Impact Factor ISSN: 1449-1907

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2021; 18(7):1532-1540. doi:10.7150/ijms.53728 This issue Cite
Research Paper
Neutrophil Extracellular Traps (NETs) and Vasculitis
Borros Arneth1 ![]() , Rebekka Arneth2
, Rebekka Arneth2
1. Institute of Laboratory Medicine and Pathobiochemistry, Molecular Diagnostics, University Hospital of the Universities of Giessen and Marburg UKGM, Justus Liebig University Giessen, Giessen, Germany.
2. Clinic of Nephrology, Internal Medicine, University Hospital of the Universities of Giessen and Marburg UKGM, Justus Liebig University Giessen, Giessen, Germany.
Received 2020-9-27; Accepted 2021-1-8; Published 2021-2-3
Abstract

Background: Neutrophil extracellular traps (NETs) have been implicated in host immune responses. Attempts have been made to examine how NETs affect the pathogenesis of complications such as autoimmune and vascular disorders.
Aim: This study aimed to explore the relationship between NETs and vasculitis.
Material and Methods: The current study entailed the searching of PsycINFO, PubMed, Web of Science, and CINAHL for articles related to the research topic. The search terms and phrases included “vasculitis,” “NETs,” “neutrophil extracellular traps,” “NETosis,” and “pathogenesis.” The search was limited to articles published between 2009 and 2019.
Results: Researchers have shown that NETs contribute to the pathogenesis of vasculitis through different mechanisms and processes, including renal failure and vascular damage. The protective effects of NETs have also been highlighted.
Discussion: Overall, some scholars have shown the effectiveness of using DNase I and the PAD4 inhibitor Cl-amidine to treat vasculitis by restricting NET formation. However, observations have been noted in only animal experimental models.
Conclusion: Neutrophil hyperactivity and its role in vasculitis are not yet fully understood. More studies aiming to determine the accurate function of NETs in vasculitis pathogenesis, particularly in humans, should be undertaken. Intensive research on NETs and vasculitis can increase the knowledge of medical practitioners and contribute to the development of new treatment methods to enhance patient outcomes in the future.
Keywords: neutrophil extracellular traps, vasculitis
Introduction
Neutrophils comprise approximately 57% of circulating white blood cells and are regarded as the most important effector cells in the innate immune system [1]. Neutrophils are normally recognized by their lobulated nuclei and short lifespans compared to those of other subpopulations of white blood cells [2,3]. When a pathogen enters the body, neutrophils act as the first line of response. They are directed to the infection or inflammation site, where they recruit and activate other types of immune cells to initiate the appropriate response [4-6]. While exerting their primary defense response, neutrophils can phagocytose disease-causing pathogens and release granules that contain proteases and antimicrobial peptides [7,8]. Researchers have recently reported that neutrophils can attack and restrain pathogens directly through the release of neutrophil extracellular traps (NETs) in a process commonly referred to as NETosis [3]. According to Linkermann et al., NETosis involves the release of extracellular DNA by host neutrophils [3]. Initially, NETosis was considered to coincide with the death of cells in the body, but recent investigations have revealed that it is a regulated cell death pathway [9]. Interestingly, NETosis is a caspase-independent process [10]. Researchers have yet to reach a consensus on whether NETosis is an anti-inflammatory or immunogenic process because it involves diverse pathways, some of which are still being studied [10]. The situation is further complicated by the fact that NET formation can take place even in the absence of necrosis. In other words, the development of NETs in humans occurs independently without the signaling of RIPK3 and MLKL, and the process is therefore independent of necroptosis [11,12]. Nevertheless, NETosis is considered a critical process that helps eliminate disease-causing pathogens.
Several previous studies have focused on NET formation as a process by which the body identifies and kills invading pathogens. In such studies, researchers noted that NETosis appeared to be a form of programmed cell death that was primarily initiated by nicotinamide adenine dinucleotide phosphate (NADPH) oxidase activation [13,14]. NADPH oxidase activation could be linked to nuclear membrane breakdown, chromatin decondensation, or the mixing of granule constituents and chromatin. Furthermore, NETosis is reported to be largely dependent on other molecules and substances, such as neutrophil elastases, peptidyl arginine deiminase (PAD), and myeloperoxidase (MPO) [15]. More recently, there have been sustained attempts to examine the link between NETosis and specific conditions, such as cancer, autoimmune disorders, and arteriosclerosis [16]. This interest stems from the realization that neutrophils are not the only subpopulation of cells capable of releasing extracellular traps; research evidence shows that other cells, such as mast cells, monocytes, and basophils, also have this ability even if it is rarely used [17]. Furthermore, the realization that NET formation can occur independently of cell death has motivated researchers to conduct further research.
The possible association between NET formation and vasculitis has also been explored in recent studies. Systemic vasculitis refers to a broad spectrum of disorders characterized by idiopathic vascular inflammation [18] and is categorized based on the size of the affected vessels. Irrespective of the nature of the disorder, both its pathogenesis and prognosis are potentially associated with NET formation [18]. Therefore, further investigations are required to understand the underlying correlation between NETs and vasculitis and possible therapeutic strategies for treating this disease. This systematic review aimed to explore the association between NETs and vasculitis.
Methodology
The current study examined the relationship between NETs and vasculitis by reviewing the existing body of research evidence, which entailed analyzing the results of investigations related to the topic of interest. The articles used were obtained from PsycINFO, PubMed, Web of Science, and CINAHL using the search terms and phrases “vasculitis,” “NETs,” “neutrophil extracellular traps,” “NETosis,” and “pathogenesis.” The search was limited to articles published in the four electronic databases between 2009 and 2019. The abstracts of the available articles were carefully reviewed to determine their quality and appropriateness (Figure 1).
PRISMA flow diagram.
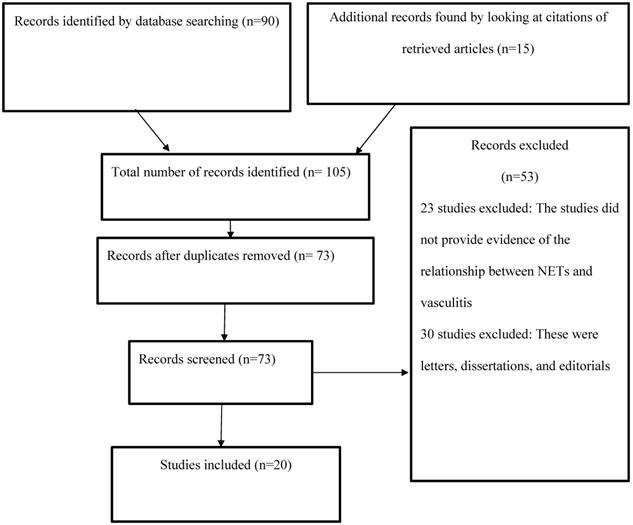
Results
At the end of the search and review process, the final list of articles consisted of animal model studies, clinical trials, experimental studies, and clinical reviews (Table 1). The articles were carefully examined to explore the relationship between NETs and vasculitis. An analysis of studies revealed that NETs are considered an important component of the innate immune system that helps fight disease [1,14,16]. The evidence gathered from the studies highlights the significant role of NETs in the adaptive immune response and their possible interaction with lymphocytes and antigen-presenting cells in lymph nodes and sites of inflammation [16,18]. Other studies revealed that NETs can induce antibody production and affect the actions of other molecules, such as proproliferative ligands [19,20]. Therefore, the analysis and elucidation of NETosis and regulatory mechanisms that affect innate immunity in patients with vasculitis can provide novel therapies for disease management.
Summary of the selected studies
| Study | Design | Results |
|---|---|---|
| Lee et al. (2017) [10] | Review | Interleukin-8, neutrophils, and anti-neutrophil cytoplasmic antibodies (ANCAs) are inflammatory molecules that play an important role in NET formation. The researchers noted that increased exposure to NTEs-related cascades and molecules could lead to systemic organ damage and to the development of autoimmune disorders. Further research is needed to explore how the NET formation process can be targeted to help treat autoimmune diseases. |
| Cornec et al. (2016) [21] | Systematic review | The authors stated that ANCA-associated vasculitis is a disorder that is characterized by the inflammation of small and mid-sized blood vessels. Available research evidence shows that PR3-ANCAs and MPO-ANCAs can be diagnosed effectively by examining the specific genetic and epidemiologic factors associated with each of the conditions. |
| Tang et al. (2015) [22] | Experimental study | Increasing research evidence suggests that NET formation is implicated in the development of ANCA-associated vasculitis. In addition, ANCA autoantibodies and anti-lysosomal membrane protein-2 (LAMP-2) antibodies have a critical pathogenic role in AAV. Targeting these molecules can help in the treatment and effective management of vasculitis. |
| Carmona-Rivera et al. (2017) [23] | Review | NET formation is implicated in the development of idiopathic autoimmune disorders. The researchers noted that NETs that were generated by levamisole appeared to be toxic to the endothelial walls. In addition, they impair endothelium vasorelaxation. Finally, the study revealed that successful stimulation of neutrophil muscarinic receptors through the use of cholinergic agonists could lead to vascular damage. |
| Nishide et al. (2017) [24] | Animal model studies | SEMA4D on the surface of neutrophils negatively control the neutrophil activation process. In patients with AAV, the proteolytic cleavage of the SEMA4D molecules can increase the neutrophil-related inflammatory responses in the body. Therefore, SEMA4D is a potential biomarker of AAV and a novel therapeutic target in the management of vasculitis. |
| Sha et al. (2016) [25] | Review | Dysregulation of the NET formation process may lead to the development of ANCA-associated vasculitis. Research evidence shows that NET formation is usually characterized by autophagy. A review of previous studies showed that autophagy can be induced by ANCAs and can increase the rate of ANCA-associated NETosis. |
| Shida et al. (2016) [26] | Clinical study | The combined stimulation of 1 µg/mL aLf. IgG and 10 nM PMA could increase the rate of NET formation. Evidence collected during the study showed that aLf enhanced NET formation. The changes were also linked to increased EGPA activity. |
| Yoshida et al. (2016) [27] | Clinical study | The researchers reported that ANCAs were linked to small-vessel vasculitis due to its role in kidney damage and the formation of NETs. The study further showed that NET induction was correlated with increased ANCA affinity for MPO in patients with ANCA-associated microscopic polyangiitis. Therefore, the targeting of NET formation markers could help in managing microscopic polyangiitis. |
| Aleman et al. (2016) [28] | Experimental study | The crosslinking of receptors like integrins did not enhance the NET formation process. It was also reported that the crosslinking of FcγRIIIb could induce NET formation in a way that is similar to the PMA-stimulation mechanism. Furthermore, the NET formation process was found to be dependent on ERK, PKC, and NADPH oxidase activation. |
| Aleyd et al. (2016) [29] | Clinical study | The researchers stated that IgA immune complexes could activate neutrophils, a trend that shows their role in the joint damage and development of vasculitis. In addition, the blocking of FcαRI reduced the rate of neutrophil activation, thus highlighting the possible link between NETs and autoimmune disorders. |
| Sandin et al. (2016) [30] | Clinical study | The study showed that the SIgA PR3-ANCA and IgA PR3-ANCA levels were higher during active ANCA-associated vasculitis than during inactive disease. The results of the study further suggest that SIgA PR3-ANCAs and IgA PR3-ANCAs are linked to disease activity in patients suffering from vasculitis. Additional research may be required to determine how the biomarkers and their isotopes can be used to monitor disease progression. |
| Kumar et al. (2015) [31] | Clinical study | Kumar et al. (2015) noted that anti-histone IgG, heparin, and recombinant-activated protein were effective in annulling severe glomerulonephritis. Combination therapy did not have a significant additional effect on the severity of the disease. The results show that NET-linked histone release can be targeted to help in the treatment of glomerulonephritis. |
| Pieterse et al. (2017) [32] | Experimental study | The results of the survey show that excessive NET formation can exceed the endothelial cell phagocytic capacity, thus causing vascular damage. In addition, it can lead to the activation of β-catenin signals and VE-cadherin degradation. |
| Westhorpe et al. (2017) [33] | Animal model study | The in vivo imaging experimental results showed the transient presence of intraglomerular NETs, an indication that high shear conditions could disrupt the NETs in the glomerular capillaries. The researchers also reported that NET dissolution by DNase I did not change the glomerular injury. The findings show that NET generation is often enhanced during glomerulonephritis. |
| Söderberg and Segelmark (2018) [34] | Review | NETs are involved in the development and progression of vasculitis. However, the NET formation process may also have a protective effect during the development of primary vasculitis. |
| van Dam et al. (2019) [35] | Clinical study | The authors noted that the induction pathways and kinetics involved in NET formation differ in patients with AAV and SLE. The successful recognition of diversity in the NET formation process helps understand the pathological role of neutrophils in the progression and activities of different autoimmune disorders. |
| Lood and Hughes (2017) [36] | Clinical study | The researchers stated that levamisole and cocaine are implicated in the development of ANCAs as they can induce the release of inflammatory NETS, BAFF, and NE autoantigens. The drug-associated release of CLAA-IgG is considered a reliable mechanism that shows the link between vasculitis and acute drug exposure among patients suffering from CLAA. Further investigations are needed to determine how the association can help in the management of ANCA-associated vasculitis and other disorders that are linked to NETosis. |
| Martinez et al. (2017) [37] | Systematic review | The author stated that the successful identification of NET formation inhibitors is critical for the management of Net-dependent diseases. The phenotypic NET assay can be developed and used to diagnose diseases that are linked to NETosis, such as vasculitis. |
| Kolaczkowska et al. (2015) [38] | Review | The study showed that neutrophils usually release NETs into the vasculature of the liver. DNase can effectively inhibit NE proteolytic activity. However, the molecule is incapable of removing the histones on the vessel walls, thus offering minimal protection against injury. The researchers further stated that the prevention or inhibition of NET formation could reduce the extent of tissue damage in the host during proteolytic activity. |
| Sondo et al. (2019) [39] | Experimental study | The researchers conducted high-content imaging analysis and identified vanilloids as an effective chemical compound that can prevent NET release and NETosis induction. Furthermore, vanilloids could reportedly reduce ROSS production and stop excessive NET formation during autoimmune disorders. |
Vasculitis and increased NETosis
The study of vasculitis pathogenesis has provided opportunities for the development of new treatment strategies. Vasculitis is a term used to indicate a broad spectrum of diseases that affect blood vessels [40], including anti-neutrophil cytoplasmic antibody (ANCA)-associated vasculitis (AAV, small-vessel vasculitis), Takayasu arteritis (large-vessel vasculitis), and giant cell arteritis [41]. These disorders can be triggered by various environmental factors and infectious pathogens that lead to vascular injury. A review of previous studies shows that vasculitis usually develops when host antibodies mistake blood vessel proteins for foreign bodies or pathogens [42]. In such instances, immune cells attack and destroy blood vessel cells. In other studies, researchers have noted that extravascular granulomatosis is a primary trigger of vasculitis in humans [43]. Therefore, the pathogenesis of vasculitis may vary depending on the type and on the triggers involved [21]. Furthermore, the disease manifestations are diverse and may include renal failure, aneurysms, visual disturbances, and glomerulonephritis. In some patients, vasculitis can lead to life-threatening complications such as pulmonary arterial hypertension, coronary heart disease, and thrombosis [44]. Consequently, there is a growing desire to explore the causes of vasculitis and to identify factors, processes, and biomarkers that can be targeted in the development of safer and more effective therapies.
The roles of NETs in the development and progression of vasculitis have been explored in previous studies. Research evidence showed that dying neutrophils around the walls of small vessels are a hallmark of AAV pathogenesis [45]. Additionally, they can attract other immune cells to the sites of inflammation or injury through the production of chemokines and cytokines [45]. However, it is not always easy to differentiate between the effects of degranulation induced by NETs and neutrophil-related cell death, as researchers contend that increased NETosis characterizes active ANCA-associated vasculitis. Furthermore, recent investigations have revealed that blood and affected tissue samples from patients with ANCA-associated vasculitis, Takayasu arteritis, and giant cell arteritis contain higher levels of NET remnants than those obtained from healthy individuals [45]. Studies have shown that PR3-ANCAs or MPO-ANCAs are found in most patients with vasculitis, and these forms of ANCAs are the primary determinants of AAV pathogenesis [46]. Nonetheless, other researchers have stated that healthcare practitioners and clinical experts need to be aware that NETs can also be mitochondrially derived and thus lack histones [47].
The first clear evidence of increased NET formation in patients with vasculitis was based on kidney biopsy samples [22,48]. Later, researchers showed that NETs also exist in skin specimens and thrombus samples obtained from people diagnosed with conditions such as Henoch-Schonlein purpura, granulomatosis with polyangiitis, microscopic polyangiitis, and eosinophilic granulomatosis [49-51]. In addition, patients with ANCA-negative MPA were reported to have increased NET levels in nerve biopsies than counterparts who did not have the disease [23,52]. The differences in NET and NET fragment levels indicate the possible role of NETosis in the pathogenesis and progression of vasculitis. In vitro studies have shown that patients with ANCA-associated vasculitis are more likely to experience spontaneous NETosis than healthy controls [53,54]. Furthermore, such patients are more likely to be affected by NET-inducing stimuli than those not suffering from this disease. In autoimmune studies, researchers have reported that NETosis can be negatively regulated by the interaction between neutrophil semaphorin D on neutrophils and plexin B2 on endothelial cells. Moreover, recombinant plexin B2 can inhibit NETosis in human cells [54-56]. These results provide a basis for understanding the possible links between NETs and the pathogenesis of ANCA-associated vasculitis.
Recent in vitro studies have shown that autoantibodies and serum immune complexes obtained from patients with ANCA-associated vasculitis are capable of inducing NETosis [24-26,56-59]. In addition, IgG from these patients can stimulate NETosis [24-26,56-59]. It has been reported that antibodies against proteinase 3 (PR3) are present in the sera of patients with Wegener's granulomatosis [24,57]. Furthermore, antibodies against MPO are present in the sera of patients with polyarteritis nodosa [24-26,56-59]. However, the underlying mechanisms that link NETosis to the development of these diseases remain unclear. A recent study by Ma et al. reported that NET formation in patients with conditions such as vasculitis could be affected by MPO-ANCA IgG and PR3-ANCA IgG [58]. Recent investigations showed that the mechanisms by which MPO-ANCA IgG causes NETosis in people with vasculitis could be linked to the degree of antibody affinity rather than to the antibody levels [25,26,57]. Furthermore, renal biopsy investigations have revealed that people with high-affinity MPO-ANCA IgG tend to show increased levels of NETosis [59]. Interestingly, some studies have documented that IgG-depleted sera from patients without ANCAs can also induce NETosis [27,59,60]. The results of such studies question the role of IgG in influencing NETosis in patients with vasculitis.
The link between NET formation and ANCA-associated vasculitis has also been examined by analyzing how immune complexes with IgG can induce NETosis by activating neutrophils through the crosslinking of the Fcγ receptor IIIb 131 in patients with MPO-ANCA-associated microscopic polyangiitis [27,28]. Recent studies have shown that aggregated immune complexes are receptor-specific and enhance the NETosis rate in patients diagnosed with conditions such as vasculitis and systemic lupus erythematosus (SLE) [28,61]. Other studies have noted that IgA immune complexes from synovial and plasma fluid samples can induce NETosis through Fc alpha receptor I [29]. The results indicate the pathogenesis of small-vessel leukocytoclastic vasculitis since IgA immune complex deposition is an indication of this disease. In addition, the findings are supported by recent investigations revealing that samples from patients with SLE and rheumatoid arthritis had high levels of PR3-ANCA IgA depending on the disease severity [30].
In addition to effects on neutrophil activation, MPO-ANCAs and PR3-ANCAs can induce NETosis in patients suffering from SLE and AAV [57]. The exact mechanism underlying the activation process remains unclear, but researchers believe that it requires the binding of autoantibodies to Fc receptors and PR3/MPO on the neutrophil surface. Recent research shows that neutrophil activation by MPO-ANCAs during NETosis can increase ROS production [57]. A review of evidence from in vitro studies showed that neutrophils from patients with vasculitis are usually more readily activated by ANCAs than those from healthy people [26,58]. Moreover, previous research revealed that vasculitis patients are more likely to show increased PR3 expression; this phenomenon can be explained by disruptions in the epigenetic silencing of the MPO and PR3 genes in affected patients. The epitope affinity and specificity of the involved autoantibodies are important concerning the development of AAV [60]; the primary hypothesis is that neutrophil activation by ANCAs and the subsequent induction of NETs are affected by the affinity of the autoantibodies.
The current review makes it evident that NETs play a key role in the initiation and progression of AAV. There is compelling research evidence that NETs are directly involved in vessel inflammation [27,28]; this process entails endothelial cell damage and complement system activation. In some instances, NETs are indirectly involved in the pathogenesis of AAV through the generation of MPO-ANCAs and PR3-ANCAs [61]. The vicious cycle affects the development of AAV by activating neutrophils in the body. Importantly, ANCA-linked pathogenicity is influenced by the epitope specificity and affinity of autoantibodies [29]. Furthermore, NET formation in patients with AAV may be increased by drugs, age-associated epigenetic changes, and infections. The increased NET formation should be balanced by clearance mechanisms, such as those involving DNases and autoantibodies with ANCA specificity [29,30]. The data imply that under unfavorable conditions, people develop pathogenic autoantibodies that can activate neutrophils, creating a vicious cycle that causes vessel wall inflammation and AAV.
Another interesting reported finding regarding the link between NETs and vasculitis is that neutrophils obtained from people suffering from AAV are less likely to undergo apoptosis than those obtained from healthy individuals; this result suggests that such neutrophils are more susceptible to other forms of cell death depending on their cellular state and contextual factors [45]. For instance, it has been reported that neutrophils from patients with AAV release NETs more spontaneously than those from healthy people [22,62,63]. In addition, low-density granulocytes (LDGs), a subpopulation of neutrophils, show a stronger association than normal-density neutrophils with the increased spontaneous production of NETs [64]. LDGs are categorized as a primary source of NETs in patients diagnosed with AAV [63]. Furthermore, recent studies on LDGs show that they can promote the increased expression of mRNAs associated with different alarmins and immunostimulatory bactericidal proteins more than normal-density neutrophils [45,65]. Such findings provide a basis for examining and understanding the pathogenesis of AAV and the possible link between this disease and NETs.
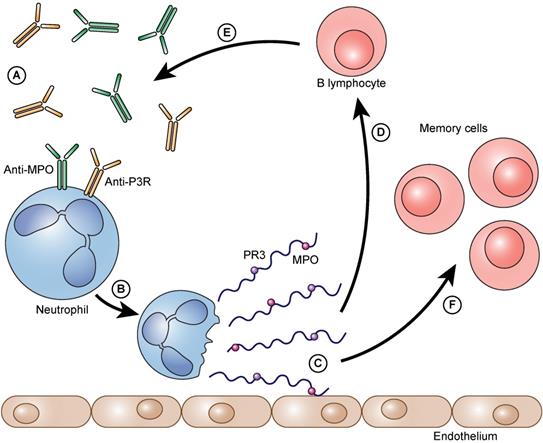
The process as a vicious circle. Auto-antibodies bind to neutrophil granulocytes and trigger NET formation (A). Neutrophils form NETs on the vascular endothelium (B). Immunogenic proteins such as PR3 and/or MPO are released (C). B- lymphocytes arrive and are immunized against these proteins (D). They form even more auto-antibodies against these proteins (PR3 and/or MPO) (E). The auto-antibodies that are formed can then, in turn, bind to neutrophils and can accelerate the further formation of NETs. Besides the immunized and thereby activated B lymphocytes also memory cells (memory B lymphocytes and memory T lymphocytes) are formed (F).
NETs can directly or indirectly cause vascular damage and worsen the symptoms of ANCA-associated vasculitis. For instance, the release of noxious molecules such as degrading enzymes can induce endothelial cell apoptosis and cause deterioration of the basement membrane [30,66]. Histones may be released during NETosis and have toxic effects on endothelial cells [66]. Although available research suggests that NETs can be phagocytized by endothelial cells, excessive amounts of NETs may promote vascular leakage and hinder interactions among endothelial cells [31]. Moreover, NET formation is associated with the transformation of endothelial cells to mesenchymal cells, which interferes with normal glomerular function [30]. When the resulting vasculitis is not managed, the alternative complement pathway can be activated, resulting in tissue fibrosis [31,32,67,68].
Finally, the presence of renal injury among AAV patients is also regarded as an indicator of the role of NETs in disease development. Research shows that patients suffering from small-vessel vasculitis are likely to experience tubulointerstitial nephritis and glomerulonephritis [33,68]. Recent investigations have indicated that NETs are usually present in glomeruli [68]. Furthermore, researchers have used animal model studies to show that NET formation can cause glomerular vasculitis by activating anti-glomerular basement membrane (GBM) antibodies [32,33,36,68]. In addition, NET formation is believed to lead to tubulointerstitial injury and epithelial tubular cell hypoxia. In some instances, NETs release histones from endothelial cells, thus causing necrosis and necroinflammation [69-72]. Moreover, NETs can worsen tubulointerstitial injury and compromise glomerular blood flow. The findings show that NETs contribute to the development of vasculitis by causing renal injury and increasing the risk of hemorrhage (Table 2).
Summary of the findings
| Event | Associated Disease |
|---|---|
| Dying neutrophils surrounding the walls of small vessels [45,46] | Small-vessel vasculitis such as AAV [45,46] |
| PR3-ANCAs or MPO-ANCAs are found in most patients with vasculitis [46] | AAV pathogenesis [46] |
| Presence of NET fragments in kidney biopsies [22,48] | Henoch-Schonlein purpura, granulomatosis with polyangiitis, microscopic polyangiitis, and eosinophilic granulomatosis [22,48] |
| Antibodies against PR3 [24,57] | Wegener's granulomatosis [24,57] |
| Antibodies against MPO [24] | Polyarteritis nodosa [24] |
| Increased levels of MPO-ANCA IgG and PR3-ANCA IgG [58] | AAV [58] |
| Activation of neutrophils via Fcγ receptor IIIb [27,28] | ANCA-associated microscopic polyangiitis [27,28] |
| Presence of aggregated immune complexes [28] | Vasculitis, SLE, and small-vessel leukocytoclastic vasculitis [28] |
| High levels of PR3-ANCA IgA [30] | SLE and rheumatoid arthritis [30] |
| Vascular damage and endothelial cell apoptosis [30,66] | AAV [30,66] |
| Increased NET formation rate [33,68] | Tubulointerstitial nephritis and glomerulonephritis [33,68] |
| Activation of glomerular basement membrane (GBM) antibodies during NET formation [32,68] | Tubulointerstitial injury and epithelial tubular cell hypoxia [32,68] |
| Histone release during NET formation [30,66] | Tubulointerstitial injury and AAV [30,66] |
Protective effect of NETs
Recent research projects have shown that NETs can protect patients from the adverse effects of vasculitis [18]. One experimental study reported that saliva could induce the formation of NETs. However, the induction capacity was diminished in patients suffering from a form of vasculitis known as Bechet's disease, which is usually characterized by acute to chronic genital and mouth ulcers [18]. In this study, the authors argued that the absence of NETs diminished protection against bacteria in the mucous membrane, thus promoting ulcer formation. In another animal model study, NETs were shown to provide a platform for degrading proinflammatory mediators [71,72]. Under some circumstances, NETs tend to impair GM-CSF/IL-4-linked dendritic cell differentiation and promote the activation of macrophage phenotypes [73]. Macrophage cell populations help reduce inflammation and promote autoantibody production in response to the disease-causing pathogens in question [56]. Significant amounts of research evidence show that NETs may contain alarmins such as HMGB1 and LL39 and enhance the rate of autoantibody production in people with vasculitis [34,73,74]. The actual mechanisms by which these processes protect against vasculitis need to be studied further to determine their therapeutic potential.
Discussion
Vasculitis is a severe condition that can lead to adverse outcomes, including death, if not well managed [34,75]. Over the years, attempts have been made to understand and study the possible causes of vasculitis and the underlying pathogenesis [75,76]. Investigations have shown that this disease is characterized by the abnormal attack of blood vessel cells following foreign pathogen invasion [35,36]. Evidence gathered in this study reveals that NETs can lead to the emergence and progression of vasculitis through a wide range of pathways, such as vascular damage and renal complications [34]. More recently, however, NETs were reported to potentially have protective effects, reducing the risk of vasculitis and lessening the severity of symptoms [34]. Therefore, NETs can be regarded as an important biomarker for detecting and monitoring vasculitis activity. Furthermore, they provide an avenue for developing new therapeutic strategies.
For instance, several approaches for treating vasculitis that involve blocking the adverse effects of NETs have been examined in vivo. Antihistamine therapy is one technique that works by promoting the production of antihistamine antibodies [34,36]. Heparin and activated protein C-based treatment have been shown to be effective in the management of anti-GBM-induced vasculitis [31]. Similar results were reported in studies that analyzed the possibility of using the PAD4 inhibitor Cl-amidine to treat vasculitis, as this inhibitor blocks histone citrullination during NETosis [37,77]. Additionally, mouse model studies involving the use of PAD4 to manage vasculitis have shown that the technique could effectively minimize the generation of NET-mediated inflammation [34]. This process is hypothesized to entail controlling ROS signaling and NETosis using tetrahydroisoquinoline, a compound that can regulate PAD4 and NETosis, via inhibition of the latter and interfering with ROS production [34].
Other researchers have noted that DNA degradation, ROS scavenging, and mitochondrial ROS inhibition might also help manage vasculitis [34,77]. The inhibitory processes are not well understood but are believed to be related to the upregulation of superoxide dismutase 3, the inhibition of mitochondrial ROS, and neutrophil phagocytosis [38,39,70,78]. Based on studies on the protective effects of NETs, researchers have hypothesized that compounds such as the anti-thyroid drug propylthiouracil (PTU) could help in the management of vasculitis by promoting autoantibody production to neutralize disease-causing pathogens and the treatment of hyperthyroidism [34,79]. PTU can induce the formation of NETs that hinder DNase I degradation, thus reducing disease severity [34,39,75,80].
Nonetheless, despite all these outcomes, minimal attention has been directed at exploring the role of NETs as biomarkers of vasculitis activity. In addition, only a few studies have investigated how NETs can be used to monitor disease progression and the treatment response [34]. The present systematic review highlighted that the NET and NET-related protein levels were increased in samples from patients with vasculitis compared to those from healthy people. However, researchers and healthcare practitioners have yet to develop an assay based on NET biomarkers that can be used to monitor vasculitis progression in the clinic [34]. The situation is further complicated by the existence of a standard approach for measuring NET levels in patients [34,35,76]. Nevertheless, there is a need to carry out further research on the utility of NETs as biomarkers for vasculitis and to determine the measures and parameters necessary to analyze treatment responses.
Conclusion
From the review, it is evident that NETs play an essential role in the pathogenesis of vasculitis. The literature has shown that patients with vasculitis have more NET components than healthy people. Such an observation concludes the cause of chronic inflammation, which is one of the fundamental elements sought by medical practitioners when diagnosing vasculitis. It is currently believed that preventing the generation of structures can contribute to favorable clinical results for patients with vasculitis. Reports from studies have shown that currently, DNase I and the PAD4 inhibitor CI-amidine could impact the formation of NETs through restriction, which can help minimize the development of vasculitis. However, these observations have thus far been made using animal experimental models. As a result, in humans, neutrophil hyperactivity and its role in vasculitis are not yet fully understood. Therefore, more studies aiming to determine the accurate role of NETs in vasculitis pathogenesis should be undertaken, particularly in humans. Intensive research on NETs and vasculitis can increase the knowledge of medical practitioners and contribute to the development of new treatment methods to enhance patient outcomes in the future.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Pillay J, den Braber I, Vrisekoop N, Kwast LM, de Boer RJ, Borghans JAM, Tesselaar K, Koenderman L. In vivo labeling with 2H2O reveals a human neutrophil lifespan of 5.4 days. Blood. 2010Jul29;116(4):625-7
2. Remijsen Q, Kuijpers TW, Wirawan E, Lippens S, Vandenabeele P, Vanden Berghe T. Dying for a cause: NETosis, mechanisms behind an antimicrobial cell death modality. Cell Death Differ. 2011Apr;18(4):581-8
3. Linkermann A, Stockwell BR, Krautwald S, Anders H-J. Regulated cell death and inflammation: an auto-amplification loop causes organ failure. Nat Rev Immunol. 2014;14(11):759-67
4. Fang C, Wei X, Wei Y. Mitochondrial DNA in the regulation of innate immune responses. Protein Cell. 2016Jan;7(1):11-6
5. Nakazawa D, Shida H, Kusunoki Y, Miyoshi A, Nishio S, Tomaru U, Atsumi T, Ishizu A. The responses of macrophages in interaction with neutrophils that undergo NETosis. J Autoimmun. 2016Feb;67:19-28
6. Ingelsson B, Söderberg D, Strid T, Söderberg A, Bergh A-C, Loitto V, Lotfi K, Segelmark M, Spyrou G, Rosén A. Lymphocytes eject interferogenic mitochondrial DNA webs in response to CpG and non-CpG oligodeoxynucleotides of class C. Proc Natl Acad Sci U S A. 2018;115(3):E478-87
7. Zhang Y, Chen X, Gueydan C, Han J. Plasma membrane changes during programmed cell deaths. Cell Res. 2018;28(1):9-21
8. Xie Y, Hou W, Song X, Yu Y, Huang J, Sun X, Kang R, Tang D. Ferroptosis: process and function. Cell Death Differ. 2016Mar;23(3):369-79
9. Gupta S, Kaplan MJ. The role of neutrophils and NETosis in autoimmune and renal diseases. Nat Rev Nephrol. 2016;12(7):402-13
10. Lee KH, Kronbichler A, Park DD-Y, Park Y, Moon H, Kim H, Choi JH, Choi Y, Shim S, Lyu IS. et al. Neutrophil extracellular traps (NETs) in autoimmune diseases: A comprehensive review. Autoimmun Rev. 2017Nov;16(11):1160-73
11. Amini P, Stojkov D, Wang X, Wicki S, Kaufmann T, Wong WW-L, Simon H-U, Yousefi S. NET formation can occur independently of RIPK3 and MLKL signaling. Eur J Immunol. 2016Jan;46(1):178-84
12. Rosazza T, Warner J, Sollberger G. NET formation - mechanisms and how they relate to other cell death pathways. FEBS J. 2020 Oct 12
13. Jorch SK, Kubes P. An emerging role for neutrophil extracellular traps in noninfectious disease. Nat Med. 2017Mar7;23(3):279-87
14. Kessenbrock K, Krumbholz M, Schönermarck U, Back W, Gross WL, Werb Z, Gröne H-J, Brinkmann V, Jenne DE. Netting neutrophils in autoimmune small-vessel vasculitis. Nat Med. 2009Jun;15(6):623-5
15. Papayannopoulos V, Metzler KD, Hakkim A, Zychlinsky A. Neutrophil elastase and myeloperoxidase regulate the formation of neutrophil extracellular traps. J Cell Biol. 2010Nov1;191(3):677-91
16. Wang Y, Li M, Stadler S, Correll S, Li P, Wang D, Hayama R, Leonelli L, Han H, Grigoryev SA, Allis CD, Coonrod SA. Histone hypercitrullination mediates chromatin decondensation and neutrophil extracellular trap formation. J Cell Biol. 2009Jan26;184(2):205-13
17. Goldmann O, Medina E. The expanding world of extracellular traps: not only neutrophils but much more. Front Immunol. 2012;3:420
18. Mohanty T, Sjögren J, Kahn F, Abu-Humaidan AHA, Fisker N, Assing K, Mörgelin M, Bengtsson AA, Borregaard N, Sørensen OE. A novel mechanism for NETosis provides antimicrobial defense at the oral mucosa. Blood. 2015Oct29;126(18):2128-37
19. Gonzalez-Aparicio M, Alfaro C. Influence of interleukin-8 and neutrophil extracellular trap (NET) formation in the tumor microenvironment: Is there a pathogenic role? J Immunol Res. 2019;2019:6252138
20. Li Y, Wang W, Yang F, Xu Y, Feng C, Zhao Y. The regulatory roles of neutrophils in adaptive immunity. Cell Commun Signal CCS. 2019;17(1):147
21. Cornec D, Cornec-Le Gall E, Fervenza FC, Specks U. ANCA-associated vasculitis - clinical utility of using ANCA specificity to classify patients. Nat Rev Rheumatol. 2016;12(10):570-9
22. Tang S, Zhang Y, Yin S-W, Gao X-J, Shi W-W, Wang Y, Huang X, Wang L, Zou L-Y, Zhao J-H. et al. Neutrophil extracellular trap formation is associated with autophagy-related signalling in ANCA-associated vasculitis. Clin Exp Immunol. 2015Jun;180(3):408-18
23. Carmona-Rivera C, Purmalek MM, Moore E, Waldman M, Walter PJ, Garraffo HM, Phillips KA, Preston KL, Graf J, Kaplan MJ. et al. A role for muscarinic receptors in neutrophil extracellular trap formation and levamisole-induced autoimmunity. JCI Insight. 2017;2(3):e89780
24. Nishide M, Nojima S, Ito D, Takamatsu H, Koyama S, Kang S, Kimura T, Morimoto K, Hosokawa T, Hayama Y. et al. Semaphorin 4D inhibits neutrophil activation and is involved in the pathogenesis of neutrophil-mediated autoimmune vasculitis. Ann Rheum Dis. 2017Aug;76(8):1440-8
25. Sha L-L, Wang H, Wang C, Peng H-Y, Chen M, Zhao M-H. Autophagy is induced by anti-neutrophil cytoplasmic Abs and promotes neutrophil extracellular traps formation. Innate Immun. 2016;22(8):658-65
26. Shida H, Nakazawa D, Tateyama Y, Miyoshi A, Kusunoki Y, Hattanda F, Masuda S, Tomaru U, Kawakami T, Atsumi T. et al. The presence of anti-lactoferrin antibodies in a subgroup of eosinophilic granulomatosis with polyangiitis patients and their possible contribution to enhancement of neutrophil extracellular trap formation. Front Immunol. 2016;7:636
27. Yoshida M, Yamada M, Sudo Y, Kojima T, Tomiyasu T, Yoshikawa N, Oda T, Yamada M. Myeloperoxidase anti-neutrophil cytoplasmic antibody affinity is associated with the formation of neutrophil extracellular traps in the kidney and vasculitis activity in myeloperoxidase anti-neutrophil cytoplasmic antibody-associated microscopic polyangiitis. Nephrol Carlton Vic. 2016Jul;21(7):624-9
28. Alemán OR, Mora N, Cortes-Vieyra R, Uribe-Querol E, Rosales C. Differential use of human neutrophil fcγ receptors for inducing neutrophil extracellular trap formation. J Immunol Res. 2016;2016:2908034
29. Aleyd E, Al M, Tuk CW, van der Laken CJ, van Egmond M. IgA complexes in plasma and synovial fluid of patients with rheumatoid arthritis induce neutrophil extracellular traps via fcαri. J Immunol Baltim Md 1950. 2016;197(12):4552-9
30. Sandin C, Eriksson P, Segelmark M, Skogh T, Kastbom A. IgA- and SIgA anti-PR3 antibodies in serum versus organ involvement and disease activity in PR3-ANCA-associated vasculitis. Clin Exp Immunol. 2016May;184(2):208-15
31. Kumar SVR, Kulkarni OP, Mulay SR, Darisipudi MN, Romoli S, Thomasova D, Scherbaum CR, Hohenstein B, Hugo C, Müller S, Liapis H. et al. Neutrophil extracellular trap-related extracellular histones cause vascular necrosis in severe GN. J Am Soc Nephrol JASN. 2015Oct;26(10):2399-413
32. Pieterse E, Rother N, Garsen M, Hofstra JM, Satchell SC, Hoffmann M, Loeven MA, Knaapen HK, van der Heijden OWH, Berden JHM. et al. Neutrophil extracellular traps drive endothelial-to-mesenchymal transition. Arterioscler Thromb Vasc Biol. 2017;37(7):1371-9
33. Westhorpe CLV, Bayard JE, O'Sullivan KM, Hall P, Cheng Q, Kitching AR, Hickey MJ. In vivo imaging of inflamed glomeruli reveals dynamics of neutrophil extracellular trap formation in glomerular capillaries. Am J Pathol. 2017Feb;187(2):318-31
34. Söderberg D, Segelmark M. Neutrophil extracellular traps in vasculitis, friend or foe? Curr Opin Rheumatol. 2018Jan;30(1):16-23
35. van Dam LS, Kraaij T, Kamerling SWA, Bakker JA, Scherer UH, Rabelink TJ, van Kooten C, Teng YKO. Intrinsically distinct role of neutrophil extracellular trap formation in anti-neutrophil cytoplasmic antibody-associated vasculitis compared to systemic lupus erythematosus. Arthritis Rheumatol Hoboken NJ. 2019;71(12):2047-58
36. Lood C, Hughes GC. Neutrophil extracellular traps as a potential source of autoantigen in cocaine-associated autoimmunity. Rheumatol Oxf Engl. 2017;56(4):638-43
37. Martinez NE, Zimmermann TJ, Goosmann C, Alexander T, Hedberg C, Ziegler S, Zychlinsky A, Waldmann H. Tetrahydroisoquinolines: New inhibitors of neutrophil extracellular trap (NET) formation. Chembiochem Eur J Chem Biol. 2017;18(10):888-93
38. Kolaczkowska E, Jenne CN, Surewaard BGJ, Thanabalasuriar A, Lee W-Y, Sanz M-J, Mowen K, Opdenakker G, Kubes P. Molecular mechanisms of NET formation and degradation revealed by intravital imaging in the liver vasculature. Nat Commun. 2015Mar26;6:6673
39. Sondo E, Bertelli R, Pesce E, Ghiggeri GM, Pedemonte N. High-content screening identifies vanilloids as a novel class of inhibitors of NET formation. Front Immunol. 2019;10:963
40. Okazaki T, Shinagawa S, Mikage H. Vasculitis syndrome-diagnosis and therapy. J Gen Fam Med. 2017;18(2):72-8
41. Yates M, Watts R. ANCA-associated vasculitis. Clin Med Lond Engl. 2017Feb;17(1):60-4
42. Flint SM, McKinney EF, Smith KGC. Emerging concepts in the pathogenesis of anti-neutrophil cytoplasmic antibody-associated vasculitis. Curr Opin Rheumatol. 2015Mar;27(2):197-203
43. Lee T, Gasim A, Derebail VK, Chung Y, McGregor JG, Lionaki S, Poulton CJ, Hogan SL, Jennette JC, Falk RJ. et al. Predictors of treatment outcomes in ANCA-associated vasculitis with severe kidney failure. Clin J Am Soc Nephrol CJASN. 2014May;9(5):905-13
44. Batra K, Chamarthy M, Chate RC, Jordan K, Kay FU. Pulmonary vasculitis: diagnosis and endovascular therapy. Cardiovasc Diagn Ther. 2018Jun;8(3):297-315
45. Söderberg D, Segelmark M. Neutrophil extracellular traps in ANCA-associated vasculitis. Front Immunol. 2016;7:256
46. Yoshida M, Sasaki M, Sugisaki K, Yamaguchi Y, Yamada M. Neutrophil extracellular trap components in fibrinoid necrosis of the kidney with myeloperoxidase-ANCA-associated vasculitis. Clin Kidney J. 2013Jun;6(3):308-12
47. Yousefi S, Simon H-U. NETosis - does it really represent nature's “suicide bomber”? Front Immunol. 2016;7:328
48. O'Sullivan KM, Lo CY, Summers SA, Elgass KD, McMillan PJ, Longano A, Ford SL, Gan P-Y, Kerr PG, Kitching AR. et al. Renal participation of myeloperoxidase in anti-neutrophil cytoplasmic antibody (ANCA)-associated glomerulonephritis. Kidney Int. 2015Nov;88(5):1030-46
49. Nakazawa D, Tomaru U, Yamamoto C, Jodo S, Ishizu A. Abundant neutrophil extracellular traps in thrombus of patient with microscopic polyangiitis. Front Immunol. 2012;3:333
50. Abreu-Velez AM, Smith JG, Howard MS. Presence of neutrophil extracellular traps and anti-neutrophil cytoplasmic antibodies associated with vasculitides. North Am J Med Sci. 2009Nov;1(6):309-13
51. Sangaletti S, Tripodo C, Chiodoni C, Guarnotta C, Cappetti B, Casalini P, Piconese S, Parenza M, Guiducci C, Vitali C. et al. Neutrophil extracellular traps mediate transfer of cytoplasmic neutrophil antigens to myeloid dendritic cells toward ANCA induction and associated autoimmunity. Blood. 2012Oct11;120(15):3007-18
52. Imamoto T, Nakazawa D, Shida H, Suzuki A, Otsuka N, Tomaru U, Ishizu A. Possible linkage between microscopic polyangiitis and thrombosis via neutrophil extracellular traps. Clin Exp Rheumatol. 2014Feb;32(1):149-50
53. Ohlsson SM, Ohlsson S, Söderberg D, Gunnarsson L, Pettersson Å, Segelmark M, Hellmark T. Neutrophils from vasculitis patients exhibit an increased propensity for activation by anti-neutrophil cytoplasmic antibodies. Clin Exp Immunol. 2014Jun;176(3):363-72
54. Sur Chowdhury C, Giaglis S, Walker UA, Buser A, Hahn S, Hasler P. Enhanced neutrophil extracellular trap generation in rheumatoid arthritis: analysis of underlying signal transduction pathways and potential diagnostic utility. Arthritis Res Ther. 2014Jun13;16(3):R122
55. Khandpur R, Carmona-Rivera C, Vivekanandan-Giri A, Gizinski A, Yalavarthi S, Knight JS, Friday S, Li S, Patel RM, Subramanian V, Thompson P. et al. NETs are a source of citrullinated autoantigens and stimulate inflammatory responses in rheumatoid arthritis. Sci Transl Med. 2013Mar27;5(178):178ra40
56. Villanueva E, Yalavarthi S, Berthier CC, Hodgin JB, Khandpur R, Lin AM, Rubin CJ, Zhao W, Olsen SH, Klinker M. et al. Netting neutrophils induce endothelial damage, infiltrate tissues, and expose immunostimulatory molecules in systemic lupus erythematosus. J Immunol Baltim Md 1950. 2011Jul1;187(1):538-52
57. Nakazawa D, Shida H, Tomaru U, Yoshida M, Nishio S, Atsumi T, Ishizu A. Enhanced formation and disordered regulation of NETs in myeloperoxidase-ANCA-associated microscopic polyangiitis. J Am Soc Nephrol JASN. 2014May;25(5):990-7
58. Ma Y-H, Ma T-T, Wang C, Wang H, Chang D-Y, Chen M, Zhao M-H. High-mobility group box 1 potentiates anti-neutrophil cytoplasmic antibody-inducing neutrophil extracellular traps formation. Arthritis Res Ther. 2016Jan6;18:2
59. Kraaij T, Bakker J, Brunini F, Pusey C, Scherer H, Toes R, Rabelink T, van Kooten C. NET-inducing capacity is a biomarker in ANCA-associated vasculitis independent of ANCA antibodies. Arthritis Rheumatol. 2016 68(Suppl 10(1)):1-15
60. Natorska J, Ząbczyk M, Siudut J, Krawiec P, Mastalerz L, Undas A. Neutrophil extracellular traps formation in patients with eosinophilic granulomatosis with polyangiitis: association with eosinophilic inflammation. Clin Exp Rheumatol. 2017 Apr;35(Suppl 103(1)):27-32
61. Kraaij T, Tengström FC, Kamerling SWA, Pusey CD, Scherer HU, Toes REM, Rabelink TJ, van Kooten C, Teng YKO. A novel method for high-throughput detection and quantification of neutrophil extracellular traps reveals ROS-independent NET release with immune complexes. Autoimmun Rev. 2016Jun;15(6):577-84
62. Söderberg D, Kurz T, Motamedi A, Hellmark T, Eriksson P, Segelmark M. Increased levels of neutrophil extracellular trap remnants in the circulation of patients with small vessel vasculitis, but an inverse correlation to anti-neutrophil cytoplasmic antibodies during remission. Rheumatol Oxf Engl. 2015Nov;54(11):2085-94
63. Grayson PC, Carmona-Rivera C, Xu L, Lim N, Gao Z, Asare AL, Specks U, Stone JH, Seo P, Spiera RF. et al. Neutrophil-related gene expression and low-density granulocytes associated with disease activity and response to treatment in anti-neutrophil cytoplasmic antibody-associated vasculitis. Arthritis Rheumatol Hoboken NJ. 2015Jul;67(7):1922-32
64. Herteman N, Vargas A, Lavoie J-P. Characterization of circulating low-density neutrophils intrinsic properties in healthy and asthmatic horses. Sci Rep. 2017;7(1):7743
65. Kaplan MJ. Neutrophils in the pathogenesis and manifestations of SLE. Nat Rev Rheumatol. 2011Sep27;7(12):691-9
66. Korkmaz B, Horwitz MS, Jenne DE, Gauthier F. Neutrophil elastase, proteinase 3, and cathepsin G as therapeutic targets in human diseases. Pharmacol Rev. 2010Dec;62(4):726-59
67. Lin F, Wang N, Zhang T-C. The role of endothelial-mesenchymal transition in development and pathological process. IUBMB Life. 2012Sep;64(9):717-23
68. Wang H, Wang C, Zhao M-H, Chen M. Neutrophil extracellular traps can activate alternative complement pathways. Clin Exp Immunol. 2015Sep;181(3):518-27
69. Nakazawa D, Kumar SV, Marschner J, Desai J, Holderied A, Rath L, Kraft F, Lei Y, Fukasawa Y, Moeckel GW. et al. Histones and neutrophil extracellular traps enhance tubular necrosis and remote organ injury in ischemic AKI. J Am Soc Nephrol JASN. 2017Jun;28(6):1753-68
70. Jiang D, Muschhammer J, Qi Y, Kügler A, de Vries JC, Saffarzadeh M, Sindrilaru A, Beken SV, Wlaschek M, Kluth MA. et al. Suppression of neutrophil-mediated tissue damage-a novel skill of mesenchymal stem cells. Stem Cells Dayt Ohio. 2016;34(9):2393-406
71. Schauer C, Janko C, Munoz LE, Zhao Y, Kienhöfer D, Frey B, Lell M, Manger B, Rech J, Naschberger E. et al. Aggregated neutrophil extracellular traps limit inflammation by degrading cytokines and chemokines. Nat Med. 2014May;20(5):511-7
72. Guimarães-Costa AB, Rochael NC, Oliveira F, Echevarria-Lima J, Saraiva EM. Neutrophil extracellular traps reprogram IL-4/GM-CSF-induced monocyte differentiation to anti-inflammatory macrophages. Front Immunol. 2017;8:523
73. Mitroulis I, Kambas K, Chrysanthopoulou A, Skendros P, Apostolidou E, Kourtzelis I, Drosos GI, Boumpas DT, Ritis K. Neutrophil extracellular trap formation is associated with IL-1β and autophagy-related signaling in gout. PloS One. 2011;6(12):e29318
74. Garcia-Romo GS, Caielli S, Vega B, Connolly J, Allantaz F, Xu Z, Punaro M, Baisch J, Guiducci C, Coffman RL. et al. Barrat FJ, Banchereau J, Pascual V. Netting neutrophils are major inducers of type I IFN production in pediatric systemic lupus erythematosus. Sci Transl Med. 2011Mar9;3(73):73ra20
75. Nakazawa D, Tomaru U, Ishizu A. Possible implication of disordered neutrophil extracellular traps in the pathogenesis of MPO-ANCA-associated vasculitis. Clin Exp Nephrol. 2013Oct;17(5):631-3
76. Mukhtyar C, Guillevin L, Cid MC, Dasgupta B, de Groot K, Gross W, Hauser T, Hellmich B, Jayne D, Kallenberg CGM. et al. EULAR recommendations for the management of primary small and medium vessel vasculitis. Ann Rheum Dis. 2009Mar;68(3):310-7
77. McDonald B, Urrutia R, Yipp BG, Jenne CN, Kubes P. Intravascular neutrophil extracellular traps capture bacteria from the bloodstream during sepsis. Cell Host Microbe. 2012Sep13;12(3):324-33
78. Papayannopoulos V, Staab D, Zychlinsky A. Neutrophil elastase enhances sputum solubilization in cystic fibrosis patients receiving DNase therapy. PloS One. 2011;6(12):e28526
79. Cao X, Lin W. Clinical study of renal impairment in patients with propylthiouracil-induced small-vessel vasculitis and patients with primary ANCA-associated small-vessel vasculitis. Exp Ther Med. 2013Jun;5(6):1619-22
80. Nakazawa D, Tomaru U, Suzuki A, Masuda S, Hasegawa R, Kobayashi T, Nishio S, Kasahara M, Ishizu A. Abnormal conformation and impaired degradation of propylthiouracil-induced neutrophil extracellular traps: implications of disordered neutrophil extracellular traps in a rat model of myeloperoxidase anti-neutrophil cytoplasmic antibody-associated vasculitis. Arthritis Rheum. 2012Nov;64(11):3779-87
Author contact
![]() Corresponding author: Borros Arneth, Institute of Laboratory Medicine and Pathobiochemistry, Molecular Diagnostics, University Hospital of the Universities of Giessen and Marburg UKGM, Justus Liebig University Giessen, Giessen, Germany, Feulgenstr. 12, 35392 Giessen, Germany. Tel.: +49 641 985 59246; Fax.: +49 641 985 41556; E-mail: borros.arnethmed.uni-giessen.de.
Corresponding author: Borros Arneth, Institute of Laboratory Medicine and Pathobiochemistry, Molecular Diagnostics, University Hospital of the Universities of Giessen and Marburg UKGM, Justus Liebig University Giessen, Giessen, Germany, Feulgenstr. 12, 35392 Giessen, Germany. Tel.: +49 641 985 59246; Fax.: +49 641 985 41556; E-mail: borros.arnethmed.uni-giessen.de.