
71. Yap JKY, Moriyama M, Iwasaki A. Inflammasomes and Pyroptosis as Therapeutic Targets for COVID-19. The Journal of Immunology. 2020;205(2):307-12 Available from: https://pubmed.ncbi.nlm.nih.gov/32493814/
72. Lee SH, Miyagi T, Biron CA. Keeping NK cells in highly regulated antiviral warfare. Trends Immunol. 2007;28(6):252-9 Available from: https://www.ncbi.nlm.nih.gov/pubmed/17466596
73. Brandstadter JD, Yang Y. Natural killer cell responses to viral infection. J Innate Immun. 2011;3(3):274-9 Available from: https://www.ncbi.nlm.nih.gov/pubmed/21411975
74. McMahon CW, Raulet DH. Expression and function of NK cell receptors in CD8+ T cells. Current Opinion in Immunology, Elsevier Ltd. 2001 p. 465-70. Available from: https://pubmed.ncbi.nlm.nih.gov/11498303/
75. Rapaport AS, Schriewer J, Gilfillan S, Hembrador E, Crump R, Plougastel BF. et al. The Inhibitory Receptor NKG2A Sustains Virus-Specific CD8(+) T Cells in Response to a Lethal Poxvirus Infection. Immunity. 2015;43(6):1112-24 Available from: https://www.ncbi.nlm.nih.gov/pubmed/26680205
76. Merino A, Zhang B, Dougherty P, Luo X, Wang J, Blazar BR. et al. Chronic stimulation drives human NK cell dysfunction and epigenetic reprograming. J Clin Invest. 2019;129(9):3770-85 Available from: https://www.ncbi.nlm.nih.gov/pubmed/31211698
77. Florez-Alvarez L, Hernandez JC, Zapata W. NK Cells in HIV-1 Infection: From Basic Science to Vaccine Strategies. Front Immunol. 2018;9:2290. Available from: https://www.ncbi.nlm.nih.gov/pubmed/30386329
78. Mikulak J, Oriolo F, Zaghi E, di Vito C, Mavilio D. Natural killer cells in HIV-1 infection and therapy. AIDS. 2017;31(17):2317-30 Available from: https://www.ncbi.nlm.nih.gov/pubmed/28926399
79. Lima JF, Oliveira LMS, Pereira NZ, Duarte AJS, Sato MN. Polyfunctional natural killer cells with a low activation profile in response to Toll-like receptor 3 activation in HIV-1-exposed seronegative subjects. Sci Rep. 2017;7(1):524. Available from: https://www.ncbi.nlm.nih.gov/pubmed/28373665
80. Bortolotti D, Gentili V, Rizzo S, Rotola A, Rizzo R. SARS-CoV-2 Spike 1 Protein Controls Natural Killer Cell Activation via the HLA-E/NKG2A Pathway. Cells. 2020 9(9). Available from: https://pubmed.ncbi.nlm.nih.gov/32859121/
81. Haanen JB, Cerundolo V. NKG2A, a New Kid on the Immune Checkpoint Block. Cell. 2018;175(7):1720-2 Available from: https://www.ncbi.nlm.nih.gov/pubmed/30550781
82. Grifoni A, Weiskopf D, Ramirez SI, Mateus J, Dan JM, Moderbacher CR. et al. Targets of T Cell Responses to SARS-CoV-2 Coronavirus in Humans with COVID-19 Disease and Unexposed Individuals. Cell. 2020;181(7):1489-1501 e15. Available from: https://www.ncbi.nlm.nih.gov/pubmed/32473127
83. Blanco-Melo D, Nilsson-Payant BE, Liu WC, Uhl S, Hoagland D, Møller R. et al. Imbalanced Host Response to SARS-CoV-2 Drives Development of COVID-19. Cell. 2020;181(5):1036-1045.e9
84. Manches O, Frleta D, Bhardwaj N. Dendritic cells in progression and pathology of HIV infection. Trends Immunol. 2014;35(3):114-22 Available from: https://www.ncbi.nlm.nih.gov/pubmed/24246474
85. Lau YL, Peiris JS, Law HK. Role of dendritic cells in SARS coronavirus infection. Hong Kong Med J. 2012;18(Suppl 3):28-30 Available from: https://www.ncbi.nlm.nih.gov/pubmed/22865220
86. Law HK, Cheung CY, Sia SF, Chan YO, Peiris JS, Lau YL. Toll-like receptors, chemokine receptors and death receptor ligands responses in SARS coronavirus infected human monocyte derived dendritic cells. BMC Immunol. 2009;10:35. Available from: https://www.ncbi.nlm.nih.gov/pubmed/19505311
87. Campana P, Parisi V, Leosco D, Bencivenga D, della Ragione F, Borriello A. Dendritic Cells and SARS-CoV-2 Infection: Still an Unclarified Connection. Cells, NLM (Medline). 2020 Available from: /pmc/articles/PMC7564940/
88. Giannessi F, Aiello A, Franchi F, Percario ZA, Affabris E. The role of extracellular vesicles as allies of HIV, HCV and SARS viruses. Viruses, MDPI AG. 2020 Available from: /pmc/articles/PMC7291340/
89. Piguet V, Sattentau Q. Dangerous liaisons at the virological synapse. J Clin Invest. 2004;114(5):605-10 Available from: https://www.ncbi.nlm.nih.gov/pubmed/15343375
90. Roberts KL, Manicassamy B, Lamb RA. Influenza A virus uses intercellular connections to spread to neighboring cells. J Virol. 2015;89(3):1537-49 Available from: https://www.ncbi.nlm.nih.gov/pubmed/25428869
91. Yang ZY, Huang Y, Ganesh L, Leung K, Kong WP, Schwartz O. et al. pH-dependent entry of severe acute respiratory syndrome coronavirus is mediated by the spike glycoprotein and enhanced by dendritic cell transfer through DC-SIGN. J Virol. 2004;78(11):5642-50 Available from: https://www.ncbi.nlm.nih.gov/pubmed/15140961
92. Garcia E, Piguet V. Virological Synapse for Cell-Cell Spread of Viruses. In: Cell-Cell Channels. Springer New York. 2006 p. 288-97. Available from: /pmc/articles/PMC7121336/
93. Marzi A, Gramberg T, Simmons G, Moller P, Rennekamp AJ, Krumbiegel M. et al. DC-SIGN and DC-SIGNR interact with the glycoprotein of Marburg virus and the S protein of severe acute respiratory syndrome coronavirus. J Virol. 2004;78(21):12090-5 Available from: https://www.ncbi.nlm.nih.gov/pubmed/15479853
94. Thépaut M, Luczkowiak J, Vivès C, Labiod N, Bally I, Lasala F. et al. DC/L-SIGN recognition of spike glycoprotein promotes SARS-CoV-2 trans-infection and can be inhibited by a glycomimetic antagonist. PLOS Pathogens. 2021;17(5):e1009576. Available from: https://dx.plos.org/10.1371/journal.ppat.1009576
95. Rogers KJ, Brunton B, Mallinger L, Bohan D, Sevcik KM, Chen J. et al. IL-4/IL-13 polarization of macrophages enhances Ebola virus glycoprotein-dependent infection. PLoS Negl Trop Dis. 2019;13(12):e0007819. Available from: https://pubmed.ncbi.nlm.nih.gov/31825972/
96. Prokunina-Olsson L, Alphonse N, Dickenson RE, Durbin JE, Glenn JS, Hartmann R. et al. COVID-19 and emerging viral infections: The case for interferon lambda. J Exp Med. 2020 217(5). Available from: https://www.ncbi.nlm.nih.gov/pubmed/32289152
97. Bindoli S, Felicetti M, Sfriso P, Doria A. The amount of cytokine-release defines different shades of Sars-Cov2 infection. Exp Biol Med (Maywood). 2020 1535370220928964. Available from: https://www.ncbi.nlm.nih.gov/pubmed/32460624
98. Misra DP, Agarwal V, Gasparyan AY, Zimba O. Rheumatologists' perspective on coronavirus disease 19 (COVID-19) and potential therapeutic targets. Clin Rheumatol. 2020;39(7):2055-62 Available from: https://www.ncbi.nlm.nih.gov/pubmed/32277367
99. Kim YM, Shin EC. Type I and III interferon responses in SARS-CoV-2 infection. Experimental and Molecular Medicine, Springer Nature. 2021 p. 750-60. Available from: https://doi.org/10.1038/s12276-021-00592-0
100. Channappanavar R, Fehr AR, Zheng J, Wohlford-Lenane C, Abrahante JE, Mack M. et al. IFN-I response timing relative to virus replication determines MERS coronavirus infection outcomes. Journal of Clinical Investigation. 2019;129(9):3625-39 Available from: https://pubmed.ncbi.nlm.nih.gov/31355779/
101. Liu L, Wei Q, Lin Q, Fang J, Wang H, Kwok H. et al. Anti-spike IgG causes severe acute lung injury by skewing macrophage responses during acute SARS-CoV infection. JCI Insight. 2019 4(4). Available from: https://www.ncbi.nlm.nih.gov/pubmed/30830861
102. Wang SF, Tseng SP, Yen CH, Yang JY, Tsao CH, Shen CW. et al. Antibody-dependent SARS coronavirus infection is mediated by antibodies against spike proteins. Biochemical and Biophysical Research Communications. 2014;451(2):208-14 Available from: https://pubmed.ncbi.nlm.nih.gov/25073113/
103. Yip MS, Leung HL, Li PH, Cheung CY, Dutry I, Li D. et al. Antibody-dependent enhancement of SARS coronavirus infection and its role in the pathogenesis of SARS. Hong Kong Med J. 2016;22(3 Suppl 4):25-31 Available from: https://www.ncbi.nlm.nih.gov/pubmed/27390007
104. Bournazos S, Gupta A, Ravetch J v. The role of IgG Fc receptors in antibody-dependent enhancement. Nature Reviews Immunology, Nature Research. 2020 p. 633-43. Available from: https://doi.org/10.1038/
105. Yasui F, Kai C, Kitabatake M, Inoue S, Yoneda M, Yokochi S. et al. Prior Immunization with Severe Acute Respiratory Syndrome (SARS)-Associated Coronavirus (SARS-CoV) Nucleocapsid Protein Causes Severe Pneumonia in Mice Infected with SARS-CoV. The Journal of Immunology. 2008;181(9):6337-48 Available from: https://pubmed.ncbi.nlm.nih.gov/18941225/
106. Kruize Z, Kootstra NA. The Role of Macrophages in HIV-1 Persistence and Pathogenesis. Front Microbiol. 2019;10:2828. Available from: https://www.ncbi.nlm.nih.gov/pubmed/31866988
107. Lopez P, Koh WH, Hnatiuk R, Murooka TT. HIV Infection Stabilizes Macrophage-T Cell Interactions To Promote Cell-Cell HIV Spread. J Virol. 2019 93(18). Available from: https://www.ncbi.nlm.nih.gov/pubmed/31270227
108. Ledford H. COVID reinfections are unusual — but could still help the virus to spread. Nature. 2021 Jan 14
109. Mukherjee A, Anand T, Agarwal A, Singh H, Chatterjee P, Narayan J. et al. SARS-CoV-2 re-infection: Development of an epidemiological definition from India. Epidemiology and Infection. 2021 149. Available from: https://pubmed.ncbi.nlm.nih.gov/33766185/
Author contact
![]() Corresponding author: Vicky Nicolaidou, Department of Life and Health Sciences, School of Sciences and Engineering, University of Nicosia, Cyprus. Telephone: +357 22 841676; Fax: +357 22 357481; Email: nicolaidou.vac.cy
Corresponding author: Vicky Nicolaidou, Department of Life and Health Sciences, School of Sciences and Engineering, University of Nicosia, Cyprus. Telephone: +357 22 841676; Fax: +357 22 357481; Email: nicolaidou.vac.cy