Impact Factor ISSN: 1449-1907
- Issue 9; 2026
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Volume 23; 2026
- Past Issues
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
GFRα Related Molecules and...
GFRα-mediated signaling...
GFRα-induced Oncogenesis
GFRα and neural invasion
GFRα and Treatment...
Conclusions
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2022; 19(4):659-668. doi:10.7150/ijms.64133 This issue Cite
Review
The Emerging Portrait of Glial Cell Line-derived Neurotrophic Factor Family Receptor Alpha (GFRα) in Cancers
Qingshang Li, Zhijun Cao ![]() , Shuliang Zhao
, Shuliang Zhao ![]()
Division of Gastroenterology and Hepatology, Ren Ji Hospital, School of Medicine, Shanghai Jiao Tong University; Shanghai Institute of Digestive Disease; State Key Laboratory for Oncogenes and Related Genes, Key Laboratory of Gastroenterology & Hepatology, Ministry of Health. 145 Middle Shandong Road, Shanghai, China.
Received 2021-6-21; Accepted 2022-3-6; Published 2022-3-28
Abstract

Glial cell line-derived neurotrophic factor family receptor alpha (GFRα) members have been widely connected to the mechanisms contributing to cell growth, differentiation, cell migration and tissue maturation. Here we review GFRα biological functions and discussed the evidence indicating whether GFRα signaling complex present novel opportunities for oncogenic intervention and treatment resistance. Thus, our work systematically reviewed the emerging role of GFRα family members in cancers, and provided novel insights for further researches.
Keywords: GFRα1, GDNF, cancer, neural invasion, treatment resistance
Introduction
The glial cell line-derived neurotrophic factors (GDNFs), a family of neurotrophic factors, were initially thought to be able to regulate the growth, survival, and differentiation of neural-derived cell types. However, it is becoming increasingly clear that these factors and their receptors are also widely found to express across many different cancers with further research.
The GDNF family ligands (GFLs) function through a glycosyl-phosphatidylinositol-(GPI) anchored coreceptor, GDNF family receptor alpha (GFRα), and rearranged during transfection (RET), a well-known receptor tyrosine kinase involved in kidney development, spermatogonial stem cell maintenance, and the development and maintenance of the sympathetic, parasympathetic, and enteric nervous systems [1, 2]. Based on whether it cooperates with the second receptor RET, GFRα has also been widely linked to the mechanisms that contribute to cell growth, differentiation and migration and tissue maturation. However, abnormal expression or aberrant activation of these molecules may convert normal growth signals to undesirable signals inducing overgrowth, becoming an important contributor to a variety of human cancers. Importantly, increasing numbers of novel reports suggest that the GFRα-mediated signaling pathway acts as an oncogenic promoter related to tumor proliferation, invasion, and metastasis as well as treatment resistance. Thus, the role of GFRα is more complicated than originally assumed, and it is necessary to revisit and review the role played by this versatile molecule in tumors.
GFRα Related Molecules and Signal Pathways
Interactions of GFRα with GFLs and RET
The GFRα family consists of four members, GFRα1, GFRα2, GFRα3 and GFRα4, located roughly extracellular and anchored to the plasma membrane by glycosyl-phosphatidyl-inositol (GPI). As the main component, extracellular structure contains some cysteine-rich repeats domains marked as D1-D2-D3 in GFRα1-3, and D2-D3 in GFRα4 (Figure 1a). Although these receptors are structurally similar, they determine specificity for four ligands—GDNF, Neurturin (NRTN), Artemin (ARTN) and Persephin (PSPN). However, the relationships among the GFLs and GFRα proteins are not strictly unique, and the ligands and receptors can cross-interact; the preferred GFRα coreceptor for GDNF is GFRα1, although GDNF also weakly binds to GFRα2 and GFRα3 [3]. In addition, NRTN and ARTN crosstalk with GFRα1 to activate RET. it is reported ARTN could also combine and activate both GFRα1 and GFRα3 [4]. PSPN not only binds GFRα4 but also signals in neurons mediated by GFRα1 [5]. When GFLs bind with GFRα, they form complexes and associate with the RET receptor, subsequently activating downstream signaling.
The GFRα and GFRα-mediated signaling pathways. a The GFRα family consists of four members, GFRα1, GFRα2, GFRα3 and GFRα4, which are tethered to the plasma membrane through GPI anchors containing CRDs. They have four characteristic ligands, namely, GDNF, NRTN, ARTN and PSPN. Two GFL monomers form an entangled homodimer to corresponding GFRα coreceptors. After GFLs and GFRα bind, the complexes associate with RET, a transmembrane tyrosine kinase coreceptor, forming a GFL-GFRα-RET ternary complex. b RET-dependent GFRα signaling is activated via phosphorylation of GFRα on multiple intracellular tyrosines. Two signal transduction pathways contribute to GFL-induced RET activation: via membrane-bound GFRα (cis-signaling) and soluble GFRα (trans-signaling) molecules released from nearby cells. Only the Ras/MAPK and PI3K/Akt signaling pathways are represented in the figure. c The presence of GDNF promotes the association of CFRα with NCAM, resulting in activation of the NCAM-mediated Fyn-FAK-MAPK signaling pathway. Other non-RET receptors of GFRα need further study. LICAM, ligand-induced cell adhesion molecules; NCAM, Neural cell adhesion molecule.
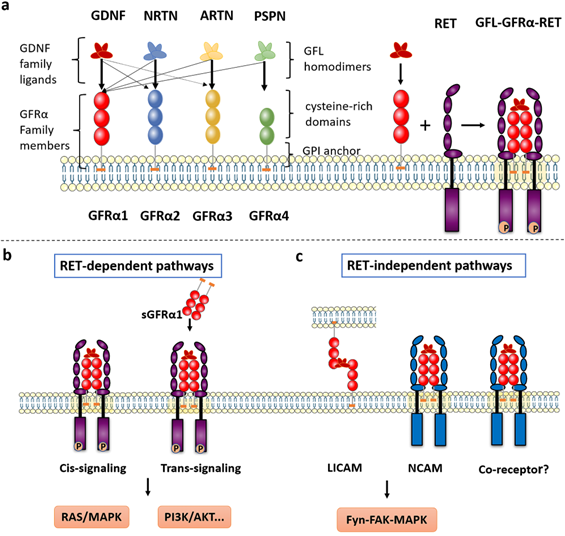
The crystal structure of GDNF was first reviewed 20 years ago [6], and other GFLs were subsequently identified [7, 8]. GFLs have a relatively conserved monomeric structure consisting of an α-helical heel region, a cystine knot core motif, and pairs of antiparallel β-strand fingers. These fingers are crucial to interact with GFRα and activate RET. Currently, the two GFL monomers are thought to be arranged structurally in a “handshake”-like head-to-tail orientation to form an entangled homodimer [9, 10]. On one side is a central region of GFRα comprising the D2 and D3 domains, which has been identified as a core region necessary for biochemical interaction with both GDNF and RET [11].
The RET receptor is a transmembrane tyrosine kinase with three regions: extracellular domain containing four cadherin like domains followed by cysteine rich domain, single pass transmembrane domain and tyrosine kinase domain [12]. RET isoforms, which differ by 51, 43, and 9 amino acids in the C-terminus, are referred to as RET51, RET43, and RET9, respectively. The two major isoforms, RET51 and RET9, are highly conserved over a broad range of species and exert different physiological functions [13, 14]. On the extracellular side of the GFRα-RET interaction, the GFL-GFRα complex associates with RET's large extracellular domain and promotes complex dimerization to form the GFL-GFRα-RET ternary complex. CLD1 and CLD2 pack together to form a clamshell-shaped structure and indirectly trap the GFL-GFRα complex, while CLD4 and CRD participate in the assembly of the signal complex [9, 15, 16]. The interaction of ARTN with GFRa3 occurs through the protruding tips of fingers 1 and 2 in ARTN inserting into a pocket in the center of a triangle of a helices in the D2 domain of GFRa3, which can be described as a small hydrophobic core surrounded by a much larger halo of charged and hydrophilic interactions [17].
The consensus is that the ternary complex conforms to a stoichiometric ratio of 2:2:2 (GFL2:GFRα2:RET2) (Figure 1a) and that RET interacts with GFL/GFRα via two hypothetical modes [18]. In the first mode, GFLs form homodimers and bind with two specific GFRα proteins, after which RET is recruited to a lipid raft membrane subdomain. After GFL-GFRα bind to each other, conformational changes in the CLD1-mediated dimerization cap facilitate RET dimerization and autophosphorylation [1, 19]. In the second mode, GFRα first recruits RET to establish a preformed receptor complex that is subsequently bound by the GFL homodimer [9].
GFRα-mediated signaling pathways
Upon interaction, RET-dependent GFRα signaling is activated via phosphorylation of RET on multiple intracellular serine and tyrosine residues, including Ser696, Tyr687, Tyr905, Tyr1015, Tyr1062, and Tyr1096 (in the RET51 isoform only), among others [1]. These residues facilitate direct interactions with signaling molecules; for example, Tyr905 binds with growth factor receptor-bound protein 7/10 (GRB7/10), Tyr1015 with phospholipase C γ (PLC-γ), and Tyr1096 with GRB2‑associated binding protein 2 (GAB2) [20]. Tyr1062 is the most well-characterized signaling hub for multiple adaptors containing a phosphotyrosine-binding domain (PTB) or SRC homology 2 (SH2) domain, such as fibroblast growth factor receptor substrate 2 (FRS2), downstream of kinase (DOK) family proteins (DOK1/4/5/6), and Enigma [21]. Next, several well-known downstream signaling pathways are induced, including the phosphatidylinositol-3-kinase (PI3K)/protein kinase B (AKT), RAS/mitogen-activated protein kinase (MAPK), PLC-γ, and c-Jun N-terminal kinase (JNK) pathways, which lead to the survival, proliferation, differentiation, and migration of cells and potentially to oncogenesis [22]. Notably, activation of RET occurs predominantly when its co-receptor GFRα bound to GFLs. Additionally, two signal transduction models contribute to GFL-induced RET activation: via membrane-bound GFRα (cis-signaling) and soluble GFRα (sGFRα, trans-signaling) molecules released from nearby cells [23, 24] (Figure 1b). The cis-signaling model is the classical pathway, where a cell expresses both RET and GFRα and both are stimulated in an autonomous fashion. In contrast, during activation via trans-signaling, soluble GFRα released from the membrane of neighboring cells presents GFLs to cells expressing only RET, and RET phosphorylation is then activated both inside and outside lipid rafts [22].
The differential expression of GFRα1 and RET in many tissues suggests that the presence of RET-independent pathways should pay more attention. A report indicated that GFRα1 was coimmunoprecipitated with SRC in the absence of RET suggests that GDNF signaling can pass through lipid rafts, but it is not clear how a direct interaction occurs owing to the opposite, seemingly mutually exclusive, positions of these proteins.
According to these findings, the Met tyrosine kinase receptor may be a candidate as a new transmembrane receptor to link Src with GDNF-GFRα1 [25].
Neural cell adhesion molecule (NCAM) is a homophilic binding glycoprotein playing critical roles in cell-cell adhesion, neurite outgrowth, and synaptic plasticity [26]. Interestingly, GFRα, as a coreceptor for GDNF, interferes with NCAM function by silencing NCAM homophilic interactions and NCAM-mediated cell adhesion [27] (Figure 1c). When GDNF is lacking, GFRα inhibits NCAM-NCAM interactions as a negative regulator (short-range). By contrast, the presence of GDNF promotes the association of CFRα and NCAM, resulting in activation of the NCAM-mediated Fyn-FAK-MAPK signaling pathway (long-range) [23]. Regarding cell adhesion molecules, GDNF can induce the association of membrane-bound GFRα from non-same cells (trans-homophilic interactions), allowing interaction between neuronal and glial cells. Therefore, a new role for GFRα proteins can be described, in which these proteins act as ligand-induced cell adhesion molecules (LICAMs) that influence extracellular crosstalk [28] (Figure 1c).
GFRα-induced Oncogenesis
Breast cancer
GFRα1 expression is upregulated in a significant proportion of human breast cancers [29-31]. Abundant expression of GFRα1 was confirmed in tissues of luminal A breast cancer, which comprise 70% of breast cancer cases, while minimal or no expression was observed in normal human breast tissue. Expression of GFRα1 or GFRα3, particularly bound with ARTN, has been consistently associated with poor survival outcomes, so these proteins can serve as prognostic markers in specific subtypes of mammary carcinoma [32]. In recent, a positive feedback loop was demonstrated between a GFRα1 and a certain gene. On the one hand, GFRα1 was identified as a target protein of ST3 beta-galactoside alpha-2,3-sialyltransferase 1 (ST3GAL1), which regulates the GDNF/GFRα1/RET pathway in breast cancer cells by mediating O-linked sialylation of GFRα1 and facilitating its interaction with RET. On the other hand, GFRα1-mediated signaling was found to stimulate the transcription of ST3GAL1 through the AKT/Sp1 pathway [33]. In addition, inhibition of the RET receptor decreases the growth and metastatic potential of ER+ breast cancer cells. In other words, GDNF-mediated GFR/RET activation promotes breast cancer proliferation and migration. Mechanistically, this activation might rescue cells from the antiproliferative effects of endocrine therapy and stimulate the expression of cytokines, especially the inflammatory cytokine IL6 [34]. Although RET is extensively involved in the development of breast cancer, GFRα is indispensable and irreplaceable in driving endocrine resistance, thus contributing to cell survival [35]. Moreover, anti-GFRα1 antibodies display robust therapeutic activity in clinically relevant cell line-derived xenograft models [36]. Therefore, high expression of GFRα1 is associated with poor prognosis in patients with high-grade breast cancers [37, 38].
Osteosarcoma
The GFRα1-dependent pathway has often been related to treatment resistance in tumors. After treating osteosarcoma cells with cisplatin, a widely used anticancer drug, it induces the overexpression of GFRα1, which promotes autophagy to lead to enhanced osteosarcoma cell survival via the SRC-AMPK signaling axis. Moreover, GFRα1 is involved in chemoresistance in osteosarcoma independent of RET and its major ligand GDNF, as confirmed by Mihwa Kim [39, 40]. These investigations suggest that GFRα1 could be a therapeutic target for the prevention of chemoresistance in osteosarcoma.
Pancreatic cancer
Recently, the importance of the PC-promoting role of GFLs and GFRα has become more prominent and better understood. The expression of GFRα and GFLs is barely detectable in normal pancreatic tissues, but both are upregulated overall in PC [41, 42]. Increased NRTN/GFRα-2 levels in PC promote an aggressive pancreatic cancer cell (PCC) phenotype, enhancing PC invasiveness. In addition, GFRα-2 but not NRTN is associated with the sensation of severe abdominal pain in PC patients [42]. The mechanism may be related to the transmission of neural signals.
GFRα1/RET receptor complex promotes the proliferation and invasion of PCC by binding to GDNF in an autocrine/paracrine manner [43]. The results of adhesion and invasion assays revealed that the enhanced expression and associated increase in the adhesive and invasive abilities of PCCs were inhibited by GFRα1 blockade [44]. Apurinic/apyrimidinic endonuclease 1 (Ape1/Ref-1)-induced GFRα1 protein expression via nuclear factor kappa B (NF-κB) contributes to GDNF-induced Matrix metalloproteinase-9 (MMP-9) expression, which strongly correlates with the desmoplastic reaction and lymphoid invasion; this mechanism might partially underlie the invasive behavior of PCCs [45]. In the tumor microenvironment, GFRα1 was demonstrated to be released by nerves, enhancing perineural invasion (PNI) and serving as a guidance signal for cancer cell migration. Notably, GDNF expression, RET phosphorylation, and MAPK pathway activity were found to be increased in a dose-dependent manner after exposure to soluble GFRα1 [46].
Both ARTN and its receptor complex GFRα3/RET were found to be overexpressed in PC, not only in primary cancer cells but also the surrounding tissues [47]. These mediators can promote the motility and invasiveness of MIA PaCa-2 cells. When ARTN treatment was administered, MMP-2 expression increases, and E-cadherin expression decreases [48]. Most notably, ARTN/GFRα3 increases the migration and invasion of PCCs in a manner like GDNF/GFRα1 [42, 47].
Prostate cancer
GDNF and GFR α 1 are secreted by the increased nerves in the peritumoral stroma of prostate cancer to create a perineural niche where RET signaling can occur. These factors are secreted via paracrine signaling, and some prostate cell lines can also express and specifically secrete GFRα1, perhaps via an autocrine mechanism [49]. In prostate cancer, GFRa1 plays a limiting role that supports GDNF/RET signaling to activate both the PI3K/AKT and MAPK/ERK pathways through phosphorylation of RET on Tyr1062, enhancing proliferation in vitro and tumor growth in vivo [1, 49]. Furthermore, GDNF stimulation increased the proliferation rate of prostate cancer cells and activated the signal pathway through GFRα1/SRC pathway, which was related to the expression level of GFRα1, but not related to RET. In addition, GFRα1/SRC activation can promote homing of resistant prostate cancer cells to a microenvironment with augmented growth-promoting and resistance-inducing properties [50]. Despite a report indicating coimmunoprecipitation of GFRα1 and SRC, whether they interact directly needs further verification due to their positions on opposite sides of the lipid bilayer.
Neuroblastoma
GFRα2 is upregulated in neuroblastoma cells and tissues, and its overexpression promotes neuroblastoma cell proliferation. As revealed by a recent study using colony formation assays and western blot analysis, GFRα2 interacts with phosphatase and tensin homolog (PTEN), a tumor suppressor that inhibits the well-known PI3K/AKT pathway. Consequently, GFRα2 promotes neuroblastoma cell proliferation by activating the PI3K/AKT pathway [51]. GFRα1 is a direct target of Ape1/Ref-1 in Neuro2a mouse neuroblastoma cells. Ape1/Ref-1 expression causes the clustering of GFRα1 in lipid rafts in response to GDNF, contributing to phosphorylation of AKT and PLCγ-1 and stimulating cell proliferation [52]. Another report [53] showed that the inhibitor of PLC-γ blocks the pro-survival effect of GDNF on the spinal motoneurons in vitro, but it's an indirect data. There are several studies indicating that GDNF may activate PLC-γ signaling pathway, but additional work is needed to answer this question.
Colorectal cancer
GDNF and NRTN were highly expressed in colorectal cells, whereas the coreceptor GFRα1 and RET were expressed in the surrounding ganglia and glial cells [54]. Increased expression of GDNF enhances β1 integrin expression via signaling through RET/GFRα1 in colorectal cancer cell lines, thus strongly influencing adhesion to and invasion of the extracellular matrix (ECM). Subsequently, these cancer cells exhibit increased invasive ability and malignancy [55]. According to a recent report, demethylation of GFRα1 is a frequent event during colorectal cancer development, and high dmGFRα1 levels can result in GFRA1 overexpression and significantly increase cancer malignancy [56]; similar results were also observed in gastric cancer [57]. Further research showed that GFRα1 enhances proliferation probably by activating the AKT and ERK pathways; thus, GFRα1 might be a marker for poor prognosis in colorectal cancer [56].
Gastric cancer
In addition, genome-wide DNA methylation analysis showed that methylation changes in GFRα1 are positively correlated with gastric carcinoma metastasis [57]. Similarly, the GFRα3 promoter region was shown to be markedly hypermethylated in almost all gastric tumors [58]. However, whether these changes can strongly influence the relevant phenotypes is less clear.
Lung cancer
ARTN, RET, and GFRα3 have been demonstrated to be upregulated in non-small cell lung carcinoma (NSCLC) cells compared with their normal counterparts, while high ARTN expression also enhances the migration and invasion of NSCLC cells. The oncogenic effect of ARTN is correlated with BCL2 expression, and these two phenomena may be causally related. Notably, both GFRα3 and GFRα1 are expressed in H1299 cells, whereas GFRA3 is expressed only in H1975 cells [59].
Other cancers
In acute myeloid leukemia cells, RET signaling was observed to be activated via ARTN/GFRα3 and NRTN/GFRα2 ligand/coreceptor complexes, and mTORC1-mediated suppression of autophagy was identified as a downstream pathway [60]. The differential activity of GFRα pathways in different cancers are shown in Table 1. More details on study methods or antibody specificity of above reviewed literatures are listed in Supplementary Table 1.
Differential activity of GFRα pathways in different cancers
| Cancer | Ligands | Receptors | Primary pathways | Main effects | Refs |
|---|---|---|---|---|---|
| Breast cancer | GDNF | GFRα1/RET | PI3K/AKT, ST3GAL1 | Proliferation | [33] |
| FAK/STAT | Migration | [34] | |||
| ERK | Endocrine therapy resistance | [35] | |||
| ARTN | GFRα1, GFRα3 | - | Worse survival outcome | [32] | |
| Osteosarcoma | - | GFRα1 | SRC-AMPK, NFκB | Autophagy and chemoresistance | [39, 40] |
| Pancreatic cancer | NRTN | GFRα2 | - | Severe cancer pain and neuroplasticity | [42] |
| GDNF | GFRα1 | Ape1/Ref-1, MMP-9 | Invasion | [45] | |
| soluble GFRα1/RET | MAPK/ERK | Perineural invasion | [46] | ||
| ARTN | GFRα3/RET | MMP-2, ECM | Invasiveness | [48] | |
| Prostate cancer | GDNF | GFRα1/RET | PI3K/AKT, MAPK/ERK | Proliferation and invasion | [49] |
| GDNF | GFRα1 | SRC/ERK | Proliferation and treatment resistance | [50] | |
| Neuroblastoma | - | GFRα2 | PI3K/AKT, PTEN | Proliferation | [51] |
| GDNF | GFRα1 | PLCγ-1, AKT, Ape1/Ref-1 | [52] | ||
| Colorectal cancer | GDNF | GFRα1/RET | RAS/PI3K/AKT RAS-RAF1-MEK1/2-ERK1/2 | Proliferation and survival | [56] |
| Lung cancer | ARTN | GFRα3/RET | Bcl-2 | Proliferation and invasion | [59] |
| Acute myeloid leukemia | NRTN | GFRα2 | mTORC1 | Autophagy suppression | [60] |
| ARTN | GFRα3 |
PI3K, phosphatidylinositol 3 kinase; AKT, protein kinase B; FAK, focal adhesion kinase; STAT signal transducer and activator of transcription; ERK, extracellular-signal-regulated kinase; SRC, AMPK, Adenosine 5'-monophosphate (AMP)-activated protein kinase; NFкB, nuclear factor-kappa beta; Ape1/Ref-1, apurinic/apyrimidinic endonuclease 1; MMP-9, matrix metalloproteinases-9; MAPK, mitogen activate protein kinase; ECM, extracellular matrix proteins; PLC-γ, phospholipase C γ; MEK, mitogen-activated protein kinase kinase.
GFRα and neural invasion
Tumor invasion and migration are major reasons for poor prognosis and are frequently the cause of cancer-related deaths. These unfavorable behaviors are closely associated with the interaction between tumor cells and the tumor microenvironment. The most remarkable role of GFL-GFRα signaling in cancers is modulating the relationship between the tumor and its surroundings. Indeed, the interaction between the two can form a reciprocal loop, leading to enhanced tumor cell malignancy.
Cancer spreads via three classical mechanisms: direct invasion of surrounding tissue, lymphatic spread and hematogenous spread. However, a fourth route of spreading, neural dissemination, should be highlighted. The presence of PNI is a key feature most strongly associated with poor prognosis and high recurrence in colorectal cancer, gastric cancer, oral squamous cell carcinoma (OSCC), and pancreatic cancer [61].
GFRα serves as a coreceptor with RET on the surface of cancer cells to activate downstream signaling, cancer cell migration, and PNI. During PNI, a soluble form of GFRα1 released by normal nerves facilitates neural tracking regardless of GFRα1 expression in cancer cells [62]. Migration of human pancreatic adenocarcinoma MiaPaCa-2 cells toward nerve-secreted GDNF, phosphorylation of RET, and MAPK pathway activity are increased dose-dependently upon exposure to soluble GFRα1. Even though GFRα1 expression varies widely in different cancer cells, both GFRα1 and its ligand GDNF can be released from the tumor microenvironment and cooperate to facilitate cancer invasion [46]. According to another report, the expression of RET and GRFα1 is higher in tumor tissues of patients with neuroinvasive pancreatic carcinoma than in normal tissues. In an in vitro Matrigel coculture model of dorsal root ganglion and PCCs, nerve-secreted GDNF induced polarized neurotrophic migration of cancer cells (PNMCs) along the nerve axons, whereas deficiency of this mediator reduced the ability to attract cancer cells. Potentially, the MAPK pathway might be stimulated by GDNF-GFRα1-RET signaling to mediate nerve invasion [63]. Accordingly, systemic therapy with pyrazolopyrimidine-1, a tyrosine kinase inhibitor targeting RET, suppresses and abolishes nerve invasion toward the spinal cord and prevents further damage [63, 64]. Via an alternative pathway, increased expression of NRTN and GFRα2 by cancer cells has been linked to nerve invasion and severe pain, indicating a poor prognosis for these patients with pancreatic ductal adenocarcinoma [42]. Moreover, in in vivo and in vitro experiments, overexpression of ARTN not only promoted the proliferation of PCCs but also enhanced their ability to invade peripheral organs, nerves and lymph nodes [65].
PNI is another prominent characteristic of head and neck cancers, occurring in as many as 5-90% of patients [66]. GDNF-increased cancer cell aggressive behavior was markedly reduced in oral cancer when pharmacological inhibitors or neutralizing antibodies inhibited MMP-9 (matrix metalloprotein 9) and MMP-13, an enzyme family destroying the histological barrier of tumor cell invasion. Further protein assays revealed that GDNF also increases ERK, p38 and JNK phosphorylation and AP-1 DNA binding activity to facilitate the interactive invasion and growth of cancer cells and nerves [67]. Similarly, in colorectal cancer cell lines, β1 integrin expression is enhanced by increased GDNF expression via signaling through RET/GFRα1, notably influencing adhesion to and invasion of the ECM. Consequently, these cancer cells exhibit increased invasive ability and malignancy [55].
GFRα and Treatment Resistance
Chemoresistance
Autophagy is a self-eating mechanism to maintain cellular homeostasis in cell survival in adverse environments, such as those established by irradiation, cytotoxicity and hypoxia [68]. GFRα1-induced cancer cell autophagy is a recently identified novel regulatory mechanism of osteosarcoma chemoresistance. In two osteosarcoma cell lines, MG-63 and U-2 OS, GFRα1 expression was upregulated at both the transcriptional and translational levels following treatment with cisplatin, as evaluated by measuring the expression and phosphorylation levels of NFκB. Overexpression of GFRα1 decreased cisplatin-induced apoptosis, accompanied by increased autophagy, and significantly promoted cell proliferation. Further molecular studies showed that GFRα1 overexpression is mediated through the SRC-AMPK signaling axis and enhances the expression of downstream molecules including beclin1, etc. The results of animal experiments confirmed that the mechanism by which cancer cells survive through chemical resistance may be GFR 1-mediated autophagy [39, 40] (Figure 2).
GFRα and treatment resistance. Chemoresistance: Cisplatin stimulates overexpression of GFRα1 via NFκB phosphorylation and decreases cisplatin-induced apoptosis, accompanied by increased autophagy, and significantly promotes cell proliferation through the SRC-AMPK signaling axis. Endocrine resistance: GFLs/GFRα/RET and ER signaling participate in intricate crosstalk via the PI3K/mTOR and RAS/MEK/ERK pathways in breast cancer. Endocrine therapy promotes the expression of GFLs, resulting in a vicious loop of RET signaling. Hypoxia resistance: Hypoxia directly activates ARTN transcription via HIF-1α, and the ARTN-dependent AKT pathway is then activated to trigger expansion of the CSC population. The solid arrows indicate the known and direct interactions between signaling molecules; the broken arrows indicate interactions requiring further investigation. The red arrows indicate the main GFRα signaling pathway.
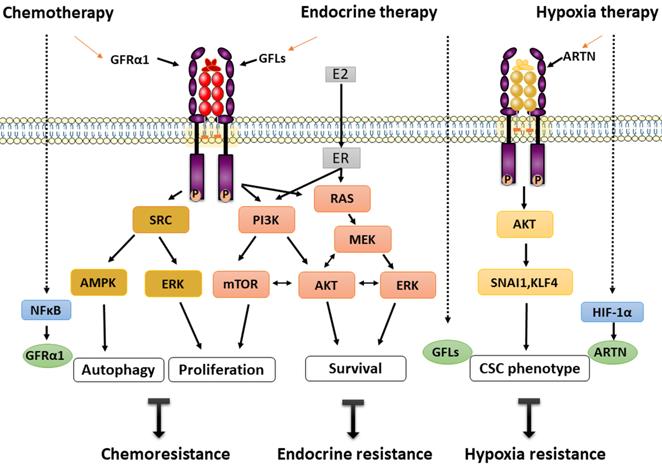
Similarly, transcription of GDNF in PSC27 prostate cancer cells was found to increase by several fold following exposure to cytotoxic agents. DNA damage caused by those drugs induced abundant GDNF secretion from cells in the tumor microenvironment, which then stimulated the growth of stromal cells and prostate cancer cells through an autocrine/paracrine loop via the SRC/ERK pathway. Additionally, tumor cells become resistant to mitoxantrone and docetaxel chemotherapy, which leads to acquired treatment resistance and can be induced by exposure to GDNF. Further gene analysis indicated that overexpression of RET and GFRα1 could be considered to act via a GDNF coreceptor to increase the mitotic rate. Moreover, only GFRα1 expression correlates with migration and invasion of prostate cancer, not RET and GFRα2-4 [50]. In summary, based on the balance of autophagy and selective proliferation, re-proliferation of drug-resistant tumor cells is suggested as a mechanism underlying rapid tumor recurrence and treatment failure.
Endocrine resistance
ER+ subtypes account for the majority of breast cancers and have exhibited good outcomes after endocrine therapy, which has been a first-line treatment for decades. Many patients exhibited a survival benefit of significantly longer survival times. However, GDNF-GFRα1-RET signaling is decisive in endocrine therapy resistance in ER+ breast cancers.
In an in vitro model of MCF7 cells, GDNF-mediated signaling was enhanced and promoted the survival of aromatase inhibitor-resistant cells. However, this increased resistance was selectively reversed by the RET kinase inhibitor NVP-BBT594. Moreover, gene analysis indicated that a GDNF response gene set predicts poor prognosis and has predictive value in breast cancer [38]. Further study showed that endogenous GDNF can be produced by endocrine-resistant cells and can be secreted into the medium and activate GFRα1/RET signaling in nearby cells [35]. Other RET ligands, ARTN and NRTN, but not PSPN, can also initiate and confer endocrine resistance. For instance, acquired tamoxifen resistance was induced by an estrogen-regulated gene, ARTN, which mediated increase of BCL-2 expression and promoted radioresistance and chemoresistance by enhancing cancer stem cell (CSC)-like behavior in breast cancer cells [69, 70]. Furthermore, ARTN depletion unexpectedly reversed trastuzumab sensitivity, resulting in trastuzumab resistance in HER2-positive cells [71].
Estrogen receptor signaling pathway plays a critical role in the occurrence and development of breast cancer. When GDNF was applied to MCF7 cells as a model of ER+/GFRa1+/RET+ breast cancer, RET signaling resulted in increased ER phosphorylation predominantly via the mTOR pathway and estrogen-independent transcriptional activation of ER-dependent genes [31]. RET downstream signaling leads to ER phosphorylation through mTOR independent of PI3/AKT and via a possible compensatory mechanism through the MEK-ERK pathways [31]. The interaction of GFLs/GFRα/RET and ER signaling establishes an intricate crosstalk network in breast cancer. Another interesting hypothesis is that estrogen-induced upregulation of ARTN and GDNF promotes tumorigenesis, which leads to activation of RET-related signaling, a vicious circle [31] (Figure 2).
Collectively, ER+ breast cancer cells may be “poised” for GFRα/RET-mediated endocrine resistance [35]. However, because the understanding of this unfavorable phenomenon is gradually increasing, novel corresponding targeted treatments are rapidly emerging.
Hypoxia resistance
Hypoxia, a major feature of solid tumors, commonly develops owing to dramatic cell proliferation and inadequate blood supply, which increases patient treatment resistance and favors tumor progression [72]. Recently, accumulating evidence has indicated that ARTN is closely associated with a higher clinical stage and poor prognosis of hepatocellular carcinoma (HCC) patients. ARTN was shown to enhance the tumorigenicity of HCC cells in vitro by reducing apoptosis and increasing epithelial-mesenchymal transition (EMT) and in vivo by promoting xenograft tumor growth and metastasis. Moreover, hypoxia directly activates ARTN transcription via hypoxia-inducible factor-1α (HIF-1α), and the ARTN-dependent AKT pathway is then activated to induce expansion of the CSC population. A novel HIF-1α/ARTN/AKT axis mediating hypoxia-induced EMT and CSC promotion in HCC cells is thus formed (Figure 2). Herein, ARTN is considered not only a hypoxia-responsive factor but also an indispensable factor for hypoxia-induced cell expansion in HCC [73]. Via this mechanism, ARTN facilitates cancer cell evasion of hypoxia-related therapies and is thus a valuable potential therapeutic target.
Conclusions
Herein, we reviewed GFRα biology and physiology and discussed the evidence indicating whether GFRα signaling complex present novel opportunities for oncogenic intervention. The GFRα family constitutes a group of four structurally related receptors that have historically been regarded to play developmental roles in the kidney and neuronal system. More recently, however, they have been credited with additional developmental functions during cancer progression. A literature review indicated that the GFRα family, consisting of GFRα1-4, is involved in breast cancer, colorectal cancer, prostate cancer, lung cancer, gastric cancer, and many other tumors, thus exhibiting a diverse oncogenic portfolio. Additionally, GFRα is prominently involved in mediating tumor peripheral infiltration and treatment resistance.
However, many questions about the role of GFRα1 signaling in tumor progression need to be studied and resolved. For example, 1) what are the regulatory factors and mechanisms underlying the differential expression of GFRα in tumors of different tissue types? 2) Are additional unrecognized coreceptors, interacting proteins or crosstalk pathways involved in GFRα signaling? 3) What is the clinical effect of GFRα1 as a therapeutic target in different tumors? Looking forward, a further understanding of the mechanisms involving GFRα family members may provide critical strategies toward the discovery of novel potential approaches for long-term tumor treatment.
Supplementary Material
Supplementary table.
Acknowledgements
This project was supported by the National Natural Science Foundation of China (Grant No. 81972655), the Program for Young Eastern Scholar at Shanghai Institutions of Higher Learning (Grant No. QD2016004), and the Shanghai Science and Technology Commission Research Project (Grant No. 14441903103) and Shanghai Municipal Key Clinic Specialty.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Mulligan LM. RET revisited: expanding the oncogenic portfolio. Nat Rev Cancer. 2014;14:173-86
2. Mulligan LM. GDNF and the RET Receptor in Cancer: New Insights and Therapeutic Potential. Frontiers in physiology. 2018;9:1873
3. Kawai K, Takahashi M. Intracellular RET signaling pathways activated by GDNF. Cell and tissue research. 2020;382:113-23
4. Baloh RH, Tansey MG, Lampe PA, Fahrner TJ, Enomoto H, Simburger KS. et al. Artemin, a novel member of the GDNF ligand family, supports peripheral and central neurons and signals through the GFRalpha3-RET receptor complex. Neuron. 1998;21:1291-302
5. Sidorova YA, Matlik K, Paveliev M, Lindahl M, Piranen E, Milbrandt J. et al. Persephin signaling through GFRalpha1: the potential for the treatment of Parkinson's disease. Molecular and cellular neurosciences. 2010;44:223-32
6. Eigenbrot C, Gerber N. X-ray structure of glial cell-derived neurotrophic factor at 1.9 A resolution and implications for receptor binding. Nature structural biology. 1997;4:435-8
7. Kotzbauer PT, Lampe PA, Heuckeroth RO, Golden JP, Creedon DJ, Johnson Jr EM. et al. Neurturin, a relative of glial-cell-line-derived neurotrophic factor. Nature. 1996;384:467-70
8. Milbrandt J, Sauvage FJD, Fahrner TJ, Baloh RH, Johnson EM. Persephin, a Novel Neurotrophic Factor Related to GDNF and Neurturin. Neuron. 1998;20:245-53
9. Xinquan W. Structural studies of GDNF family ligands with their receptors—Insights into ligand recognition and activation of receptor tyrosine kinase RET. Biochimica et biophysica acta. 2013;1834:2205-12
10. Ibáñez CF, Paratcha G, Ledda F. RET-independent signaling by GDNF ligands and GFRα receptors. Cell and tissue research. 2020;382:71-82
11. Scott RP, Ibanez CF. Determinants of Ligand Binding Specificity in the Glial Cell Line-derived Neurotrophic Factor Family Receptor alpha s? Journal of Biological Chemistry. 2001;276:1450-8
12. Arighi E, Borrello MG, Sariola H. RET tyrosine kinase signaling in development and cancer. Cytokine & growth factor reviews. 2005;16:441-67
13. Tahira T, Ishizaka Y, Itoh F, Sugimura T, Nagao M. Characterization of ret proto-oncogene mRNAs encoding two isoforms of the protein product in a human neuroblastoma cell line. Oncogene. 1990;5:97-102
14. Myers SM, Eng C, Ponder BA, Mulligan LM. Characterization of RET proto-oncogene 3' splicing variants and polyadenylation sites: a novel C-terminus for RET. Oncogene. 1995;11:2039-45
15. Sandmark J, Dahl G, Oster L, Xu B, Johansson P, Akerud T. et al. Structure and biophysical characterization of the human full-length neurturin-GFRa2 complex: A role for heparan sulfate in signaling. J Biol Chem. 2018;293:5492-508
16. Parkash V, Leppänen VM, Virtanen H, Jurvansuu JM, Bespalov MM, Sidorova YA. et al. The structure of the glial cell line-derived neurotrophic factor-coreceptor complex: insights into RET signaling and heparin binding. J Biol Chem. 2008;283:35164-72
17. Wang X, Baloh RH, Milbrandt J, Garcia KC. Structure of artemin complexed with its receptor GFRalpha3: convergent recognition of glial cell line-derived neurotrophic factors. Structure. 2006;14:1083-92
18. Airaksinen MS, Saarma M. The GDNF family: signalling, biological functions and therapeutic value. Nature reviews Neuroscience. 2002;3:383-94
19. Kjaer S, Hanrahan S, Totty N, McDonald NQ. Mammal-restricted elements predispose human RET to folding impairment by HSCR mutations. Nat Struct Mol Biol. 2010;17:726-31
20. Uchida M EA, Fukuda T, Kurokawa K, Maeda K, Kodama Y, Asai N, Hasegawa T, Shimono Y, Jijiwa M, Ichihara M, Murakumo Y, Takahashi M. Dok-4 regulates GDNF-dependent neurite outgrowth through downstream activation of Rap1 and mitogen-activated protein kinase. Journal of cell science. 2006;119:3067-77
21. Ai K, Murakumo Y, Jijiwa M, Kurokawa K, Itoh Y, Kodama Y. et al. Analysis of DOK-6 function in downstream signaling of RET in human neuroblastoma cells. Cancer Sci. 2010;101:1147-55
22. Fielder GC, Yang WS, Razdan M, Li Y, Liu DX. The GDNF Family: a Role in Cancer? Neoplasia (New York, NY). 2017;20:99-117
23. Paratcha G LF. GDNF and GFRα: a versatile molecular complex for developing neurons. Trends in neurosciences. 2008;310-391
24. Paratcha G, Ledda F, Baars L, Coulpier M, Besset V, Anders J. et al. Released GFRalpha1 potentiates downstream signaling, neuronal survival, and differentiation via a novel mechanism of recruitment of c-Ret to lipid rafts. Neuron. 2001;29:171-84
25. Popsueva A PD, Arighi E, Meng X, Angers-Loustau A, Kaplan D, Saarma M, Sariola H. GDNF promotes tubulogenesis of GFRalpha1-expressing MDCK cells by Src-mediated phosphorylation of Met receptor tyrosine kinase. J Cell Biol. 2003;161:119-29
26. Nielsen J GK, Li S, Kulahin N, Soroka V, Rasmussen KK, Bock E, Berezin V. Role of Glial Cell Line-Derived Neurotrophic Factor (GDNF)-Neural Cell Adhesion Molecule (NCAM) Interactions in Induction of Neurite Outgrowth and Identification of a Binding Site for NCAM in the Heel Region of GDNF. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2009;29:11360-76
27. Paratcha G LF, Ibáñez CF. The Neural Cell Adhesion Molecule NCAM Is an Alternative Signaling Receptor for GDNF Family Ligands. Cell. 2003;1130-879
28. Ledda F PG, Sandoval-Guzmán T, Ibáñez CF. GDNF and GFRα1 promote formation of neuronal synapses by ligand-induced cell adhesion. Nature neuroscience. 2007;10:293-300
29. Boulay A, Breuleux M, Stephan C, Fux C, Brisken C, Fiche M. et al. The Ret receptor tyrosine kinase pathway functionally interacts with the ERalpha pathway in breast cancer. Cancer Res. 2008;68:3743-51
30. Esseghir S, Todd SK, Hunt T, Poulsom R, Isacke CMJCR. A Role for Glial Cell Derived Neurotrophic Factor Induced Expression by Inflammatory Cytokines and RET/GFR 1 Receptor Up-regulation in Breast Cancer. 2008; 67: 11732-41.
31. Morandi A, Plaza-Menacho I, Isacke CM. RET in breast cancer: functional and therapeutic implications. Trends in molecular medicine. 2011;17:149-57
32. Wu ZS, Pandey V, Wu WY, Ye S, Zhu T, Lobie PE. Prognostic significance of the expression of GFRalpha1, GFRalpha3 and syndecan-3, proteins binding ARTEMIN, in mammary carcinoma. BMC Cancer. 2013;13:34
33. Fan TC, Yeo HL, Hsu HM, Yu JC, Ho MY, Lin WD. et al. Reciprocal feedback regulation of ST3GAL1 and GFRA1 signaling in breast cancer cells. Cancer Lett. 2018;434:184-95
34. Gattelli A, Nalvarte I, Boulay A, Roloff TC, Schreiber M, Carragher N. et al. Ret inhibition decreases growth and metastatic potential of estrogen receptor positive breast cancer cells. EMBO molecular medicine. 2013;5:1335-50
35. Horibata S, Rice EJ, Mukai C, Marks BA, Sams K, Zheng H. et al. ER-positive breast cancer cells are poised for RET-mediated endocrine resistance. PLoS One. 2018;13:e0194023
36. Bhakta S, Crocker LM, Chen Y, Hazen M, Schutten MM, Li D. et al. An Anti-GDNF Family Receptor Alpha 1 (GFRA1) Antibody-Drug Conjugate for the Treatment of Hormone Receptor-Positive Breast Cancer. Mol Cancer Ther. 2018;17:638-49
37. Spanheimer PM, Park JM, Askeland RW, Kulak MV, Woodfield GW, De Andrade JP. et al. Inhibition of RET increases the efficacy of antiestrogen and is a novel treatment strategy for luminal breast cancer. Clinical cancer research: an official journal of the American Association for Cancer Research. 2014;20:2115-25
38. Morandi A, Martin LA, Gao Q, Pancholi S, Mackay A, Robertson D. et al. GDNF-RET signaling in ER-positive breast cancers is a key determinant of response and resistance to aromatase inhibitors. Cancer Res. 2013;73:3783-95
39. Kim M, Jung JY, Choi S, Lee H, Morales LD, Koh JT. et al. GFRA1 promotes cisplatin-induced chemoresistance in osteosarcoma by inducing autophagy. Autophagy. 2017;13:149-68
40. Kim M, Kim DJ. GFRA1: A Novel Molecular Target for the Prevention of Osteosarcoma Chemoresistance. Int J Mol Sci. 2018 19
41. Zeng Q, Cheng Y, Zhu Q, Yu Z, Wu X, Huang K. et al. The relationship between overexpression of glial cell-derived neurotrophic factor and its RET receptor with progression and prognosis of human pancreatic cancer. The Journal of international medical research. 2008;36:656-64
42. Wang K, Demir IE, D'Haese JG, Tieftrunk E, Kujundzic K, Schorn S. et al. The neurotrophic factor neurturin contributes toward an aggressive cancer cell phenotype, neuropathic pain and neuronal plasticity in pancreatic cancer. Carcinogenesis. 2014;35:103-13
43. H L, Q M, J L. High glucose promotes cell proliferation and enhances GDNF and RET expression in pancreatic cancer cells. Mol Cell Biochem. 2011;347:95-101
44. Funahashi H, Okada Y, Sawai H, Takahashi H, Matsuo Y, Takeyama H. et al. The role of glial cell line-derived neurotrophic factor (GDNF) and integrins for invasion and metastasis in human pancreatic cancer cells. Journal of surgical oncology. 2005;91:77-83
45. Kim MH, Kim HB, Acharya S, Sohn HM, Jun JY, Chang IY. et al. Ape1/Ref-1 induces glial cell-derived neurotropic factor (GDNF) responsiveness by upregulating GDNF receptor alpha1 expression. Mol Cell Biol. 2009;29:2264-77
46. He S, Chen CH, Chernichenko N, He S, Bakst RL, Barajas F. et al. GFRalpha1 released by nerves enhances cancer cell perineural invasion through GDNF-RET signaling. Proc Natl Acad Sci U S A. 2014;111:E2008-17
47. GO C, NA G, M E, AG K, MN W, T G. et al. The neurotrophic factor artemin promotes pancreatic cancer invasion. Annals of surgery. 2006;244:274-81
48. LX M, YH C, XX W, ZJ D, LC F, H Z. et al. Neurotrophic artemin promotes motility and invasiveness of MIA PaCa-2 pancreatic cancer cells. Asian Pac J Cancer Prev. 2012;13:1793-7
49. Ban K, Feng S, Shao L, Ittmann M. RET Signaling in Prostate Cancer. Clinical cancer research: an official journal of the American Association for Cancer Research. 2017;23:4885-96
50. RM H, JM L, LA G-S, I C, S Z, R C. et al. DNA damage induces GDNF secretion in the tumor microenvironment with paracrine effects promoting prostate cancer treatment resistance. Oncotarget. 2015;6:2134-47
51. Li Z, Xie J, Fei Y, Gao P, Xie Q, Gao W. et al. GDNF family receptor alpha 2 promotes neuroblastoma cell proliferation by interacting with PTEN. Biochemical and Biophysical Research Communications. 2019;510:339-44
52. Kang MY, Kim KY, Yoon Y, Kang Y, Kim HB, Youn CK. et al. Ape1/Ref-1 Stimulates GDNF/GFRalpha1-mediated Downstream Signaling and Neuroblastoma Proliferation. Korean J Physiol Pharmacol. 2009;13:349-56
53. Fu R, Wang LQ, Chu GL, Zhou LH. Involvement of phospholipase C-γ in the pro-survival role of glial cell line-derived neurotrophic factor in developing motoneurons in rat spinal cords. Molecular medicine reports. 2012;6:805-10
54. Qiao S, Iwashita T, Ichihara M, Murakumo Y, Yamaguchi A, Isogai M. et al. Increased expression of glial cell line-derived neurotrophic factor and neurturin in a case of colon adenocarcinoma associated with diffuse ganglioneuromatosis. Clinical neuropathology. 2009;28:105-12
55. A F, H F, H S, M S, Y O, H T. et al. The relationship between GDNF and integrins in human colorectal cancer cell activity. Hepatogastroenterology. 2007;54:1398-402
56. Ma WR, Xu P, Liu ZJ, Zhou J, Gu LK, Zhang J. et al. Impact of GFRA1 gene reactivation by DNA demethylation on prognosis of patients with metastatic colon cancer. World journal of gastroenterology. 2020;26:184-98
57. Z L, J Z, Y G, L P, J Z, L G. et al. Large-scale characterization of DNA methylation changes in human gastric carcinomas with and without metastasis. J Clinical cancer research. 2014;20:4598-612
58. LL E, J K, VN K, J T, QY E, GP B. et al. GFRA3 promoter methylation may be associated with decreased postoperative survival in gastric cancer. BMC cancer. 2016;16:225
59. JZ T, XJ K, J K, GC F, M S, JK P. et al. Artemin-stimulated progression of human non-small cell lung carcinoma is mediated by BCL2. Molecular cancer therapeutics. 2010;9:1697-708
60. Rudat S, Pfaus A, Cheng YY, Holtmann J, Scholl C. RET-mediated autophagy suppression as targetable co-dependence in acute myeloid leukemia. Leukemia. 2018
61. Kuol N, Stojanovska L, Apostolopoulos V, Nurgali K. Role of the nervous system in cancer metastasis. Journal of Experimental & Clinical Cancer Research. 2018;37:5
62. Paratcha G, Ledda F, Baars L, Coulpier M, Besset V, Anders J. et al. Released GFRα1 Potentiates Downstream Signaling, Neuronal Survival, and Differentiation via a Novel Mechanism of Recruitment of c-Ret to Lipid Rafts. Neuron. 2001
63. Gil Z, Cavel O, Kelly K, Brader P, Rein A, Gao SP. et al. Paracrine Regulation of Pancreatic Cancer Cell Invasion by Peripheral Nerves. JNCI: Journal of the National Cancer Institute. 2010;102:107-18
64. Carlomagno F, Vitagliano D, Guida T, Napolitano M, Santoro M. The kinase inhibitor PP1 blocks tumorigenesis induced by RET oncogenes. Cancer Research. 2002;62:1077-82
65. Gao L BH, Wang Y, Zhang J, Zhu M. Neurotrophic Factor Artemin Promotes Invasiveness and Neurotrophic Function of Pancreatic Adenocarcinoma In vivo and In vitro. Pancreas. 2015;44:134-43
66. Chen SH, Zhang BY, Zhou B, Zhu CZ, Sun LQ, Feng YJ. Perineural invasion of cancer: a complex crosstalk between cells and molecules in the perineural niche. American journal of cancer research. 2019;9:1-21
67. Chuang JY TC, Chang SW, Chiang IP, Huang SM, Lin HY, Yeh WL, Lu DY. Glial cell line-derived neurotrophic factor induces cell migration in human oral squamous cell carcinoma. Oral oncology. 2013;49:1103-12
68. Boya P RF, Codogno P. Emerging regulation and functions of autophagy. Nature Cell Biology. 2013;15:1017 -
69. Kang J, Qian PX, Pandey V, Perry JK, Miller LD, Liu ET. et al. Artemin is estrogen regulated and mediates antiestrogen resistance in mammary carcinoma. Oncogene. 2010;29:3228-40
70. Banerjee A, Qian P, Wu Z-S, Ren X, Steiner M, Bougen NM. et al. Artemin Stimulates Radio- and Chemo-resistance by Promoting TWIST1-BCL-2-dependent Cancer Stem Cell-like Behavior in Mammary Carcinoma Cells. Journal of Biological Chemistry. 2012;287:42502-15
71. Ding K BA, Tan S, Zhao J, Zhuang Q, Li R, Qian P, Liu S, Wu ZS, Lobie PE, Zhu T. Artemin, a Member of the Glial Cell Line-derived Neurotrophic Factor Family of Ligands, Is HER2-regulated and Mediates Acquired Trastuzumab Resistance by Promoting Cancer Stem Cell-like Behavior in Mammary Carcinoma Cells. Journal of Biological Chemistry. 2014;289:16057-71
72. Brahimi-Horn MC CJ, Pouysségur J. Hypoxia and cancer. Journal of Molecular Medicine. 2007;85:1301-7
73. Zhang M, Zhang W, Wu Z, Liu S, Zhu T. Artemin is hypoxia responsive and promotes oncogenicity and increased tumor initiating capacity in hepatocellular carcinoma. Oncotarget. 2015;7:3267-82
Author contact
![]() Corresponding authors: Shuliang Zhao and Zhijun Cao, Renji Hospital, No.160 Pu Jian Ave, Shanghai 200127, China. E-mail: shuliangzhaocom and caozj_renjicom.
Corresponding authors: Shuliang Zhao and Zhijun Cao, Renji Hospital, No.160 Pu Jian Ave, Shanghai 200127, China. E-mail: shuliangzhaocom and caozj_renjicom.