Impact Factor ISSN: 1449-1907
- Issue 9; 2026
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Volume 23; 2026
- Past Issues
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and methods
Results
Discussion
Conclusions
Abbreviations
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2021; 18(6):1456-1464. doi:10.7150/ijms.51256 This issue Cite
Research Paper
LY3214996 relieves acquired resistance to sorafenib in hepatocellular carcinoma cells
Yongfang Ma1,2*, Ruyue Xu1,2*, Xueke Liu1,3*, Yinci Zhang1,2*, Li Song1,2, Shuyu Cai1,2, Shuping Zhou1, Yinghai Xie1, Amin Li1,2, Weiya Cao1,2, Xiaolong Tang1,2 ![]()
1. Medical school, Anhui University of Science and Technology, Huainan 232001, China.
2. Institute of Environment-friendly Materials and Occupational Health of Anhui University of Science and Technology (Wuhu), Wuhu, 241003, China.
3. Department of Clinical Laboratory Medicine, The Fourth Affiliated Hospital, Zhejiang University School of Medicine, Yiwu 322000, China.
*These authors contributed equally to this work.
Received 2020-7-29; Accepted 2021-1-4; Published 2021-1-29
Abstract

Background: Sorafenib, an oral multi-kinase inhibitor of rapidly accelerated fibrosarcoma; vascular endothelial growth factor receptor-2/3, platelet-derived growth factor receptor, c-Kit, and Flt-3 signaling, is approved for treatment of advanced hepatocellular carcinoma (HCC). However, the benefit of sorafenib is often diminished because of acquired resistance through the reactivation of ERK signaling in sorafenib-resistant HCC cells. In this work, we investigated whether adding LY3214996, a selective ERK1/2 inhibitor, to sorafenib would increase the anti-tumor effectiveness of sorafenib to HCC cells.
Methods: The Huh7 cell line was used as a cell model for treatment with sorafenib, LY3214996, and their combination. Phosphorylation of the key kinases in the Ras/Raf/MAPK and PI3K/Akt pathways, protein expression of the cell cycle, and apoptosis migration were assessed with western blot. MTT and colony-formation assays were used to evaluate cell proliferation. Wound-healing assay was used to assess cell migration. Cell cycle and apoptosis analyses were conducted with flow cytometry.
Results: LY3214996 decreased phosphorylation of the Ras/Raf/MAPK and PI3K/Akt pathways, including p-c-Raf, p-P90RSK, p-S6K and p-eIF4EBP1 activated by sorafenib, despite increased p-ERK1/2 levels. LY3214996 increased the anti-proliferation, anti-migration, cell-cycle progression, and pro-apoptotic effects of sorafenib on Huh7R cells.
Conclusions: Reactivation of ERK1/2 appears to be a molecular mechanism of acquired resistance of HCC to sorafenib. LY3214996 combined with sorafenib enhanced the anti-tumor effects of sorafenib in HCC. These findings form a theoretical basis for trial of LY3214996 combined with sorafenib as second-line treatment of sorafenib-resistant in advanced HCC.
Keywords: sorafenib, LY3214996, hepatocellular carcinoma, Ras/Raf/MAPK pathway, acquired resistance
Introduction
Hepatocellular carcinoma (HCC) is the sixth commonest cancer and the fourth leading cause of cancer-related death in the world, as reported by the International Agency for Research on Cancer [1-2]. HCC, which occurs usually in the setting of chronic liver disease and cirrhosis, makes up about 90% of all cases of primary liver cancer. The curative treatment options for HCC are surgical resection, liver transplantation, and local ablative therapies. Although these measures are effective for early-stage HCC, around 80% of cases are diagnosed in advanced stages when the treatment options are limited. Thus, the prognosis of advanced HCC is poor, with median overall survival time less than one year [3]. Systemic therapy is a hopeful but unproven approach for extending the lives of HCC patients.
Sorafenib, an oral multi-target, multi-kinase inhibitor, is the most widely used systemic chemotherapy for HCC. It was approved as a first-line agent for unresectable or advanced disease by the United States Food and Drug Administration in 2007 [4-5]. On the one hand, sorafenib inhibits the receptor tyrosine kinases c-Kit and Flt-3 [6] and the serine/threonine kinases, including Raf kinases involved in the Raf/MEK/ERK pathway [7], which leads to inhibition of tumor-cell proliferation. On the other hand, sorafenib inhibits the receptor tyrosine kinase, including vascular endothelial growth factor receptor-2/3 and platelet-derived growth factor receptor, which inhibits tumor angiogenesis [8]. Despite encouraging progress in the treatment of HCC with sorafenib, response rates for patients with advanced disease are poor, largely because of acquired resistance to the drug. Other obstacles, including epithelial-mesenchymal transition, Ras/Raf/MAPK, PI3K/Akt, JAK/STAT pathways, and hypoxia also contribute to the failure of sorafenib in HCC patients [9-11]. Hence, new treatment options for advanced HCC are needed.
Recently, aberrant rapidly accelerated fibrosarcoma (Raf) /mitogen-activated protein kinase, MEK/extracellular signal-regulated kinase, and ERK signaling pathway activation have been identified as central for cancer growth, motility, and survival. The Ras/Raf/MAPK cascade participates in cell-cycle regulation, apoptosis, and cell differentiation [15]. The cascade also plays an important role in apoptosis by phosphorylating apoptosis-regulating factors, such as Bad, Bim, Mcl-1, caspase 9, and the controversial Bcl-2 [16]. Consequently, drugs have been developed to inhibit Ras/Raf/MAPK signals in cancer, and Raf and MEK inhibitors have been approved for clinical use [17-18]. The combination of Raf /MEK inhibition seems to improve progression-free survival compared to survival with Raf or MEK monotherapy [19]. Despite the success of these treatments, almost all HCC patients develop resistance to the drugs [20]. Many studies on mechanisms of resistance to Raf/MEK inhibition have been described. The most common mechanisms activate MAPK pathways through multiple methods, including alternate splicing B-RAF, N-RAS, or MEK1/2 mutations, upregulation of MAPK38, clinical drug resistance mechanisms, and/or receptor tyrosine kinase signaling [21-22]. Since the pathway from Raf to MEK to ERK is linear, ERK1/2 itself has been a target, and all these changes are focused on the continuous activation of ERK. Hence, the clinical development of small-molecule ERK inhibitors has aroused interest. LY3214996, a potent, selective, ATP-competitive ERK inhibitor, inhibits the pharmacodynamic biomarker, phospho-P90RSK, which may be the mechanism of its antitumor activities [23].
In this study, we investigated the mechanism of acquired resistance to sorafenib in HCC, and we combined LY3214996 with sorafenib to restore chemosensitivity of drug-resistant HCC cells. Our results provide theoretical basis for combination therapy of sorafenib and ERK1/2 inhibitors in treatment of HCC.
Materials and methods
Reagents and antibodies
Sorafenib was obtained from MedChem Express (Monmouth Junction, NJ, USA). ERK1/2 inhibitor (LY3214996) was purchased from Selleckchem (Houston, TX, USA). Antibodies to cyclin D1 and phospho-Rb were purchased from Abcam Biological Technology (USA). Phospho-ERK1/2 Kit (#9911) and antibodies to ERK1/2, Caspase-3, Cleaved Caspase-3, Caspase-9, Cleaved Caspase-9, PARP, Cleaved PARP, PI3K110β, Akt, phospho-Akt (Ser473), mTOR, phospho-mTOR (Ser2481), Bim, Bad, Bak, Bax, P70S6K, S6K, β-actin, phospho-S6K, phospho-P70S6K and secondary horseradish peroxidase (HRP)-conjugated goat anti-rabbit and anti-mouse antibodies were purchased from Cell Signal Technology (Danvers, MA, USA).
Cell lines and cell culture
Huh7, a human HCC cell line, was purchased from American Type Culture Collection (Rockville, MD, USA). Huh7R, an acquired sorafenib-resistant HCC cell line, was established from Huh7 cells as follows: When the cells were in the logarithmic growth phase, the culture medium was changed, and a lower concentration of sorafenib was added for 24 h. Cell inheritance was performed, and stimulation with this concentration was repeated until it was stable. When the sorafenib concentration reached 4-5 times the IC50 value of the sensitive strain, the resistant strain was obtained. The two cell lines were cultured in RPMI-1640 (Hyclone, Salt Lake City, UT, USA), supplemented with 15% fetal bovine serum (Sijiqing Bioengineering Materials, Hangzhou, China) and incubated at 37 °C in 5% CO2.
Cell viability assay
Cells (10,000 cells per well) were seeded into 96-well plates, allowed to adhere overnight, and exposed to a range of drug concentrations. After 12, 24, 48, and 72 h, 10 µl of 5 mg/ml MTT dissolved in PBS was added to each well and incubated for 4 h at 37 °C. The medium was aspirated, and 100 µl of dimethyl sulfoxide was added to each well and shaken for 15 min. Absorbance was measured at 490 nm. IC50 value was calculated by GraphPad Prism Version 5.0 software.
Colony-formation assay
Cells were plated in six-well plates at a density of 2×103 cells per well for 24 h, and drug experiments as previously designed were carried out. After two weeks, when cellular clones were visible, the cells were fixed in 4% paraformaldehyde for 15 min, and then stained with 0.1% crystal violet for 30 min. The 6-well plates were thoroughly washed with water for counting of the clones. The rate of colony formation reflects two important characteristics of cell population: dependence and proliferation ability. All experiments were done in triplicate.
Wound-healing assay
Cells (2×105 cells/well) were plated overnight in 6-well plates with 15% fetal bovine serum medium. Injured cells were removed by washing with PBS. Serum-free medium containing drugs was added to corresponding wells and incubated at 37 °C in a humidified atmosphere in 5% CO2. The width of the scratched area after 24 and 48 h was recorded by pictures taken with a light microscope.
Cell-cycle assay
Cells were digested with 0.25% trypsin-EDTA after being incubated with various drug combinations for 24 h, washed with PBS three times, and fixed with 70% pre-cooled ethanol. Before testing, the cells were rewashed by PBS, 50 μg/ml propidium iodide, and 100 μg/ml RNase A were added and reacted in the dark for 30 min at 37 °C. Flow cytometry analysis was conducted (with BD FACSCalibur, USA); the distribution of cells at cell-cycle stages was assessed with ModFit Version 3.0 software (Verity Software House, Topsham, ME).
Apoptosis assay
After drug treatment for 24 h, cells from the experimental groups were collected. The cells were washed with cold PBS, resuspended in 500 µL binding buffer containing 5 µl propidium iodide and 10 µl FITC-labelled Annexin-V, and incubated for 15 min in the dark at room temperature. Cell apoptosis was detected with Flow cytometry analysis (BD Biosciences), and apoptotic rate was measured with FlowJo Version 7.6.1 software (FlowJo, Ashland, OR).
Western blot
Whole cell lysates were obtained with the combination of radio immunoprecipitation assay buffer (Beyotime, Jiangsu, China) and a mixture of protease and phosphatase inhibitor (Beyotime, Jiangsu, China). Protein concentration was measured with BCA protein assay kit (Biosharp, Hefei, China). Supernatants per sample were aliquoted, mixed with loading buffer, separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), and transferred to a polyvinylidene fluoride membrane. The membranes were blocked with 5% skim-milk, primary antibodies were applied and reacted at 4 °C for 24 h, and the secondary antibody was applied and reacted for 1 h at 37 °C. Visualization of the signals was clarified with Western ECL substrate (Bio-Rad, Hercules, CA). Protein expression was quantified with Image J Version 1.48 software (NIH, Bethesda, MD) with β-actin as a loading control.
Statistical analysis
All in vitro assay results represent three replicates of three independent experiments performed under the same conditions. All data were expressed as mean values ± standard deviation. Statistical tests were performed on the paired values with Student's t-test; p<0.05 was considered statistically significant.
Results
PI3K/Akt and Ras/Raf/MAPK pathways were stably activated in Huh7R cells
To learn the activation status of PI3K/Akt and Ras/Raf/MAPK pathways in Huh7R and Huh7 cells, we analyzed the phosphorylation levels of the main kinases of the related pathways in the two cells lines. Western blot results revealed that the expression of p-Akt (Ser473), p-mTOR (Ser2481), p-S6K, p-eIF4EBP1, p-c-Raf (Ser289), p-MEK (Ser217/221), p-ERK (Thr202/Tyr204), and p-P90RSK (Ser380) in Huh7R cells was higher than in Huh7 cells (Fig. 1A and B), a result that was evidence that the above pathways were activated in Huh7R cells. Next, Huh7R cells were treated with sorafenib for 0 h, 12 h, 24 h, 36 h, 48 h, and 72 h at the same concentration to assess for the activated form of the two pathways. Western blot results showed that the phosphorylation levels of major kinases in the two pathways were decreased at 12 h compared with 0 h, reached the lowest level at 24 h, and were in stable activation from 24 h to 72 h (Fig. 1C and D).
PI3K/Akt and Ras/Raf/MAPK pathways were stably activated in Huh7R cells. (A and B) Major kinase levels of PI3K/Akt and Ras/Raf/MAPK pathways in Huh7 and Huh7R cells were examined by western blot. (C and D) Kinase expression levels in related pathways in Huh7R cells treated with sorafenib at various times were detected by western blot. Data expressed as mean ± SD of three independent experiments (*p<0.05; **p<0.01; ***p<0.001).
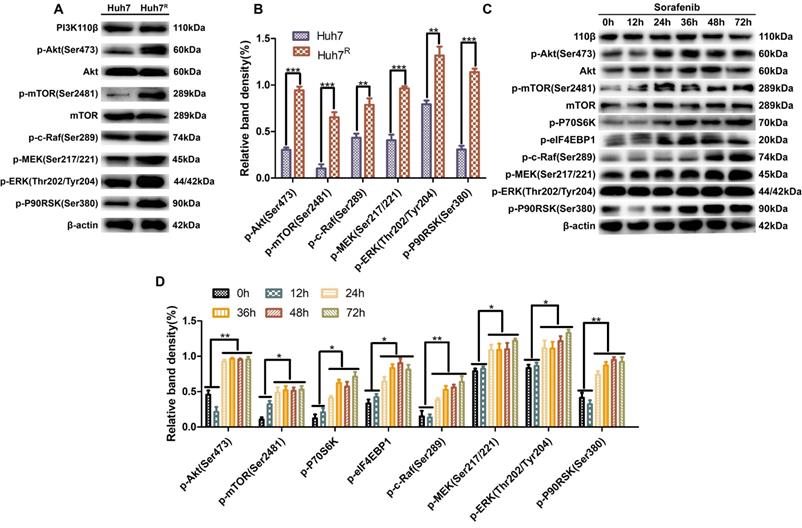
LY3214996 enhanced sorafenib anti-proliferative effects in Huh7R cells
To detect the inhibitory effects on Huh7R cells treated with sorafenib, LY3214996 or a combination of the two, we measured cell viabilities by MTT assays. IC50s of sorafenib on Huh7 and Huh7R cells were obtained (Fig. 2A). There was a dose- and time-dependent reduction in the cell viability of Huh7R cells treated with sorafenib, LY3214996, and the combination of the two drugs (Fig. 2B and C). Compared with sorafenib or LY3214996 monotherapy, the combination had a stronger inhibiting effect on cell viability according to IC50 values. The results of colony formation assays also showed that sorafenib plus LY3214996 had stronger inhibitory effect on clone formation than did the control group and any single-agent treatment (Fig. 2D and E), which confirmed that LY3214996 enhanced the effects of sorafenib to suppress cell viability in Huh7R cells.
LY3214996 enhanced sorafenib anti-proliferative effects in Huh7R cells. (A) MTT assays detected cell viability of Huh7 and Huh7R cells incubated with various concentrations of sorafenib for 24 h. (B) Cell viability of Huh7R cells treated with sorafenib, LY3214996, and the combination after 24 h were assessed with MTT assays. (C) Cell viability of Huh7R cells treated with sorafenib, LY3214996, or the combination for 12 h, 24 h, and 48 h determined with MTT assays. (D and E) Colony formation assays were used to determine the clonogenic capacity of Huh7R cells treated with sorafenib, LY3214996, or a combination of the two. Data are expressed as mean ± SD of three independent experiments (*p<0.05; **p<0.01; ***p<0.001).
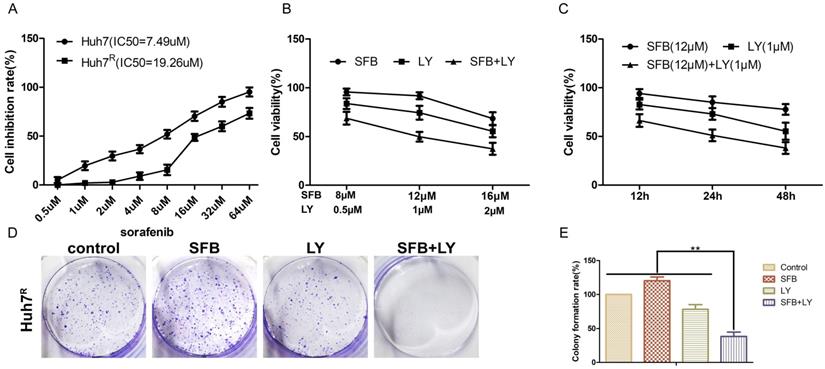
LY3214996 increased sorafenib inhibition of Huh7R cellular migration
To verify whether LY3214996 enhanced the inhibitory effect of sorafenib on Huh7R cell migration, Huh7R cells were treated with sorafenib, LY3214996, or a combination of the two for 12 h and 24 h, using the wound healing test. Compared to the control group, the wound size of cells treated with sorafenib or LY3214996 were 92.4% and 72.1% at 12 h and 67.3% and 61.4% at 24 h, respectively (p<0.05). The migration inhibition rates of LY3214996 combined with sorafenib were 94.1% at 12 h and 93.9% at 24 h (Fig. 3A and B), which was significantly higher than that of either single agent group (p<0.05). Western blot results revealed that after treatment with sorafenib combined with LY3214996, the expressions of MMP-2 and MMP-9 in Huh7R cells were significantly lower than with monotherapy treatment (Fig. 3C and D) (p<0.05). These results indicated that the LY3214996 increased sorafenib inhibition of Huh7R cell migration.
LY3214996 increased sorafenib inhibition of Huh7R cell migration. (A and B) Wound healing assays were carried out to assess cell migration of Huh7R cells treated with sorafenib, LY3214996, or the combination of the two for 12 h and 24 h. (C and D) Related proteins of tumor-cell migration (MMP-2 and MMP-9) were detected by western blot. (E) A proposed working model of how LY3214996 acts on the Ras/Raf/MAPK pathway to affect migration of Huh7R cells. Data are expressed as mean ± SD of three independent experiments (* p < 0.05; **p < 0.01; ***p < 0.001).
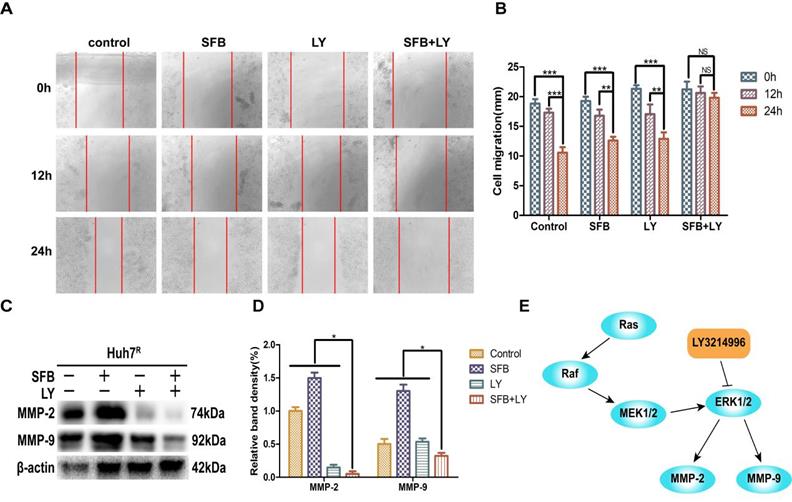
LY3214996 combined with sorafenib induced cell-cycle arrest in the G0/G1 phase
Abnormal cell-cycle progression causes unrestricted cell division, leading to continuous proliferation, which is a key driver of carcinogenesis [24]. Thus, we assessed the effects of each treatment on the cell-cycle distribution by flow cytometry. After treatment for 24 h, the G0/G1 phase cell number percentages of cells treated with sorafenib plus LY3214996 (62.0%) (p<0.05) were more than with sorafenib (45.4%) or LY3214996 alone (50.0%) and the control group (40.0%) (Fig. 4A and B). Moreover, the combined treatment significantly increased the percentage of sub-G1 phase cells compared with the percentage in cells treated with either drug alone, which is evidence that the combination of two the drugs inhibited cell proliferation and promoted apoptosis.
LY3214996 combined with sorafenib induced cell-cycle arrest in the G0/G1 phase. (A) and (B) Huh7R cells accumulated at the G0/G1 phase after exposure to sorafenib (12 µM), LY3214996(1 µM), or the combination (sorafenib 12 µM, LY3214996 1 µM) for 24 h were measured by flow cytometry. (C) and (D) Cell- cycle protein expression (p-GSK3β, cyclin D1, and p-Rb) were quantified with western blot. Data expressed as mean ± SD of three independent experiments (*p<0.05; **p<0.01; ***p<0.001).
Western blot results showed that the expression of cyclin D1 and p-Rb, two of the proteins that promote cell-cycle progression from G1 to S phase, were suppressed in LY3214996-treated Huh7R cells. Furthermore, sorafenib combined with LY3214996 treatment led to decreased levels of cyclin D1 and p-Rb in the Huh7R cells. Besides, the expression level of upstream protein p-GSK-3β was decreased after the combined treatment (Fig. 4C and D). These data indicate that combined sorafenib and LY3214996 treatment resulted in a pronounced cell-cycle arrest G0/G1 phase.
LY3214996 increased sorafenib-induced apoptosis in Huh7R cells
To investigate the effect of combination treatment on Huh7R apoptosis, Huh7R cells were incubated with sorafenib (12 µM), LY3214996 (1 µM), or the combination, and the cells were harvested after 24 h and analyzed with flow cytometry. As illustrated in Figure 5, compared with the control group, the apoptosis rates of cells treated with sorafenib or LY3214996 were 5.8% and 11% (p<0.05), respectively; the apoptosis rate of cells treated with combined drugs was 17.7%, which was higher than with either single agent or control values (Fig. 5A and B) (p<0.05).
LY3214996 increased sorafenib-induced apoptosis in Huh7R cells. (A) and (B) Apoptosis-related protein expression of Bcl-xL, Bax, Bak, Bad, Bim, caspase-3, -7, -9, and PARP were measured with western blot. (C) and (D) Cells were treated with sorafenib (12 µM), LY3214996 (1 µM), or in combination for 24 h. Apoptosis of Huh7R cells was detected by flow cytometry with annexin V-FITC/propidium iodide staining. (E) Signal mechanism model of LY3214996 inducing mitochondrial apoptosis in Huh7R cells. Data expressed as mean ± SD of three independent experiments (*p<0.05; **p<0.01; ***p<0.001).
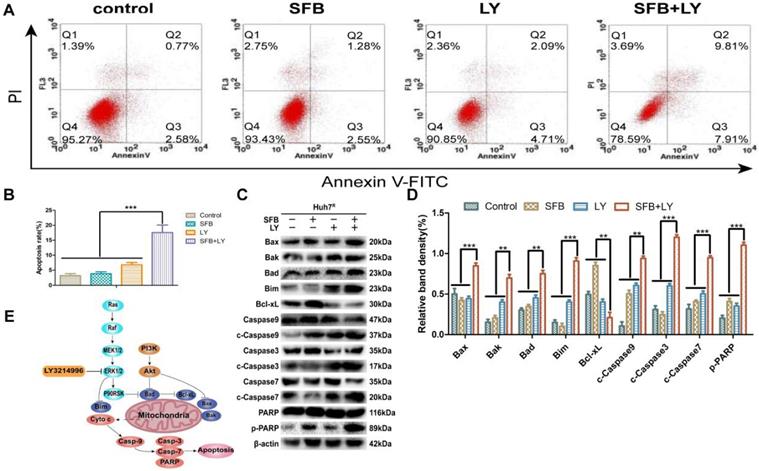
Immunoblotting analysis was performed to assay for apoptosis-related molecules to help understand the mechanism of the mitochondrial apoptotic pathway. The results revealed that levels of anti-apoptotic protein (Bcl-xL) in the combined treatment group were significantly down-regulated (p<0.05), while the key death-effector proteins (Bax and Bak), BH3-only proteins (Bad, Bim), and downstream proteins (caspase-3, -7, -9 and PARP) were significantly increased (Fig. 5C and D). These results suggest that sorafenib has a pro-apoptotic effect on Huh7R cells when co-treated with LY3214996.
LY3214996 enhanced anti-tumor effect by blocking Ras/Raf/MAPK pathway in Huh7R cells
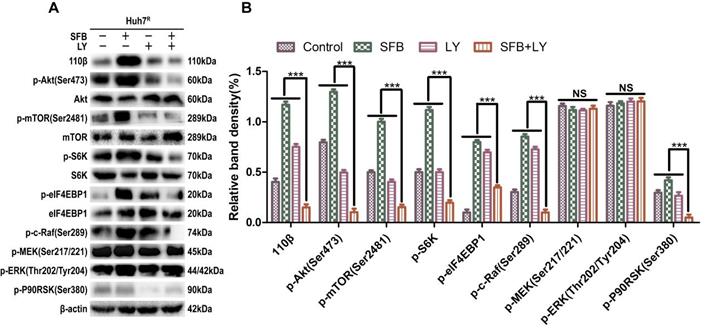
Next, we explored further the anti-tumor mechanism of LY3214996 combined with sorafenib in Huh7R cells. Western blot was performed to detect the levels of the major proteins, Ras/Raf/MAPK, in Huh7R cells treated with LY3214996 or sorafenib monotherapy or in combination for 24 h. The results revealed that sorafenib treatment increased the values of p-Akt, p-mTOR, p-c-Raf, p-ERK1/2, p-S6K, p-eIF4EBP1, and p-P90RSK compared with values in the control group. However, the combination of sorafenib and LY3214996 led to suppression of p-Akt, p-mTOR, p-S6K, p-c-Raf, and p-P90RSK compared with control or sorafenib and LY3214996 treatment alone. Interestingly, p-ERK1/2 and p-MEK1/2 had an opposite result (Fig. 6A and B). Reactivation of p-ERK1/2 mediated by LY3214996 may be due to inhibition of p-c-Raf (S289/S296/S301), which are phosphorylation sites that inhibit feedback activation of ERK, an effect that suggests that the increase in p-ERK1/2 is due to the activation of c-Raf [25]. These results indicate that LY3214996 enhanced anti-tumor effect by blocking the Ras/Raf/MAPK pathway and interfering with crosstalk of the PI3K/Akt pathway.
LY3214996 increases the anti-tumor effect by blocking Ras/Raf/MAPK pathway in Huh7R cells. (A) and (B) After incubation with sorafenib (12 µM), LY3214996 (1 µM), or combination for 24 h, cells were lysed, and the designated proteins of Ras/Raf/MAPK and PI3K/Akt pathways were detected by western blot analysis. Data expressed as mean ± SD of three independent experiments (*p<0.05; **p<0.01; ***p<0.001).
Discussion
Chemotherapy is an important option for patients with advanced HCC, even though the available chemotherapeutic drugs have limited benefits and are accompanied by side effects [26]. Sorafenib, as an oral multi-targeted kinase inhibitor, is well tolerated and significantly prolongs patient survival, which makes it the first choice for patients with advanced HCC [27]. However, in many clinical studies and in practice, acquired resistance to sorafenib has resulted in a low response rate and limited clinical efficacy after a few treatment cycles [28]. Therefore, it is necessary to explore agents that can overcome the resistance and improve the anti-tumor effect of sorafenib.
Activation of the PI3K/Akt pathway is highly related to the resistance of HCC to sorafenib [29-30]. Sorafenib exerts its anti-tumor effect mainly by inhibiting the Ras/Raf/MAPK pathway, but crosstalk activates the PI3K/Akt pathway during this process [31]. Despite feedback activation of p-ERK1/2, LY3214996 still inhibits the downstream signals of ERK signaling [32]. In this study, we found that the levels of p-Akt and p-ERK1/2 in Huh7R cells were higher than in Huh7 cells, while p-Akt and p-P90RSK were downregulated by a combination treatment of LY3214996 and sorafenib. This result suggested that that the PI3K/Akt pathway was activated with sorafenib treatment, and inhibited ERK1/2 could suppress Akt activation. Due to ERK or RSK activation directly mediating the PI3K/Akt/mTOR pathway [33-34], leading to compensated proliferation and anti-apoptotic effects, blocking the up-regulation of PI3K/Akt/mTOR pathway induced by ERK activation is an effective strategy to decrease sorafenib resistance and improve efficacy of the drug.
Some studies have shown that sorafenib-resistant HCC cells have enhanced migration and invasion ability by activating epithelial-mesenchymal transition after long-term sorafenib treatment [35-36]. MMP2 and MMP9 participate in angiogenesis by destroying basal-layer molecules and remodeling the extracellular matrix during angiogenesis [37]. The MMPs in the extracellular matrix participate in the acquisition of migration characteristics and make it easier for tumor cells to invade surrounding tissues and metastasize to secondary sites [38]. It has been reported that ERK1/2 can regulate MMP-2 and MMP-9 expression [39]. Similar results were found in our study, and when Huh7R cells were exposed to the combination of LY3214996 and sorafenib, the wound size in wound-healing assays was significantly more than with sorafenib or LY3214996 monotherapy, and the expression levels of MMP-2 and MMP-9 were correspondingly reduced.
PI3K/Akt signaling is closely related to the mitochondrial apoptosis pathway. P-Akt phosphorylated Bad, a member of the Bcl-2 family, which was followed by Bcl-xL induced anti-apoptosis [40]. LY3214996 plus sorafenib treatment inhibited p-Akt and p-P90RSK but upregulated Bad and Bim and induced a cascade of mitochondrial apoptotic proteins, caspase-9, caspase-3, and caspase-7. Thus, sorafenib and LY3214996 inhibited the Ras/Raf/MAPK and PI3K/Akt pathways, leading to activation of the mitochondrial apoptotic pathway. LY3214996 evidently enhanced the anti-tumor effect of sorafenib by inhibiting the Ras/Raf/MAPK pathway to promote the mitochondrial apoptotic pathway.
LY3214996 has induced G1 cell-cycle arrest in cell lines of melanoma, colorectal cancer, pancreatic cancer, and non-small cell lung cancer [32]. Our results revealed that combined treatment of LY3214996 and sorafenib suppressed the expression of cyclin D1 and p-Rb, then increased the proportion of cells in the G0/G1 phase to enhance the anti-proliferative effect of sorafenib on Huh7R cells, as others have reported also [28].
Overall, we found that acquired resistance to sorafenib in Huh7R cells resulted in limited effectiveness in treatment. One of the most important mechanisms of this result was the activation of PI3K/Akt and Ras/Raf/MAPK pathways. Inhibition of ERK simultaneously blocked the downstream MAPK pathway and crosstalk with PI3K/Akt, which provides a theoretical basis for overcoming sorafenib resistance in patients with advanced HCC. Since the experiment was limited to in vitro studies, our future work will focus on in vivo research, and more signaling pathways will be assessed. We believe that sorafenib combined with LY3214996 can be a promising strategy in the treatment of sorafenib-resistant HCC.
Conclusions
The present study demonstrated that PI3K/Akt and MAPK/ERK pathways are abnormally activated in sorafenib-resistant HCC cells. Sorafenib combined with LY3214996 inhibited these two pathways to enhance anti-tumor effects by inducing apoptosis and inhibiting cell proliferation. These findings form a theoretical basis for trial of LY3214996 combined with sorafenib as second-line treatment of sorafenib-resistant, advanced HCC.
Abbreviations
HCC: hepatocellular carcinoma; SFB: sorafenib; MEK: mitogen-activated protein kinase; ERK: extracellular signal-regulated kinase; HRP: horseradish peroxidase; PBS: phosphate buffered saline; FITC: fluorescein isothiocyanate; SDS-PAGE: sodium dodecyl sulfate-polyacrylamide gel electrophoresis.
Acknowledgements
The National Natural Science Fund of China (82071862, 81872017, 81572431), University Natural Science Research Project of Anhui Province (KJ2018ZD011, KJ2018A0097, KJ2019A0093), Anhui Provincial Science and Technology program (1604a0802094, 202004j07020053) and the Institute of Environment-friendly Materials and Occupational Health of Anhui University of Science and Technology (Wuhu) (ALW2020YF17) funded this research.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Ferlay J, Colombet M, Soerjomataram I. et al. Estimating the global cancer incidence and mortality in 2018: GLOBOCAN sources and methods. Int J Cancer. 2019;144:1941-53
2. Li X, Li C, Zhang L. et al. The significance of exosomes in the development and treatment of hepatocellular carcinoma. Mol Cancer. 2020;19:1
3. De Sanctis V, Soliman AT, Daar S. et al. A Concise Review on the Frequency, Major Risk Factors and Surveillance of Hepatocellular Carcinoma (HCC) in β-Thalassemias: Past, Present and Future Perspectives and the ICET-A Experience. Mediterr J Hematol Infect Dis. 2020;12:e2020006
4. Llovet JM, Ricci S, Mazzaferro V. et al. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med. 2008;359:378-90
5. Faivre S, Rimassa L, Finn RS. Molecular therapies for HCC: Looking outside the box. J Hepatol. 2020;72:342-52
6. Mokdad AA, Hester CA, Singal AG. et al. Management of hepatocellular in the United States. Chin Clin Oncol. 2017;6:21
7. Zhang Y, Xie C, Li A. et al. PKI-587 enhances chemosensitivity of oxaliplatin in hepatocellular carcinoma through suppressing DNA damage repair pathway (NHEJ and HR) and PI3K/AKT/mTOR pathway. Am J Transl Res. 2019;11:5134-49
8. Méndez-Blanco C, Fondevila F, García-Palomo A. et al. Sorafenib resistance in hepatocarcinoma: role of hypoxia-inducible factors. Exp Mol Med. 2018;50:1-9
9. Ray EM, Sanoff HK. Optimal therapy for patients with hepatocellular carcinoma and resistance or intolerance to sorafenib: challenges and solutions. J Hepatocell Carcinoma. 2017;4:131-8
10. Zhu YJ, Zheng B, Wang HY. et al. New knowledge of the mechanisms of sorafenib resistance in liver cancer. Acta Pharmacol Sin. 2017;38:614-22
11. Goyal L, Muzumdar MD, Zhu AX. Targeting the HGF/c-MET pathway in hepatocellular carcinoma. Clin Cancer Res. 2013;19:2310-8
12. Buscà R, Pouysségur J, Lenormand P. ERK1 and ERK2 Map Kinases: Specific Roles or Functional Redundancy? Front Cell Dev Biol. 2016;4:53
13. Tang X, Li A, Xie C. et al. The PI3K/mTOR dual inhibitor BEZ235 nanoparticles improve radiosensitization of hepatoma cells through apoptosis and regulation DNA repair pathway. Nanoscale Res Lett. 2020;15:63
14. Asati V, Mahapatra DK, Bharti SK. PI3K/Akt/mTOR and Ras/Raf/MEK/ERK signaling pathways inhibitors as anticancer agents: Structural and pharmacological perspectives. Eur J Med Chem. 2016;109:314-41
15. Tang X, Chen L, Li A. et al. Anti-GPC3 antibody-modified sorafenib-loaded nanoparticles significantly inhibited HepG2 hepatocellular carcinoma. Drug Deliv. 2018;25:1484-94
16. Steelman LS, Pohnert SC, Shelton JG. et al. JAK/STAT, Raf/MEK/ERK, PI3K/Akt and BCR ABL in cell cycle progression and leukemogenesis. Leukemia. 2004;18:189-218
17. Caunt CJ, Sale MJ, Smith PD. et al. MEK1 and MEK2 inhibitors and cancer therapy: the long and winding road. Nat Rev Cancer. 2015;15:577-92
18. Holderfield M, Deuker MM, McCormick F. et al. Targeting RAF kinases for cancer therapy: BRAF-mutated melanoma and beyond. Nat Rev Cancer. 2014;14:455-67
19. Flaherty KT, Infante JR, Daud A. et al. Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. N Engl J Med. 2012;367:1694-703
20. Trunzer K, Pavlick AC, Schuchter L. et al. Pharmacodynamic effects and mechanisms of resistance to vemurafenib in patients with metastatic melanoma. J Clin Oncol. 2013;31:1767-74
21. Johannessen CM, Boehm JS, Kim SY. et al. COT drives resistance to RAF inhibition through MAP kinase pathway reactivation. Nature. 2010;468:968-72
22. Wagle N, Van Allen EM, Treacy DJ. et al. MAP kinase pathway alterations in BRAF-mutant melanoma patients with acquired resistance to combined RAF/MEK inhibition. Cancer Discov. 2014;4:61-8
23. Zhou LB, Zhou YQ, Zhang XY. Blocking VEGF signaling augments interleukin-8 secretion via MEK/ERK/1/2 axis in human retinal pigment epithelial cells. Int J Ophthalmol. 2020;13:1039-45
24. Asghar U, Witkiewicz AK, Turner NC. et al. The history and future of targeting cyclin-dependent kinases in cancer therapy. Nat Rev Drug Discov. 2015;14:130-46
25. Dougherty MK, Müller J, Ritt DA. et al. Regulation of Raf-1 by direct feedback phosphorylation. Mol Cell. 2005;17:215-24
26. Kumari R, Sharma A, Ajay AK. et al. Mitomycin C induces bystander killing in homogeneous and heterogeneous hepatoma cellular models. Mol Cancer. 2009;8:87
27. Llovet JM, Ricci S, Mazzaferro V. et al. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med. 2008;359:378-90
28. Niu L, Liu L, Yang S. et al. New insights into sorafenib resistance in hepatocellular carcinoma: Responsible mechanisms and promising strategies. Biochim Biophys Acta Rev Cancer. 2017;1868:564-70
29. Li A, Zhang R, Zhang Y. et al. BEZ235 increases sorafenib inhibition of hepatocellular carcinoma cells by suppressing the PI3K/AKT/mTOR pathway. Am J Transl Res. 2019;11:5573-85
30. Gan H, Chen L, Sui X. et al. Enhanced delivery of sorafenib with anti-GPC3 antibody-conjugated TPGS-b-PCL/Pluronic P123 polymeric nanoparticles for targeted therapy of hepatocellular carcinoma. Mater Sci Eng C Mater Biol Appl. 2018;91:395-403
31. Mendoza MC, Er EE, Blenis J. The Ras-Erk and Pi3k-mtor pathways: cross-talk and compensation. Trends Biochem Sci. 2011;36:320-8
32. Bhagwat SV, McMillen WT, Cai S. et al. ERK Inhibitor LY3214996 Targets ERK Pathway-Driven Cancers: A Therapeutic Approach Toward Precision Medicine. Mol Cancer Ther. 2020;19:325-36
33. Carriere A, Romeo Y, Acosta-Jaquez HA. et al. Erk1/2 phosphorylate raptor to promote Ras-dependent activation of mtor complex 1 (Mtorc1). J Biol Chem. 2011;286:567-77
34. Zoncu R, Efeyan A, Sabatini DM. Mtor: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol. 2011;12:21-35
35. van Malenstein H, Dekervel J, Verslype C. et al. Long-term exposure to sorafenib of liver cancer cells induces resistance with epithelial-to-mesenchymal transition, increased invasion and risk of rebound growth. Cancer Lett. 2013;329:74-83
36. Chow AK, Ng L, Lam CS. et al. The Enhanced metastatic potential of hepatocellular carcinoma (HCC) cells with sorafenib resistance. PLoS One. 2013;8:e78675
37. Singh D, Srivastava SK, Chaudhuri TK. et al. Multifaceted role of matrix metalloproteinases (MMPs). Front Mol Biosci. 2015;2:19
38. Tang X, Lyu Y, Xie D. et al. Therapeutic Effect of Sorafenib-Loaded TPGS-b-PCL Nanoparticles on Liver Cancer. J Biomed Nanotechnol. 2018;14:396-403
39. Cepeda MA, Evered CL, Pelling JJH. et al. Inhibition of MT1-MMP proteolytic function and ERK1/2 signalling influences cell migration and invasion through changes in MMP-2 and MMP-9 levels. J Cell Commun Signal. 2017;11:167-79
40. Liu X, Xie C, Li A. et al. BEZ235 enhances chemosensitivity of paclitaxel in hepatocellular carcinoma through inhibiting the PI3K/Akt/mTOR pathway. Am J Transl Res. 2019;11:7255-71
Author contact
![]() Corresponding author: Dr. Xiaolong Tang, Anhui University of Science and Technology, 168 Taifeng Street, Huainan 232001, China. E-mail: txljd2006com.
Corresponding author: Dr. Xiaolong Tang, Anhui University of Science and Technology, 168 Taifeng Street, Huainan 232001, China. E-mail: txljd2006com.