Impact Factor ISSN: 1449-1907
- Issue 9; 2026
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Volume 23; 2026
- Past Issues
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Neutrophils
Macrophages
Other innate immune cells in AP
Conclusions and future prospects
Abbreviations
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2021; 18(2):534-545. doi:10.7150/ijms.51618 This issue Cite
Review
The Role of Pancreatic Infiltrating Innate Immune Cells in Acute Pancreatitis
Cheng Peng, Zhiqiang Li ![]() , Xiao Yu
, Xiao Yu ![]()
Department of Hepatopancreatobiliary Surgery, Third Xiangya Hospital, Central South University, Changsha 410013, Hunan, China.
Received 2020-8-6; Accepted 2020-11-13; Published 2021-1-1
Abstract
Acute pancreatitis (AP) is a leading cause of gastrointestinal-related hospital admissions with significant morbidity and mortality. Although the underlying pathophysiology of AP is rather complex, which greatly limits the treatment options, more and more studies have revealed that infiltrating immune cells play a critical role in the pathogenesis of AP and determine disease severity. Thus, immunomodulatory therapy targeting immune cells and related inflammatory mediators is expected to be a novel treatment modality for AP which may improve the prognosis of patients. Cells of the innate immune system, including macrophages, neutrophils, dendritic cells, and mast cells, represent the majority of infiltrating cells during AP. In this review, an overview of different populations of innate immune cells and their role during AP will be discussed, with a special focus on neutrophils and macrophages.
Keywords: acute pancreatitis, immune cell, inflammation, macrophage, neutrophil
Introduction
Acute pancreatitis (AP) is a leading cause of gastrointestinal-related hospital admissions [1]. There are regional and ethnic differences in the etiology of AP, overall, gallstones and alcohol abuse are the two most common causes of AP, while hypertriglyceridemia ranks third place with an increasing trend. Other less common causes include hypercalcemia, cigarette smoking, drug reactions, genetic factors, etc. [2]. Around 80% of cases of AP are mild AP (MAP) [3], with only interstitial changes of the pancreas, which can usually be relieved within 2 weeks, while patients with severe AP (SAP) are associated with persistent organ failure and multi-organ dysfunction die [4]. Currently, it is generally accepted that AP initiates local inflammation and injury in the pancreas; then the inflammation is amplified due to the inflammatory cascade, which ultimately leads to systemic inflammatory response syndrome (SIRS), multiple organ dysfunction syndromes (MODS) or even multiple organ failure (MOF). However, due to an obscure pathophysiological mechanism, AP remains considerable incidence and mortality [5, 6]. Therefore, it is essential to explore the pathogenesis of AP, especially the mechanisms of inflammation and immune response activation.
Immune responses are an integral part of the pathogenesis of pancreatitis [7]. Evidence shows that the excessive systemic inflammation associated with AP is a consequence of uncontrolled or dysregulated activation of the immune system [8]. In the early phase of AP, the pancreatic acinar cell injury occurs in an aseptic environment since the uninflamed pancreas is a sterile organ, which leads to proinflammatory mediators release, immune cells infiltration and sterile inflammation [7]. Therefore, pathogen-associated molecular patterns (PAMPs) play no role in the recruitment and activation of immune cells in early AP [9]. Instead, the necrotized pancreatic acinar cells release various kinds of danger-associated molecular patterns (DAMPs), including high-mobility group box protein 1 (HMGB1), self-DNA, and so on [10, 11]. Subsequently, these DAMPs activate the pattern recognition receptors (PRRs) of infiltrating immune cells to produce more inflammatory mediators, which in turn promote more immune cell infiltration and aggravate inflammation [12].
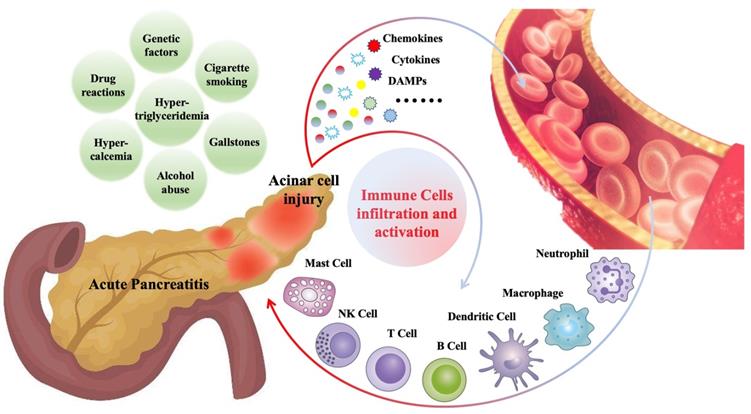
Various immune cells begin to infiltrate the pancreas within minutes after the onset of AP and are closely related to the severity and prognosis of AP [8, 13]. These infiltrated immune cells include innate immune cells, such as macrophages [14], neutrophils [15], dendritic cells [16], mast cells [17], natural killer cells (NK cells) [18], as well as adaptive immune cells, such as T lymphocytes [19], B lymphocytes [19]. Cells of the innate immune system like neutrophil and macrophage represent the majority of infiltrating cells. Generally, immune cell infiltration, as a defense mechanism, is beneficial for disease recovery. However, in some cases, the pathogenic factors cannot be cleared in the short term, and the immune cells related inflammatory response will persist and amplify, which may further aggravate pancreatic damage and contribute to systemic inflammation [20, 21]. Figure 1 is a summary of common causes of AP and mechanisms of immune cell infiltration during AP.
Gallstones, alcohol abuse, hypertriglyceridemia, hypercalcemia, cigarette smoking, drug reactions and genetic factors are the common causes of AP. During AP, injured pancreatic acinar cells release a series of pro-inflammatory mediators (cytokines, chemokines, DAMPs, etc.), which promote the recruitment and activation of immune cells, including innate immune cells such as neutrophils, macrophages, dendritic cells, mast cells, NK cells, and adaptive immune cells such as T cells and B cells.
In this review, an overview of different populations of innate immune cells and their role during AP will be discussed, with a special focus on neutrophils and macrophages.
Neutrophils
Neutrophils and inflammation
Neutrophils, a kind of polymorphonuclear cells with the rod or lobulated nuclei, originating from bone marrow hematopoietic stem cells, accounting for 60-70% of the circulating leucocytes and among the first cells recruited to the inflammatory site [22]. Neutrophils have long been recognized as potent pathogen scavengers, whose cytoplasmic granules contain bactericidal substances such as myeloperoxidase (MPO), acid phosphatase, alkaline phosphatase, lysozyme, and defensins. However, In recent years, increasing evidence suggests that neutrophils also play an important role in sterile inflammation, they are required to clear cell debris produced in the process of tissue damage to restore cell homeostasis [22-25]. During an infection or sterile inflammation, damaged tissue cells may produce chemokines, such as CXC chemokine ligands 1 (CXCL1), CXC chemokine ligands 2 (CXCL2), and CXC chemokine ligands 8 (CXCL8) [22, 26], which can activate neutrophils. Then, under the interaction of a series of integrins including very late antigen-4 (VLA-4), macrophage-1 antigen (Mac-1), lymphocyte function-associated antigen-1 (LFA-1) [26], selectin (P-selectin, E-selectin) [27] and adhesion molecule such as intercellular adhesion molecule-1 (ICAM-1) [28], neutrophils ultimately reach the inflammatory site. Besides, PAMPs released by pathogens and DAMPs released by damaged tissue cells can also directly activate PRRs on neutrophils, and then recruit neutrophils to the inflammatory site [29].
It is generally accepted that the clearance of pathogens and cell fragments is accomplished by phagocytosis and degranulation, two classical functions of neutrophils. However, in 2004, Brinkmann first reported that the production of neutrophil extracellular traps (NETs) also enables neutrophils to do this [30]. NETs are weblike structures released by activated neutrophils into extracellular space, with DNA as its scaffold, which is decorated with histones, MPO, neutrophil elastase (NE), cathepsin G, calreticulin, protease 3, HMGB1, etc. [30, 31]. Since the formation of NETs is usually accompanied by the death of neutrophils, this process is also called NETosis [32]. NETs were initially thought to be a mechanism by which neutrophils clear pathogens, for example, the replication of staphylococcus aureus, salmonella typhimurium, streptococcus pneumoniae, and shigella flexneri can be inhibited by NETs [30, 33-35]. But its critical role in sterile inflammation has also been revealed in recent years, such as deep venous thrombosis [36], lung cancer [37], arthritis [38], AP [25, 39, 40], endogenous particulate-related injury [41], glioma [31], liver ischemia-reperfusion injury [42], ischemic stroke [43], etc.
In summary, neutrophils are important effector cells in the maintenance of immune surveillance and their clearance of pathogens effectively limit the spread of infection and the development of sepsis [44]. Also, neutrophils can be recruited to the sterile inflammatory sites, clearing necrotic tissue and cells, and promoting tissue repair [45]. However, the infiltration of neutrophils is a double-edged sword, lack of resolution or persistent local inflammation may lead to a more aggressive neutrophil response [46] characterized by the destruction of normal tissue and uncontrolled systemic inflammation.
Neutrophils infiltration in the pathogenesis of AP and therapy
A substantial number of studies have suggested that neutrophils are involved in the pathogenesis of AP [25, 27, 28, 39, 40, 47-76] (Figure 2). Measurements of MPO levels in serum [27] or pancreatic tissue [49, 54, 61] can reflect the number and activity of infiltrating neutrophils. The commonly used methods include commercial detection kits [61], enzyme-linked immunosorbent assay (ELISA) [27, 49], and immunofluorescence [54]. In addition, for mice and rats AP models, MPO-positive cells, or Ly6G-positive cells in pancreatic tissues, which are commonly used markers of neutrophils, can also be detected by immunohistochemistry [21, 28, 66].
During AP, damaged pancreatic acinar cells release proinflammatory mediators, leading to the recruitment, activation and functional changes of neutrophils and macrophages: (1) Neutrophils release proteases and ROS, which directly damage pancreatic tissue.The released MMP-9 is a potent trypsinogen activator. In addition, activated neutrophils can form NETs, which promotes trypsinogen activation by phosphorylating STAT3 and leads to pancreatic duct obstruction by forming aggregates. More critically, the formation of NETs will in turn promote more neutrophil infiltration. (2) Macrophages secrete inflammatory mediators MIP-1α, MCP-1, MIF and PAF to directly damage pancreatic tissue, and in turn promote more macrophage infiltration. In AP, inflammatory mediator microenvironment promotes macrophage M1 polarization and inhibits M2 polarization. M1 macrophages secrete IL-1β, IL-6, IL-12 and TNF-α to mediate pancreatic inflammation and injury. Like neutrophils, macrophages are also involved in trypsinogen activation. In addition, AIM2 or NLRP3 inflammasome activation within macrophages triggers macrophage pyroptosis, leading to the rupture of plasma membrane, maturation and release of IL-1β and IL-18, aggravating inflammation and pancreatic injury.
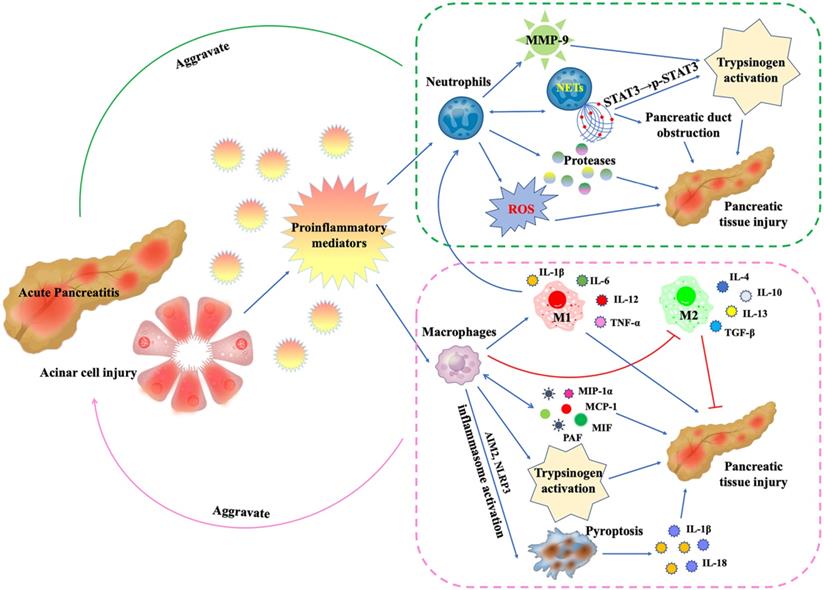
Intrapancreatic activation of trypsinogen is a key event in AP, which leads to acinar cell injury [69]. During the development of AP, neutrophils infiltrating the pancreas play a critical role in pathologic activation of trypsinogen and modulation of inflammation [69, 74]. The depletion of neutrophils by the administration of antineutrophil serum [69] or anti-Gr-1 antibody [74] exhibited a significant reduction in intrapancreatic trypsinogen activation, trypsin-activation peptide (TAP) level, and tissue damage. Neutrophil-derived matrix metalloproteinase-9 (MMP-9) is also a potent activator of trypsinogen activation in pancreatic acinar cells during AP [68]. The accumulation of reactive oxygen species (ROS) and related oxidative stress are also involved in the initiation of AP [71]. The high nicotinamide adenine dinucleotide phosphate (NADPH) oxidase activity of neutrophils makes them the main source of ROS in AP, mediating oxidative damage to the pancreas and promoting trypsinogen activation [69]. In addition, proteases released by neutrophils, such as elastase, can also contribute to tissue damage during AP [47, 58], while the deficiency of Cathepsin C alleviates AP through the reduction of neutrophil elastase activation [50]. Based on the above evidence, we can conclude that neutrophils are closely involved in the initiation and development of AP and are associated with the severity. Therefore, more and more scientists are devoted to exploring the mechanism of neutrophil infiltration in AP and looking for intervention targets.
Genetic or pharmacological inhibition of some specific gene targets, or administration of some medication, can alleviate AP through reducing neutrophils infiltration or NETs formation. The nuclear factor of activated T cells 3 (NFATc3) is an important downstream target of calcineurin and is up-regulated in AP. Knockdown of NFATc3 or the administration of the NFAT inhibitor A-285222 both significantly reduced neutrophil infiltration and pathological damage in the pancreas and lung in AP [70]. Inhibition of LFA-1, one of the integrins, by gene knockout or anti-LFA-1 antibody significantly reduced neutrophil recruitment and activation in AP, thereby reducing pancreatic injury [65]. Galectin-3, as a member of the galectins family, plays an important pro-inflammatory role in inflammatory diseases. The deletion of galectin-3 decreases early neutrophils influx in AP [51]. Inhibition of Ras signaling by farnesylthiosalicylic acid (FTS) can also attenuate SAP by reducing neutrophil infiltration [63]. Loss of tissue nonspecific alkaline phosphatase (TNAP) [56] or receptor-activity-modifying-protein-1 (RAMP1) [67] expression markedly increase neutrophil infiltration and tissue damage in AP, thus maintaining their expression may be a therapeutic strategy. Several widely studied or novel medications, including 4-phenyl butyric acid (4-PBA) [62], lactose [64], pentoxifylline [72], zileuton [27], desoxo-narchinol-A [59], Evasin-3 [76], biomimetic carbon monoxide delivery [48] and Emodin [53], can abrogate pancreatic neutrophil infiltration and associated inflammation in AP.
NETs and AP
As a special functional state of neutrophils, NETs participate in multiple processes of AP pathogenesis. A recent study has shown that NETs are also involved in the activation of trypsinogen [25]. Histones 2A, 2B, 3, and 4, the major protein of NETs structure, are the key components for increasing trypsin activity, which promotes the activation of trypsin by phosphorylating Signal Transducer and Activator of Transcription 3 (STAT3) in pancreatic acinar cells [25]. Interestingly, the formation of NETs in the pancreas and lung during AP can recruit more infiltrating neutrophils, which in turn aggravate tissue damage [25]. Moreover, NETs can directly activate neutrophils, promoting their expression of Mac-1 as well as the generation of ROS and MMP-9 [25]. NETs can also regulate systemic inflammation by upregulating HMGB1, CXCL2, and IL-6 [25]. Obstruction of the pancreatic duct can also lead to AP and determine its severity [52]. During AP, the pancreatic juice is a strong instigator of infiltrating neutrophil chromatin extrusion and thereby lead to the formation of macroscopic NETs aggregates, which may occlude pancreatic duct and drive pancreatic inflammation, inhibition of NETs formation can mitigate AP [57]. For the detection of NETs formation in AP, cell-free DNA (cfDNA) [55], MPO-DNA complex [55], or histone-DNA complex [39, 75] in blood samples can be measured by commercial kits. Besides, citrullinated histone H3 (CitH3) [39, 75] in pancreatic samples can be measured by western blot.
Peptidyl arginine deiminase 4 (PAD4) and c-Abelson (c-Abl) kinase are responsible for regulating the formation of NETs [39, 60]. Pharmacologically inhibition of PAD4 by Cl-amidine, a pan-PAD inhibitor, or inhibition of c-Abl by GZD824, a c-Abl kinase inhibitor, can reduce NETs formation and tissue Injury in SAP [39, 75]. The autophagy inhibitor chloroquine (CQ) also inhibits NETs formation, thus reduces the severity of AP [55]. Recent studies have shown that platelets also have a role in the regulation of neutrophils recruitment and NETs formation. Activated platelets induce neutrophils recruitment by secreting CXCL4 and promote NETs formation by producing inositol hexakisphosphate kinase 1 (IP6K1). Depletion of platelets, inhibition of IP6K1 by TNP, or inhibition of CXCL4 can alleviate neutrophil infiltration, NETs formation, and tissue damage in AP [40, 73].
Macrophages
Macrophages and inflammation
Macrophages include tissue-resident macrophages and migrating macrophages. Evidence has shown that tissue-resident macrophages are derived from the yolk sac and seeded before birth, which has self-renewal ability [77, 78]. The names of tissue-resident macrophages vary among different organs, for example, Kupfer cells in the liver, microglia in the brain, and osteoclasts in bone. Migrating macrophages are derived from bone marrow stem cells. Stimulated by the multi-colony stimulating factor (multi-SCF) and macrophage colony-stimulating factor (GM-CSF), bone marrow stem cells develop into granulocyte-monocyte progenitor cells, then differentiate into promonocytes and enter the blood, and finally mature into monocytes, accounting for 3-8% of the peripheral blood leukocytes. Monocytes exhibit a short half-life of 20 hours [79], then migrate to tissues and organs and differentiate into macrophages.
Macrophages can clear pathogens, tissue debris, necrotic and apoptotic cells through their effective phagocytosis, playing an important role in infectious and sterile inflammation [80]. They are not only an important component of innate immunity but also involve in regulating adaptive immune response as antigen-presenting cells (APCs) [81]. High plasticity is a hallmark of macrophages. Macrophage polarization is a phenotypical and functional change in response to microenvironment variations [80]. In the process of infection and sterile inflammation, tissue-resident macrophages play an important role in detecting PAMPs released by pathogens and DAMPs released by damaged cells and then secrete a variety of pro-inflammatory mediators to recruit more tissue-resident macrophages and circulating monocytes to the inflammatory sites [82], where the cytokine microenvironment dominates the orientation of macrophage polarization [83]. Macrophages commonly exist in two distinct subsets, including classically activated (M1) and alternatively activated (M2) macrophages, the former are generally polarized by Th1 cytokines IFN-γ, TNF-α and act in a pro-inflammatory manner by secreting interleukin (IL)-1β, IL-6, IL-12 and tumor necrosis factor (TNF-α), while the latter is induced by Th2 cytokines IL-4, IL-13 and play an anti-inflammatory, immunoregulatory and pro-fibrotic role by secreting IL-4, IL-10, IL-13 and transforming growth factor-β (TGF-β) [80, 82, 83]. It is worth mentioning that M2 macrophages can be further subdivided in M2a, M2b, M2c, and M2d based on the applied stimuli and the resultant transcriptional changes [84]. M2a macrophages, also known as wound healing macrophages, promote tissue repair and regeneration by secreting the profibrotic factor TGF-β [85]. M2b is a regulatory macrophage that can secrete both pro-inflammatory and anti-inflammatory mediators [85]. M2c macrophages are anti-inflammatory macrophages, mainly secrete anti-inflammatory cytokine IL-10, and can efficiently phagocytose apoptotic cells [85]. M2d macrophages represent a novel M2 subset that is also known as tumor-associated macrophages (TAMs) and mainly plays a role in angiogenesis and metastasis of tumors [86]. Macrophage polarization imbalance, especially the type in which the ratio of M1 to M2 is significantly increased, is one of the mechanisms underlying the aggravation of many inflammatory diseases, including autoimmune myocarditis [87], inflammatory root resorption [88], brain ischemic injury [89], acute lung injury [90], AP [91], intestinal ischemia/reperfusion injury [92] and so on. M1 macrophages exacerbate tissue damage and impair tissue regeneration by secreting a large quantity of pro-inflammatory cytokines to induce inflammatory cascade, triggering ROS-mediated inflammation through activation of NADPH oxidase [82], as well as activating neutrophils [93].
Since the initial description of NETs appeared in 2004 [30], it is now known that macrophages are also able to produce “extracellular trap” structures through METosis [94]. METs function to immobilize and kill some pathogenic microorganisms, but may also play a role in disease pathology [95-97]. Inadequate resolution and degradation of METs, as well as prolonged exposure of self-antigens comprising METs may contribute to inflammation and autoimmune diseases [98], such as autoimmune arthritis [95], atherothrombosis [96], rhabdomyolysis-induced acute kidney injury [97].
In recent years, the critical role of macrophage pyroptosis in inflammatory diseases has been revealed. Pyroptosis is a rapid, proinflammatory, programmed cell necrosis mediated by gasdermin family proteins (GSDMA, GSDMB, GSDMC, GSDMD, GSDME, DFNB59), characterized by cellular swelling, membrane disruption, and release of proinflammatory cellular contents, such as IL-1β, IL-18 and HMGB1 [99, 100]. Macrophage pyroptosis is involved in and aggravates many inflammatory diseases, such as knee osteoarthritis [101], sepsis [102], liver ischemia-reperfusion injury [103], multiple sclerosis [104], retinal detachment-induced photoreceptor cell death [105], while inhibition of macrophage pyroptosis can significantly reduce the severity of the disease [101-105].
Macrophages infiltration in the pathogenesis of AP and therapy
Like neutrophils, macrophages are also the main innate immune cells involved in the pathogenesis of AP, infiltrating the inflamed pancreas in the early phase of AP and promoting the development of AP [19-21, 48, 91, 106-128] (Figure 2). Detection of F4/80 positive cells or CD68 positive cells in the pancreas by immunofluorescence or immunohistochemistry is the main method to detect pancreatic infiltrating macrophages in mice [67, 106, 129].
Damaged pancreatic acinar cells recruit macrophages by releasing DAMPs and proinflammatory mediators, such as monocyte chemo attractant protein-1 (MCP-1, also known as CCL2), TNF-α, etc. [107-109]. In addition, pancreatic acinar cell death-derived DNA, pancreatic elastase, lipase, pancreatitis-associated ascites, and even lipid extracts from ascites can activate infiltrating macrophages [110-112], and the degree of macrophage activation is positively correlated with the severity of AP [113, 114]. These infiltrating macrophages produce proinflammatory mediators including TNF-α, IL-1β, IL-6, IL-18, MCP-1, platelet-activating factor (PAF), macrophage inflammatory protein (MIP)-1α and macrophage migration inhibitory factor (MIF), macrophage inflammatory protein-2 (MIP-2) [115-118], thus, more macrophages are recruited, triggering and further amplifying local inflammation, aggravating tissue damage. The depletion of macrophages through the administration of clodronate liposomes significantly reduced disease severity in caerulein-induced AP model [20], suggesting an important role of macrophages in the pathogenesis of AP. Construction of caerulein-induced AP model in CCR2 (receptor of CCL2) deficient mice showed that infiltrating macrophages in the pancreas were significantly reduced and tissue damage was significantly mitigated [107]. Targeting macrophage associated inflammatory mediators in AP also achieves a good therapeutic effect. Treatment with bindarit, which blocks the synthesis of MCP-1, or injection of a plasmid expression vector containing a dominant-negative mutant MCP-1 gene (mMCP-1), significantly reduced the severity of AP [117, 119]. Prophylactic administration of anti-MIF antibody significantly improved the survival rate of mice and rat model of SAP [116]. Pretreatment with ISO-1, an inhibitor of MIF, also notably attenuated the severity of AP [120]. Blockade of MIP-2 by anti-MIP-2 antibody significantly reduced the infiltration of inflammatory cells in the pancreas and lung, and reduced tissue damage in AP [118]. For PAF, the administration of PAF receptor antagonists (PAF-RAs) could significantly reduce local and systemic events after AP [130].
Macrophages polarization and AP
With the polarization of macrophages, M1 and M2 subtypes express unique markers, such as CD40, iNOS, TNF-α, IL-1β and IL-6 of M1 macrophages, Arg-1, Clec10, Mrc1 and CD206 of M2 macrophages [91, 131], which are the common markers for detecting M1 and M2 macrophages.
Since the polarization status of macrophages is related to the progression and severity of AP, many researchers have focused on the therapeutic effect of regulating the polarization status of macrophages on AP [48, 91, 121, 122]. It is generally accepted that abdominal paracentesis drainage (APD) can alleviate AP. A recent study revealed that pancreatitis-associated ascitic fluids (PAAF) can promote macrophage M1 polarization, and the therapeutic effect of APD is partly achieved by promoting macrophage M2 polarization and inhibiting M1 polarization [121]. Adipose stem cells (ASCs) are characterized by significant immunomodulatory and regenerative ability. The proportion of anti-inflammatory M2 phenotype in pancreatic macrophages was significantly increased by the infusion of ASCs into the tail vein of SAP mice model [122]. The infusion of carbon monoxide-bound hemoglobin vesicles (CO-HbV), as a nanomedicine modality, polarized macrophages toward an M2 phenotype and in the pancreas and mitigated AP [48]. Inducing SAP in LysM+/CreDRD2fl/fl mice exhibited more M1 polarization of pancreatic macrophages and stronger inflammatory response, suggesting that myeloid-specific dopamine D2 receptor (DRD2) could protect AP by inhibiting M1 polarization [91]. Inhibition of macrophage M1 polarization in the pancreas and liver can also reduce AP-related liver injury and lung injury [123, 124].
Macrophages pyroptosis and AP
As a kind of potent pro-inflammatory cell death, pyroptosis plays an important role in local and systemic inflammation in AP. Genetic deletion of Nod-like receptor family pyrin domain-containing 3 (NLRP3), absent in melanoma-2 (AIM2), apoptosis-associated speck-like protein containing caspase recruitment domains (ASC), or Caspase-1, which are the components of the inflammasome, markedly reduced the severity of AP [109, 125, 126]. MCC950, an NLRP3 inhibitor, can also reduce the severity of AP [125]. Besides, the administration of lactate can alleviate SAP by inhibiting NLRP3 inflammasome activation in macrophages [127]. More importantly, studies have shown that pyroptosis occurs mainly in infiltrating macrophages, but not in pancreatic acinar cells during AP [109, 125]. A recent study revealed the mechanism of pyroptosis in infiltrating macrophages. The infiltrating macrophages were found to phagocytose zymogen-containing vesicles from injured acinar cells and activate trypsinogen intracellularly, which acts as DAMPs and induces NLRP3 inflammasome activation within macrophages. Inhibition of trypsinogen activation within macrophages reduced pro-inflammatory cytokine secretion and mitigated AP [21]. Interestingly, a recent study suggested that the inflamed pancreas releases exosomes containing various signaling molecules which leads to SAP associated lung injury through NLRP3 inflammasome activation and subsequent pyroptosis in alveolar macrophages (AMs). Inhibition of exosome release or uptake by GW4869 or Enoxaparin significantly reduced AMs pyroptosis and thus alleviated SAP-induced lung injury [128]. These studies indicate that targeting pyroptosis of infiltrating macrophages may be a novel and effective strategy for the treatment of AP.
Other innate immune cells in AP
Dendritic cells
Dendritic cells (DCs) originate from bone marrow hematopoietic stem cells and are the most powerful antigen-presenting cells. DCs can phagocytose and clear pathogens as well as harmful antigens to exert innate immune functions. Also, DCs can activate T and B cells through antigen presentation, and regulate immune responses by secreting various cytokines. Therefore, DCs are the link between innate and adaptive immunity [132].
In AP, infiltrating DCs seem to play a protective role in some cases [16, 133, 134], while in others, they aggravate the severity of AP [135-137]. DCs increased 100-fold in the pancreas of caerulein- or L-arginine-induced SAP mouse models, presenting the phenotype of MHC II+CD11c+, secreting IL-6, MCP-1, and TNF-a. Interestingly, rather than inducing an organ-destructive inflammation, DCs were required for promoting pancreatic viability. SAP mice that were depleted of DCs by diphtheria toxin died from severe acinar cell death within 4 days [16]. In type B coxsackieviruses (CVB)-induced AP, the inflamed pancreas secretes CCL17, which binds to CCR4 on DCs and recruits them to the site of inflammation. The infiltrating DCs triggers Th1 immune responses, thereby reducing viral load and tissue damage. CCR4 knockout resulted in a reduced recruitment of DCs, elevated viral load, and increased severity of AP [133]. Furthermore, DCs limit neutrophils infiltration and tissue damage by expressing dendritic cell natural killer lectin group receptor-1 (DNGR-1) encoded by the gene Clec9a as a feedback mechanism. Knockout of DNGR-1or inhibition of DNGR-1 by DNGR-1-blocking antibody significantly aggravated tissue damage [134]. However, increasing DCs activity by MyD88 inhibition exacerbated pancreatic inflammation [137]. Supplementation with Clostridium butyricum, a probiotic, can alleviate tissue damage and inflammation by reducing the infiltration of DCs in AP [135]. A recent study reported an interesting phenomenon that in AP, pancreatic acinar cells undergo acinar-to-dendritic cell transition, which in turn promotes the differentiation of naive CD4+T cells into CD4+/IFN-γ+Th1 and CD4+/IL-17A+Th17 cells, thereby aggravating local inflammation and tissue damage. The administration of mTOR inhibitor rapamycin or Myc inhibitor 10058-F4 can inhibit acinar-to-dendritic cell transition, thus reducing the severity of AP [136]. In summary, DCs play both pro- and anti-inflammatory roles in AP, subsequent studies are needed to further uncover their roles in AP.
Mast cells
Mast cells originate from bone marrow stem cells. Activated mast cells release cytoplasmic granules including histamine, serotonin, protease, cytokines, and chemokines in a process known as degranulation. Mast cells are best well-known for their expression of high-affinity IgE receptors and involvement in IgE-mediated type I hypersensitivity. However, recent data indicate that mast cells are also pivotal players in innate and adaptive immune responses [138]. For example, abnormal accumulation and activation of mast cells are present in AP [17, 139-143].
Elevated mast cell count and mast cell degranulation were observed in the pancreas during AP [17, 139]. Treatment with ketotifen, a mast cell stabilizer, significantly reduced pancreatic tissue damage [139]. Intraperitoneally injection of mast cell inhibitor cromolyn significantly reduced inflammation in the pancreas and lung [17]. Administration of sodium cromoglycate (SCG), another mast cell stabilizer, decreased the release of histamine and reduced plasma exudation in the pancreas, colon, and lungs, suggesting that mast cell activation is involved in the development of endothelial dysfunction in the pancreas and other distant organs in AP, which may underlie MODS [142]. In addition, the inflamed pancreas expressed and released IL-33 during AP. As an ST2 ligand, IL-33 reduced the activation of mast cells after binding with ST2 on mast cells, which is a protective mechanism for AP. ST2 knockout leads to more severe AP [143]. The activated infiltrating mast cells can also secrete IL-33 and histamine, which drives pancreatic inflammation [140]. Scopoletin reduced the severity of pancreatic and associated lung injury in AP by reducing mast cell activation and corresponding levels of IL-33 [141]. Therefore, the mast cells may hold promise as a treatment target in AP.
NK cells
NK cells originated from bone marrow lymphoid stem cells, accounting for 5%-15% of peripheral blood lymphocytes. NK cells can kill tumor cells or virus-infected cells directly or through antibody-dependent cell-mediated cytotoxicity (ADCC). Besides, they are also involved in maintaining immune homeostasis and regulating inflammation [144]. So far, studies on NK cells in AP are limited. Several studies have reported that NK cells infiltrate the pancreas during AP [18, 145]. In the adenoviral vector-mediated AP model, NK cells infiltrated the inflamed pancreas from the second day, reaching a maximum on the fourth day, and persisting until 28 days [18], however, their role in the pathogenesis of AP is not clear. More studies focused on changes in NK cells in peripheral blood. Studies have shown that peripheral blood NK cell counts in patients with AP were lower than those in healthy controls [146, 147]. The activity of NK cells and ADCC decreased in some SAP patients [148]. Compared with MAP, patients with SAP experienced a significant decrease in the number of peripheral blood NK cells in the early phase and persisted for 30 days [147, 149]. The immunosuppressive state of peripheral leukocyte and NK cell depletion is thought to be responsible for the infectious complications of AP [148, 149]. More studies are needed to explore the exact role of NK cells in AP.
Conclusions and future prospects
AP begins with abnormal activation of trypsinogen and self-digestion. Some patients experience a transition from pancreatic local inflammation to systemic inflammation even MODS. However, the current understanding of the pathogenesis of AP is still far from complete, limiting our treatment options. With the advances of research in the past 20 years, the role of innate immune cells, including neutrophils, macrophages, dendritic cells, mast cells, in the inflammatory storm of AP has become increasingly prominent, however, reports on the role of infiltrating eosinophils and basophils in AP are lacking. While promoting pancreatic injury, these innate immune cells also interact with each other and adaptive immune cells to form a huge regulatory network. A substantial of studies have shown that the intervention of immune cells through pharmacological and genetic methods has indeed alleviated AP, bringing hope for the treatment of AP. Immunotherapy, as an emerging therapy, is very promising. However, immunotherapy is the future since the underlying immunological mechanisms of AP are rather complex, a lot of works are needed to clarify the regulatory network. Unlike neoplastic diseases, the acquisition of tissue samples from patients with AP is difficult, which limits the relevant scientific research to some extent. Therefore, direct evidence of immunological alternations of AP in the human pancreas is lacking. At present, the widely used caerulein-induced mice AP model and the sodium taurocholate-induced rat AP model cannot fully correspond to the clinical AP with various etiological backgrounds. Therefore, according to the current evidence, it is difficult to answer whether AP of different etiological backgrounds shares the same immunological characteristics. This requires further exploration at the patients' tissue level or in animal models of more types of AP before conclusions can be drawn. In summary, we believe that for the treatment of AP, immunotherapy is a powerful means in the future, which has high clinical transformation value. But before that, we still have a lot of work to do.
Abbreviations
AP: acute pancreatitis; MAP: mild acute pancreatitis; SAP: severe acute pancreatitis; SIRS: systemic inflammatory response syndrome; MODS: multiple organ dysfunction syndromes; MOF: multiple organ failure; PAMPs: pathogen-associated molecular patterns; DAMPs: danger-associated molecular patterns; HMGB1: high-mobility group box protein 1; PRRs: pattern recognition receptors; NK cells: natural killer cells; MPO: myeloperoxidase; NETs: neutrophil extracellular traps; NE: neutrophil elastase; ELISA: enzyme-linked immunosorbent assay; TAP: trypsin-activation peptide; MMP-9: matrix metalloproteinase-9; ROS: reactive oxygen species; NADPH: nicotinamide adenine dinucleotide phosphate; NFATc3: nuclear factor of activated T cells 3; FTS: farnesylthiosalicylic acid; TNAP: tissue nonspecific alkaline phosphatase; RAMP1: receptor-activity-modifying-protein-1; 4-PBA: 4-phenyl butyric acid; STAT3: Signal Transducer and Activator of Transcription 3; cfDNA: cell-free DNA; CitH3: citrullinated histone H3; PAD4: Peptidyl arginine deiminase 4; c-Abl: c-Abelson; CQ: chloroquine; IP6K1: inositol hexakisphosphate kinase 1; multi-SCF: multi-colony stimulating factor; GM-CSF: macrophage colony-stimulating factor; APCs: antigen-presenting cells; TNF-α: tumor necrosis factor-α; TGF-β: transforming growth factor-β; MCP-1: monocyte chemo attractant protein-1; PAF: platelet-activating factor; MIF: macrophage migration inhibitory factor; MIP: macrophage inflammatory protein; mMCP-1: mutant MCP-1 gene; PAF-RAs: PAF receptor antagonists; APD: abdominal paracentesis drainage; PAAF: pancreatitis-associated ascitic fluids; ASCs: adipose stem cells; CO-HbV: carbon monoxide-bound hemoglobin vesicles; DRD2: dopamine D2 receptor; NLRP3: Nod-like receptor family pyrin domain-containing 3; AIM2: absent in melanoma-2; ASC: apoptosis-associated speck-like protein containing caspase recruitment domains; AMs: alveolar macrophages; DCs: dendritic cells; CVB: type B coxsackieviruses; DNGR-1: dendritic cell natural killer lectin group receptor-1; SCG: sodium cromoglycate; ADCC: antibody-dependent cell-mediated cytotoxicity; TAMs: tumor-associated macrophages; CXCL1/CXCL2/CXCL8: CXC chemokine ligands 1/2/8; VLA-4: very late antigen-4; Mac-1: macrophage-1 antigen; LFA-1: lymphocyte function-associated antigen-1; ICAM-1: intercellular adhesion molecule-1.
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (81873589).
Authors' ORCID
Cheng Peng: 0000-0002-5338-5325;
Xiao Yu: 0000-0002-3172-8606.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Gukovskaya AS, Gukovsky I, Algül H, Habtezion A. Autophagy, Inflammation, and Immune Dysfunction in the Pathogenesis of Pancreatitis. Gastroenterology. 2017;153:1212-26
2. Hines OJ, Pandol SJ. Management of severe acute pancreatitis. Bmj. 2019;367:l6227
3. van Dijk SM, Hallensleben NDL, van Santvoort HC, Fockens P, van Goor H, Bruno MJ. et al. Acute pancreatitis: recent advances through randomised trials. Gut. 2017;66:2024-32
4. Banks PA, Bollen TL, Dervenis C, Gooszen HG, Johnson CD, Sarr MG. et al. Classification of acute pancreatitis-2012: revision of the Atlanta classification and definitions by international consensus. Gut. 2013;62:102-11
5. Xiao AY, Tan ML, Wu LM, Asrani VM, Windsor JA, Yadav D. et al. Global incidence and mortality of pancreatic diseases: a systematic review, meta-analysis, and meta-regression of population-based cohort studies. Lancet Gastroenterol Hepatol. 2016;1:45-55
6. Crockett SD, Wani S, Gardner TB, Falck-Ytter Y, Barkun AN. American Gastroenterological Association Institute Guideline on Initial Management of Acute Pancreatitis. Gastroenterology. 2018;154:1096-101
7. Watanabe T, Kudo M, Strober W. Immunopathogenesis of pancreatitis. Mucosal Immunol. 2017;10:283-98
8. Gukovsky I, Li N, Todoric J, Gukovskaya A, Karin M. Inflammation, autophagy, and obesity: common features in the pathogenesis of pancreatitis and pancreatic cancer. Gastroenterology. 2013;144:1199-209.e4
9. Mayerle J, Sendler M, Hegyi E, Beyer G, Lerch MM, Sahin-Tóth M. Genetics, Cell Biology, and Pathophysiology of Pancreatitis. Gastroenterology. 2019;156:1951-68.e1
10. Hoque R, Malik AF, Gorelick F, Mehal WZ. Sterile inflammatory response in acute pancreatitis. Pancreas. 2012;41:353-7
11. Kang R, Zhang Q, Hou W, Yan Z, Chen R, Bonaroti J. et al. Intracellular Hmgb1 inhibits inflammatory nucleosome release and limits acute pancreatitis in mice. Gastroenterology. 2014;146:1097-107
12. Sarhan M, Land WG, Tonnus W, Hugo CP, Linkermann A. Origin and Consequences of Necroinflammation. Physiol Rev. 2018;98:727-80
13. Habtezion A. Inflammation in acute and chronic pancreatitis. Curr Opin Gastroenterol. 2015;31:395-9
14. Antonucci L, Fagman JB, Kim JY, Todoric J, Gukovsky I, Mackey M. et al. Basal autophagy maintains pancreatic acinar cell homeostasis and protein synthesis and prevents ER stress. Proc Natl Acad Sci U S A. 2015;112:E6166-74
15. Wan J, Yang X, Ren Y, Li X, Zhu Y, Haddock AN. et al. Inhibition of miR-155 reduces impaired autophagy and improves prognosis in an experimental pancreatitis mouse model. Cell Death Dis. 2019;10:303
16. Bedrosian AS, Nguyen AH, Hackman M, Connolly MK, Malhotra A, Ibrahim J. et al. Dendritic cells promote pancreatic viability in mice with acute pancreatitis. Gastroenterology. 2011;141:1915-26.e1 -14
17. Lopez-Font I, Gea-Sorlí S, de-Madaria E, Gutiérrez LM, Pérez-Mateo M, Closa D. Pancreatic and pulmonary mast cells activation during experimental acute pancreatitis. World J Gastroenterol. 2010;16:3411-7
18. Shifrin AL, Chirmule N, Gao GP, Wilson JM, Raper SE. Innate immune responses to adenoviral vector-mediated acute pancreatitis. Pancreas. 2005;30:122-9
19. Son A, Ahuja M, Schwartz DM, Varga A, Swaim W, Kang N. et al. Ca(2+) Influx Channel Inhibitor SARAF Protects Mice From Acute Pancreatitis. Gastroenterology. 2019;157:1660-72.e2
20. Sendler M, Dummer A, Weiss FU, Krüger B, Wartmann T, Scharffetter-Kochanek K. et al. Tumour necrosis factor α secretion induces protease activation and acinar cell necrosis in acute experimental pancreatitis in mice. Gut. 2013;62:430-9
21. Sendler M, Weiss FU, Golchert J, Homuth G, van den Brandt C, Mahajan UM. et al. Cathepsin B-Mediated Activation of Trypsinogen in Endocytosing Macrophages Increases Severity of Pancreatitis in Mice. Gastroenterology. 2018;154:704-18.e10
22. Castanheira FVS, Kubes P. Neutrophils and NETs in modulating acute and chronic inflammation. Blood. 2019;133:2178-85
23. Jorch SK, Kubes P. An emerging role for neutrophil extracellular traps in noninfectious disease. Nat Med. 2017;23:279-87
24. Huang H, Tohme S, Al-Khafaji AB, Tai S, Loughran P, Chen L. et al. Damage-associated molecular pattern-activated neutrophil extracellular trap exacerbates sterile inflammatory liver injury. Hepatology. 2015;62:600-14
25. Merza M, Hartman H, Rahman M, Hwaiz R, Zhang E, Renström E. et al. Neutrophil Extracellular Traps Induce Trypsin Activation, Inflammation, and Tissue Damage in Mice With Severe Acute Pancreatitis. Gastroenterology. 2015;149:1920-31.e8
26. Furze RC, Rankin SM. Neutrophil mobilization and clearance in the bone marrow. Immunology. 2008;125:281-8
27. Liao D, Qian B, Zhang Y, Wu K, Xu M. Inhibition of 5-lipoxygenase represses neutrophils activation and activates apoptosis in pancreatic tissues during acute necrotizing pancreatitis. Biochem Biophys Res Commun. 2018;498:79-85
28. Ferrero-Andres A, Panisello-Rosello A, Serafin A, Rosello-Catafau J, Folch-Puy E. Polyethylene Glycol 35 (PEG35) Protects against Inflammation in Experimental Acute Necrotizing Pancreatitis and Associated Lung Injury. Int J Mol Sci. 2020 21
29. Pittman K, Kubes P. Damage-associated molecular patterns control neutrophil recruitment. J Innate Immun. 2013;5:315-23
30. Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS. et al. Neutrophil extracellular traps kill bacteria. Science. 2004;303:1532-5
31. Zha C, Meng X, Li L, Mi S, Qian D, Li Z. et al. Neutrophil extracellular traps mediate the crosstalk between glioma progression and the tumor microenvironment via the HMGB1/RAGE/IL-8 axis. Cancer Biol Med. 2020;17:154-68
32. Steinberg BE, Grinstein S. Unconventional roles of the NADPH oxidase: signaling, ion homeostasis, and cell death. Sci STKE. 2007;2007:pe11
33. Beiter K, Wartha F, Albiger B, Normark S, Zychlinsky A, Henriques-Normark B. An endonuclease allows Streptococcus pneumoniae to escape from neutrophil extracellular traps. Curr Biol. 2006;16:401-7
34. Berends ET, Horswill AR, Haste NM, Monestier M, Nizet V, von Köckritz-Blickwede M. Nuclease expression by Staphylococcus aureus facilitates escape from neutrophil extracellular traps. J Innate Immun. 2010;2:576-86
35. Thammavongsa V, Missiakas DM, Schneewind O. Staphylococcus aureus degrades neutrophil extracellular traps to promote immune cell death. Science. 2013;342:863-6
36. Dyer MR, Chen Q, Haldeman S, Yazdani H, Hoffman R, Loughran P. et al. Deep vein thrombosis in mice is regulated by platelet HMGB1 through release of neutrophil-extracellular traps and DNA. Sci Rep. 2018;8:2068
37. Zhou J, Yang Y, Gan T, Li Y, Hu F, Hao N. et al. Lung cancer cells release high mobility group box 1 and promote the formation of neutrophil extracellular traps. Oncol Lett. 2019;18:181-8
38. Zhu M, Yuan K, Lu Q, Zhu Q, Zhang S, Li X. et al. Emodin ameliorates rheumatoid arthritis by promoting neutrophil apoptosis and inhibiting neutrophil extracellular trap formation. Mol Immunol. 2019;112:188-97
39. Madhi R, Rahman M, Mörgelin M, Thorlacius H. c-Abl kinase regulates neutrophil extracellular trap formation, inflammation, and tissue damage in severe acute pancreatitis. J Leukoc Biol. 2019;106:455-66
40. Madhi R, Rahman M, Taha D, Linders J, Merza M, Wang Y. et al. Platelet IP6K1 regulates neutrophil extracellular trap-microparticle complex formation in acute pancreatitis. JCI Insight. 2019
41. Desai J, Foresto-Neto O, Honarpisheh M, Steiger S, Nakazawa D, Popper B. et al. Particles of different sizes and shapes induce neutrophil necroptosis followed by the release of neutrophil extracellular trap-like chromatin. Sci Rep. 2017;7:15003
42. Arumugam S, Girish Subbiah K, Kemparaju K, Thirunavukkarasu C. Neutrophil extracellular traps in acrolein promoted hepatic ischemia reperfusion injury: Therapeutic potential of NOX2 and p38MAPK inhibitors. J Cell Physiol. 2018;233:3244-61
43. Kim SW, Lee H, Lee HK, Kim ID, Lee JK. Neutrophil extracellular trap induced by HMGB1 exacerbates damages in the ischemic brain. Acta Neuropathol Commun. 2019;7:94
44. Sônego F, Castanheira FV, Ferreira RG, Kanashiro A, Leite CA, Nascimento DC. et al. Paradoxical Roles of the Neutrophil in Sepsis: Protective and Deleterious. Front Immunol. 2016;7:155
45. Wang J. Neutrophils in tissue injury and repair. Cell Tissue Res. 2018;371:531-9
46. Kovtun A, Messerer DAC, Scharffetter-Kochanek K, Huber-Lang M, Ignatius A. Neutrophils in Tissue Trauma of the Skin, Bone, and Lung: Two Sides of the Same Coin. J Immunol Res. 2018;2018:8173983
47. Novovic S, Andersen AM, Nord M, Astrand M, Ottosson T, Jørgensen LN. et al. Activity of neutrophil elastase reflects the progression of acute pancreatitis. Scand J Clin Lab Invest. 2013;73:485-93
48. Taguchi K, Nagao S, Maeda H, Yanagisawa H, Sakai H, Yamasaki K. et al. Biomimetic carbon monoxide delivery based on hemoglobin vesicles ameliorates acute pancreatitis in mice via the regulation of macrophage and neutrophil activity. Drug Deliv. 2018;25:1266-74
49. Song G, Liu D, Geng X, Ma Z, Wang Y, Xie W. et al. Bone marrow-derived mesenchymal stem cells alleviate severe acute pancreatitis-induced multiple-organ injury in rats via suppression of autophagy. Exp Cell Res. 2019;385:111674
50. John DS, Aschenbach J, Krüger B, Sendler M, Weiss FU, Mayerle J. et al. Deficiency of cathepsin C ameliorates severity of acute pancreatitis by reduction of neutrophil elastase activation and cleavage of E-cadherin. J Biol Chem. 2019;294:697-707
51. Stojanovic B, Jovanovic I, Stojanovic BS, Stojanovic MD, Gajovic N, Radosavljevic G. et al. Deletion of Galectin-3 attenuates acute pancreatitis in mice by affecting activation of innate inflammatory cells. Eur J Immunol. 2019;49:940-6
52. Rünzi M, Saluja A, Lerch MM, Dawra R, Nishino H, Steer ML. Early ductal decompression prevents the progression of biliary pancreatitis: an experimental study in the opossum. Gastroenterology. 1993;105:157-64
53. Cui H, Li S, Xu C, Zhang J, Sun Z, Chen H. Emodin alleviates severe acute pancreatitis-associated acute lung injury by decreasing pre-B-cell colony-enhancing factor expression and promoting polymorphonuclear neutrophil apoptosis. Mol Med Rep. 2017;16:5121-8
54. Zhang Q, Tao X, Xia S, Qu J, Song H, Liu J. et al. Emodin attenuated severe acute pancreatitis via the P2X ligandgated ion channel 7/NODlike receptor protein 3 signaling pathway. Oncol Rep. 2019;41:270-8
55. Murthy P, Singhi AD, Ross MA, Loughran P, Paragomi P, Papachristou GI. et al. Enhanced Neutrophil Extracellular Trap Formation in Acute Pancreatitis Contributes to Disease Severity and Is Reduced by Chloroquine. Front Immunol. 2019;10:28
56. Gámez-Belmonte R, Hernández-Chirlaque C, Sánchez de Medina F, Martínez-Augustin O. Experimental acute pancreatitis is enhanced in mice with tissue nonspecific alkaline phoshatase haplodeficiency due to modulation of neutrophils and acinar cells. Biochim Biophys Acta Mol Basis Dis. 2018;1864:3769-79
57. Leppkes M, Maueröder C, Hirth S, Nowecki S, Günther C, Billmeier U. et al. Externalized decondensed neutrophil chromatin occludes pancreatic ducts and drives pancreatitis. Nat Commun. 2016;7:10973
58. Mayerle J, Schnekenburger J, Krüger B, Kellermann J, Ruthenbürger M, Weiss FU. et al. Extracellular cleavage of E-cadherin by leukocyte elastase during acute experimental pancreatitis in rats. Gastroenterology. 2005;129:1251-67
59. Bae GS, Kim DG, Jo IJ, Choi SB, Kim MJ, Shin JY. et al. Heme oxygenase-1 induced by desoxo-narchinol-A attenuated the severity of acute pancreatitis via blockade of neutrophil infiltration. Int Immunopharmacol. 2019;69:225-34
60. Wang Y, Li M, Stadler S, Correll S, Li P, Wang D. et al. Histone hypercitrullination mediates chromatin decondensation and neutrophil extracellular trap formation. J Cell Biol. 2009;184:205-13
61. Yang J, Su J, Xi SS, Ke XF, Zhu Y, Lin HP. et al. Human umbilical cord mesenchymal stem cells pretreated with Angiotensin-II attenuate pancreas injury of rats with severe acute pancreatitis. Biomed Pharmacother. 2019;117:109052
62. Hong YP, Deng WH, Guo WY, Shi Q, Zhao L, You YD. et al. Inhibition of endoplasmic reticulum stress by 4-phenylbutyric acid prevents vital organ injury in rat acute pancreatitis. Am J Physiol Gastrointest Liver Physiol. 2018;315:G838-g47
63. Yu C, Merza M, Luo L, Thorlacius H. Inhibition of Ras signalling reduces neutrophil infiltration and tissue damage in severe acute pancreatitis. Eur J Pharmacol. 2015;746:245-51
64. Pan LL, Deng YY, Wang R, Wu C, Li J, Niu W. et al. Lactose Induces Phenotypic and Functional Changes of Neutrophils and Macrophages to Alleviate Acute Pancreatitis in Mice. Front Immunol. 2018;9:751
65. Awla D, Abdulla A, Zhang S, Roller J, Menger MD, Regnér S. et al. Lymphocyte function antigen-1 regulates neutrophil recruitment and tissue damage in acute pancreatitis. Br J Pharmacol. 2011;163:413-23
66. Jochheim LS, Odysseos G, Hidalgo-Sastre A, Zhong S, Staufer LM, Kroiss M. et al. The neuropeptide receptor subunit RAMP1 constrains the innate immune response during acute pancreatitis in mice. Pancreatology. 2019;19:541-7
67. Jochheim LS, Odysseos G, Hidalgo-Sastre A, Zhong S, Staufer LM, Kroiss M. et al. The neuropeptide receptor subunit RAMP1 constrains the innate immune response during acute pancreatitis in mice. Pancreatology. 2019;19:541-7
68. Awla D, Abdulla A, Syk I, Jeppsson B, Regnér S, Thorlacius H. Neutrophil-derived matrix metalloproteinase-9 is a potent activator of trypsinogen in acinar cells in acute pancreatitis. J Leukoc Biol. 2012;91:711-9
69. Gukovskaya AS, Vaquero E, Zaninovic V, Gorelick FS, Lusis AJ, Brennan ML. et al. Neutrophils and NADPH oxidase mediate intrapancreatic trypsin activation in murine experimental acute pancreatitis. Gastroenterology. 2002;122:974-84
70. Awla D, Zetterqvist AV, Abdulla A, Camello C, Berglund LM, Spégel P. et al. NFATc3 regulates trypsinogen activation, neutrophil recruitment, and tissue damage in acute pancreatitis in mice. Gastroenterology. 2012;143:1352-60.e7
71. Tsai K, Wang SS, Chen TS, Kong CW, Chang FY, Lee SD. et al. Oxidative stress: an important phenomenon with pathogenetic significance in the progression of acute pancreatitis. Gut. 1998;42:850-5
72. de Campos T, Deree J, Martins JO, Loomis WH, Shenvi E, Putnam JG. et al. Pentoxifylline attenuates pulmonary inflammation and neutrophil activation in experimental acute pancreatitis. Pancreas. 2008;37:42-9
73. Wetterholm E, Linders J, Merza M, Regner S, Thorlacius H. Platelet-derived CXCL4 regulates neutrophil infiltration and tissue damage in severe acute pancreatitis. Transl Res. 2016;176:105-18
74. Abdulla A, Awla D, Thorlacius H, Regnér S. Role of neutrophils in the activation of trypsinogen in severe acute pancreatitis. J Leukoc Biol. 2011;90:975-82
75. Madhi R, Rahman M, Taha D, Mörgelin M, Thorlacius H. Targeting peptidylarginine deiminase reduces neutrophil extracellular trap formation and tissue injury in severe acute pancreatitis. J Cell Physiol. 2019;234:11850-60
76. Montecucco F, Mach F, Lenglet S, Vonlaufen A, Gomes Quinderé AL, Pelli G. et al. Treatment with Evasin-3 abrogates neutrophil-mediated inflammation in mouse acute pancreatitis. Eur J Clin Invest. 2014;44:940-50
77. Davis MJ, Tsang TM, Qiu Y, Dayrit JK, Freij JB, Huffnagle GB. et al. Macrophage M1/M2 polarization dynamically adapts to changes in cytokine microenvironments in Cryptococcus neoformans infection. mBio. 2013;4:e00264-13
78. Epelman S, Lavine KJ, Randolph GJ. Origin and functions of tissue macrophages. Immunity. 2014;41:21-35
79. Sica A, Erreni M, Allavena P, Porta C. Macrophage polarization in pathology. Cell Mol Life Sci. 2015;72:4111-26
80. Gordon S, Martinez-Pomares L. Physiological roles of macrophages. Pflugers Arch. 2017;469:365-74
81. Arango Duque G, Descoteaux A. Macrophage cytokines: involvement in immunity and infectious diseases. Front Immunol. 2014;5:491
82. Shapouri-Moghaddam A, Mohammadian S, Vazini H, Taghadosi M, Esmaeili SA, Mardani F. et al. Macrophage plasticity, polarization, and function in health and disease. J Cell Physiol. 2018;233:6425-40
83. Murray PJ. Macrophage Polarization. Annu Rev Physiol. 2017;79:541-66
84. Wang LX, Zhang SX, Wu HJ, Rong XL, Guo J. M2b macrophage polarization and its roles in diseases. J Leukoc Biol. 2019;106:345-58
85. Mantovani A, Sica A, Sozzani S, Allavena P, Vecchi A, Locati M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004;25:677-86
86. Wu H, Xu JB, He YL, Peng JJ, Zhang XH, Chen CQ. et al. Tumor-associated macrophages promote angiogenesis and lymphangiogenesis of gastric cancer. J Surg Oncol. 2012;106:462-8
87. Su Z, Zhang P, Yu Y, Lu H, Liu Y, Ni P. et al. HMGB1 Facilitated Macrophage Reprogramming towards a Proinflammatory M1-like Phenotype in Experimental Autoimmune Myocarditis Development. Sci Rep. 2016;6:21884
88. Zhang J, Liu X, Wan C, Liu Y, Wang Y, Meng C. et al. NLRP3 inflammasome mediates M1 macrophage polarization and IL-1beta production in inflammatory root resorption. J Clin Periodontol. 2020;47:451-60
89. Yang J, Zhao Y, Zhang L, Fan H, Qi C, Zhang K. et al. RIPK3/MLKL-Mediated Neuronal Necroptosis Modulates the M1/M2 Polarization of Microglia/Macrophages in the Ischemic Cortex. Cereb Cortex. 2018;28:2622-35
90. Wang J, Li R, Peng Z, Zhou W, Hu B, Rao X. et al. GTS-21 Reduces Inflammation in Acute Lung Injury by Regulating M1 Polarization and Function of Alveolar Macrophages. Shock. 2019;51:389-400
91. Han X, Ni J, Wu Z, Wu J, Li B, Ye X. et al. Myeloid-specific dopamine D2 receptor signalling controls inflammation in acute pancreatitis via inhibiting M1 macrophage. Br J Pharmacol. 2020
92. Wen S, Li X, Ling Y, Chen S, Deng Q, Yang L. et al. HMGB1-associated necroptosis and Kupffer cells M1 polarization underlies remote liver injury induced by intestinal ischemia/reperfusion in rats. Faseb j. 2020;34:4384-402
93. Lee JW, Park JW, Shin NR, Park SY, Kwon OK, Park HA. et al. Picrasma quassiodes (D. Don) Benn. attenuates lipopolysaccharide (LPS)-induced acute lung injury. Int J Mol Med. 2016;38:834-44
94. Aulik NA, Hellenbrand KM, Czuprynski CJ. Mannheimia haemolytica and its leukotoxin cause macrophage extracellular trap formation by bovine macrophages. Infect Immun. 2012;80:1923-33
95. El Shikh MEM, El Sayed R, Nerviani A, Goldmann K, John CR, Hands R. et al. Extracellular traps and PAD4 released by macrophages induce citrullination and auto-antibody production in autoimmune arthritis. J Autoimmun. 2019;105:102297
96. Pertiwi KR, de Boer OJ, Mackaaij C, Pabittei DR, de Winter RJ, Li X. et al. Extracellular traps derived from macrophages, mast cells, eosinophils and neutrophils are generated in a time-dependent manner during atherothrombosis. J Pathol. 2019;247:505-12
97. Okubo K, Kurosawa M, Kamiya M, Urano Y, Suzuki A, Yamamoto K. et al. Macrophage extracellular trap formation promoted by platelet activation is a key mediator of rhabdomyolysis-induced acute kidney injury. Nat Med. 2018;24:232-8
98. Doster RS, Rogers LM, Gaddy JA, Aronoff DM. Macrophage Extracellular Traps: A Scoping Review. J Innate Immun. 2018;10:3-13
99. Frank D, Vince JE. Pyroptosis versus necroptosis: similarities, differences, and crosstalk. Cell Death Differ. 2019;26:99-114
100. Shi J, Gao W, Shao F. Pyroptosis: Gasdermin-Mediated Programmed Necrotic Cell Death. Trends Biochem Sci. 2017;42:245-54
101. Zhang L, Xing R, Huang Z, Zhang N, Zhang L, Li X. et al. Inhibition of Synovial Macrophage Pyroptosis Alleviates Synovitis and Fibrosis in Knee Osteoarthritis. Mediators Inflamm. 2019;2019:2165918
102. Song F, Hou J, Chen Z, Cheng B, Lei R, Cui P. et al. Sphingosine-1-phosphate Receptor 2 Signaling Promotes Caspase-11-dependent Macrophage Pyroptosis and Worsens Escherichia coli Sepsis Outcome. Anesthesiology. 2018;129:311-20
103. Hua S, Ma M, Fei X, Zhang Y, Gong F, Fang M. Glycyrrhizin attenuates hepatic ischemia-reperfusion injury by suppressing HMGB1-dependent GSDMD-mediated kupffer cells pyroptosis. Int Immunopharmacol. 2019;68:145-55
104. McKenzie BA, Mamik MK, Saito LB, Boghozian R, Monaco MC, Major EO. et al. Caspase-1 inhibition prevents glial inflammasome activation and pyroptosis in models of multiple sclerosis. Proc Natl Acad Sci U S A. 2018;115:E6065-e74
105. Kataoka K, Matsumoto H, Kaneko H, Notomi S, Takeuchi K, Sweigard JH. et al. Macrophage- and RIP3-dependent inflammasome activation exacerbates retinal detachment-induced photoreceptor cell death. Cell Death Dis. 2015;6:e1731
106. Wu J, Ma X, Chen W, Yang N, Gao L, Mao W. et al. Protective effects of HTD4010, a Reg3alpha/PAP-derived peptide, in mouse model of acute pancreatitis via toll-like receptor 4 pathway. Biochem Biophys Res Commun. 2019;512:670-7
107. Tsuji Y, Watanabe T, Kudo M, Arai H, Strober W, Chiba T. Sensing of commensal organisms by the intracellular sensor NOD1 mediates experimental pancreatitis. Immunity. 2012;37:326-38
108. Xue J, Sharma V, Habtezion A. Immune cells and immune-based therapy in pancreatitis. Immunol Res. 2014;58:378-86
109. Hoque R, Sohail M, Malik A, Sarwar S, Luo Y, Shah A. et al. TLR9 and the NLRP3 inflammasome link acinar cell death with inflammation in acute pancreatitis. Gastroenterology. 2011;141:358-69
110. Gutierrez PT, Folch-Puy E, Bulbena O, Closa D. Oxidised lipids present in ascitic fluid interfere with the regulation of the macrophages during acute pancreatitis, promoting an exacerbation of the inflammatory response. Gut. 2008;57:642-8
111. Jaffray C, Mendez C, Denham W, Carter G, Norman J. Specific pancreatic enzymes activate macrophages to produce tumor necrosis factor-alpha: role of nuclear factor kappa B and inhibitory kappa B proteins. J Gastrointest Surg. 2000;4:370-7 discussion 7-8
112. Zhao Q, Wei Y, Pandol SJ, Li L, Habtezion A. STING Signaling Promotes Inflammation in Experimental Acute Pancreatitis. Gastroenterology. 2018;154:1822-35.e2
113. Liang T, Liu TF, Xue DB, Sun B, Shi LJ. Different cell death modes of pancreatic acinar cells on macrophage activation in rats. Chin Med J (Engl). 2008;121:1920-4
114. Perides G, Weiss ER, Michael ES, Laukkarinen JM, Duffield JS, Steer ML. TNF-alpha-dependent regulation of acute pancreatitis severity by Ly-6C(hi) monocytes in mice. J Biol Chem. 2011;286:13327-35
115. Makhija R, Kingsnorth AN. Cytokine storm in acute pancreatitis. J Hepatobiliary Pancreat Surg. 2002;9:401-10
116. Sakai Y, Masamune A, Satoh A, Nishihira J, Yamagiwa T, Shimosegawa T. Macrophage migration inhibitory factor is a critical mediator of severe acute pancreatitis. Gastroenterology. 2003;124:725-36
117. Bhatia M, Ramnath RD, Chevali L, Guglielmotti A. Treatment with bindarit, a blocker of MCP-1 synthesis, protects mice against acute pancreatitis. Am J Physiol Gastrointest Liver Physiol. 2005;288:G1259-65
118. Pastor CM, Rubbia-Brandt L, Hadengue A, Jordan M, Morel P, Frossard JL. Role of macrophage inflammatory peptide-2 in cerulein-induced acute pancreatitis and pancreatitis-associated lung injury. Lab Invest. 2003;83:471-8
119. Ishibashi T, Zhao H, Kawabe K, Oono T, Egashira K, Suzuki K. et al. Blocking of monocyte chemoattractant protein-1 (MCP-1) activity attenuates the severity of acute pancreatitis in rats. J Gastroenterol. 2008;43:79-85
120. Li M, Yu J, Zhao L, Mei FC, Zhou Y, Hong YP. et al. Inhibition of macrophage migration inhibitory factor attenuates inflammation and fetal kidney injury in a rat model of acute pancreatitis in pregnancy. Int Immunopharmacol. 2019;68:106-14
121. Liu RH, Wen Y, Sun HY, Liu CY, Zhang YF, Yang Y. et al. Abdominal paracentesis drainage ameliorates severe acute pancreatitis in rats by regulating the polarization of peritoneal macrophages. World J Gastroenterol. 2018;24:5131-43
122. Roch AM, Maatman TK, Cook TG, Wu HH, Merfeld-Clauss S, Traktuev DO. et al. Therapeutic Use of Adipose-Derived Stromal Cells in a Murine Model of Acute Pancreatitis. J Gastrointest Surg. 2020;24:67-75
123. Xu L, Yang F, Lin R, Han C, Liu J, Ding Z. Induction of m2 polarization in primary culture liver macrophages from rats with acute pancreatitis. PLoS One. 2014;9:e108014
124. Sun K, He SB, Qu JG, Dang SC, Chen JX, Gong AH. et al. IRF5 regulates lung macrophages M2 polarization during severe acute pancreatitis in vitro. World J Gastroenterol. 2016;22:9368-77
125. Sendler M, van den Brandt C, Glaubitz J, Wilden A, Golchert J, Weiss FU. et al. NLRP3 Inflammasome Regulates Development of Systemic Inflammatory Response and Compensatory Anti-Inflammatory Response Syndromes in Mice With Acute Pancreatitis. Gastroenterology. 2020;158:253-69.e14
126. Kang R, Chen R, Xie M, Cao L, Lotze MT, Tang D. et al. The Receptor for Advanced Glycation End Products Activates the AIM2 Inflammasome in Acute Pancreatitis. J Immunol. 2016;196:4331-7
127. Hoque R, Farooq A, Ghani A, Gorelick F, Mehal WZ. Lactate reduces liver and pancreatic injury in Toll-like receptor- and inflammasome-mediated inflammation via GPR81-mediated suppression of innate immunity. Gastroenterology. 2014;146:1763-74
128. Wu XB, Sun HY, Luo ZL, Cheng L, Duan XM, Ren JD. Plasma-derived exosomes contribute to pancreatitis-associated lung injury by triggering NLRP3-dependent pyroptosis in alveolar macrophages. Biochim Biophys Acta Mol Basis Dis. 2020;1866:165685
129. Vrolyk V, Schneberger D, Le K, Wobeser BK, Singh B. Mouse model to study pulmonary intravascular macrophage recruitment and lung inflammation in acute necrotizing pancreatitis. Cell Tissue Res. 2019;378:97-111
130. Chen C, Xia SH, Chen H, Li XH. Therapy for acute pancreatitis with platelet-activating factor receptor antagonists. World J Gastroenterol. 2008;14:4735-8
131. Bombardo M, Saponara E, Malagola E, Chen R, Seleznik GM, Haumaitre C. et al. Class I histone deacetylase inhibition improves pancreatitis outcome by limiting leukocyte recruitment and acinar-to-ductal metaplasia. Br J Pharmacol. 2017;174:3865-80
132. Puhr S, Lee J, Zvezdova E, Zhou YJ, Liu K. Dendritic cell development-History, advances, and open questions. Semin Immunol. 2015;27:388-96
133. Françozo MCS, Costa FRC, Guerra-Gomes IC, Silva JS, Sesti-Costa R. Dendritic cells and regulatory T cells expressing CCR4 provide resistance to coxsackievirus B5-induced pancreatitis. Sci Rep. 2019;9:14766
134. Del Fresno C, Saz-Leal P, Enamorado M, Wculek SK, Martínez-Cano S, Blanco-Menéndez N. et al. DNGR-1 in dendritic cells limits tissue damage by dampening neutrophil recruitment. Science. 2018;362:351-6
135. Pan LL, Niu W, Fang X, Liang W, Li H, Chen W. et al. Clostridium butyricum Strains Suppress Experimental Acute Pancreatitis by Maintaining Intestinal Homeostasis. Mol Nutr Food Res. 2019: e1801419.
136. Xu D, Xie R, Xu Z, Zhao Z, Ding M, Chen W. et al. mTOR-Myc axis drives acinar-to-dendritic cell transition and the CD4(+) T cell immune response in acute pancreatitis. Cell Death Dis. 2020;11:416
137. Ochi A, Nguyen AH, Bedrosian AS, Mushlin HM, Zarbakhsh S, Barilla R. et al. MyD88 inhibition amplifies dendritic cell capacity to promote pancreatic carcinogenesis via Th2 cells. J Exp Med. 2012;209:1671-87
138. González-de-Olano D, Álvarez-Twose I. Mast Cells as Key Players in Allergy and Inflammation. J Investig Allergol Clin Immunol. 2018;28:365-78
139. Yönetçi N, Oruç N, Ozütemiz AO, Celik HA, Yüce G. Effects of mast-cell stabilization in cerulein-induced acute pancreatitis in rats. Int J Pancreatol. 2001;29:163-71
140. Kempuraj D, Twait EC, Williard DE, Yuan Z, Meyerholz DK, Samuel I. The novel cytokine interleukin-33 activates acinar cell proinflammatory pathways and induces acute pancreatic inflammation in mice. PLoS One. 2013;8:e56866
141. Leema G, Tamizhselvi R. Protective Effect of Scopoletin Against Cerulein-Induced Acute Pancreatitis and Associated Lung Injury in Mice. Pancreas. 2018;47:577-85
142. Dib M, Zhao X, Wang XD, Andersson R. Role of mast cells in the development of pancreatitis-induced multiple organ dysfunction. Br J Surg. 2002;89:172-8
143. Ouziel R, Gustot T, Moreno C, Arvanitakis M, Degré D, Trépo E. et al. The ST2 pathway is involved in acute pancreatitis: a translational study in humans and mice. Am J Pathol. 2012;180:2330-9
144. Zitti B, Bryceson YT. Natural killer cells in inflammation and autoimmunity. Cytokine Growth Factor Rev. 2018;42:37-46
145. Ramsingh AI, Lee WT, Collins DN, Armstrong LE. Differential recruitment of B and T cells in coxsackievirus B4-induced pancreatitis is influenced by a capsid protein. J Virol. 1997;71:8690-7
146. Mylona V, Koussoulas V, Tzivras D, Makrygiannis E, Georgopoulou P, Koratzanis G. et al. Changes in adaptive and innate immunity in patients with acute pancreatitis and systemic inflammatory response syndrome. Pancreatology. 2011;11:475-81
147. Wei X, Yao W, Li H, Qian J, Xie Y, Zhang Z. et al. B and NK Cells Closely Correlate with the Condition of Patients with Acute Pancreatitis. Gastroenterol Res Pract. 2019;2019:7568410
148. Ueda T, Takeyama Y, Yasuda T, Shinzeki M, Sawa H, Nakajima T. et al. Immunosuppression in patients with severe acute pancreatitis. J Gastroenterol. 2006;41:779-84
149. Dabrowski A, Osada J, Dabrowska MI, Wereszczynska-Siemiatkowska U. Monocyte subsets and natural killer cells in acute pancreatitis. Pancreatology. 2008;8:126-34
Author contact
![]() Corresponding authors: Xiao Yu and Zhiqiang Li, Department of Hepatopancreatobiliary Surgery, Third Xiangya Hospital, Central South University, Tongzipo Road No.138, Changsha 410013 Hunan, China. E-mail addresses: yuxiaoyx4com (Xiao Yu), 1214905254com (Zhiqiang Li).
Corresponding authors: Xiao Yu and Zhiqiang Li, Department of Hepatopancreatobiliary Surgery, Third Xiangya Hospital, Central South University, Tongzipo Road No.138, Changsha 410013 Hunan, China. E-mail addresses: yuxiaoyx4com (Xiao Yu), 1214905254com (Zhiqiang Li).