Impact Factor ISSN: 1449-1907

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2020; 17(16):2427-2439. doi:10.7150/ijms.49053 This issue Cite
Research Paper
Comprehensive analysis of TPX2-related ceRNA network as prognostic biomarkers in lung adenocarcinoma
Chen Huo1, Meng-Yu Zhang1, Rui Li1, Xi-Jia Zhou1, Ting-Ting Liu1, Jian-Ping Li1, Xiao Liu1, Yi-Qing Qu2 ![]()
1. Department of Pulmonary and Critical Care Medicine, Qilu Hospital, Cheeloo College of Medicine, Shandong University, Jinan 250012, China.
2. Department of Pulmonary and Critical Care Medicine, Qilu Hospital of Shandong University, Jinan 250012, China.
Received 2020-6-3; Accepted 2020-8-13; Published 2020-9-1
Abstract
Background and aim: Competing endogenous RNA (ceRNA) is believed to play vital roles in tumorigenesis. The goal of this study was to screen prognostic biomarkers in lung adenocarcinoma (LUAD).
Methods: Common differentially expressed genes (DEGs) were collected from Gene Expression Omnibus (GEO) databases and The Cancer Genome Atlas databases (TCGA) using GEO2R and “limma” package in R, respectively. Overlapping DEGs were conducted using enrichment of functions and protein-protein interaction (PPI) network to discover significant candidate genes. By using a comprehensive analysis, we constructed an mRNA mediated ceRNA network. Survival rates were used Kaplan-Meier analysis. Statistical analysis was used to further identify the prognosis of studied genes.
Results: Integrated analysis of GSE32863 and TCGA databases, a total of 886 overlapping DEGs, including 279 up-regulated and 607 down-regulated genes were identified. Considering the highest term of candidate genes in PPI, we identified TPX2, which was enriched in cell division signaling pathway. Besides, 35 differentially expressed miRNAs (DEmiRNAs) were predicted to target TPX2 and only 7 DEmiRNAs were identified to be prognostic biomarkers in LUAD. Then, 30 differentially expressed lncRNAs (DElncRNAs) were predicted to bind these 7 DEmiRNAs. Finally, we found that 7 DElncRNAs were correlated with the overall survival (all p <0.05). Furthermore, we identified elevated TPX2 was strongly correlated with the worse survival rate among 458 samples. Univariate and multivariate cox analysis showed TPX2 may act as an independent factor for prognosis in LUAD (p <0.05). Then pathway enrichment results suggested that TPX2 may facilitate tumorigenesis by participating in several cancer-related signaling pathways in LUAD, especially in Notch signal pathway.
Conclusions: TPX2-related lncRNAs and miRNAs are related to the survival of LUAD. 7 lncRNAs, 7 miRNAs and TPX2 may serve as prognostic biomarkers in LUAD.
Keywords: Lung adenocarcinoma, TPX2, competing endogenous RNA (ceRNA), prognostic biomarkers
Introduction
Lung cancer is the main cause of cancer-related death, and lung adenocarcinoma (LUAD) is one of histologic type, which account for about half of lung cancer [1-4]. Despite the advances in new targeted therapies, chemotherapy, radiotherapy as well as surgical methods, the 5-year survival rate of LUAD is 20% [5-7], which illustrate the urgency to find biomarkers for prognosis of LUAD. So it is vital to identify potential molecular mechanisms and significant prognostic biomarkers in LUAD.
Long non-coding RNAs (lncRNAs) with longer than 200 bp could interact with mRNAs, miRNAs and circular RNAs, participating in complicated regulation network of gene expression. LncRNAs' abnormal expressions have linked with various cancers, for instance, prostate cancer, colorectal cancer and pancreatic cancer [8-12]. MiRNAs are classes of short RNA molecules with 19 to 25 nucleotides, which regarded as one of the regulators of gene expression and silenced target genes in post-transcriptional levels. Growing evidences suggested that miRNAs participate in biological process, for instance, DNA damage response, cell proliferation, cell apoptosis [13-15]. With further understanding of biological process, a new regulatory mechanism named competing endogenous RNA (ceRNA) was proposed, which provides a better insight into tumorigenesis [16]. CeRNA can bind to miRNA by miRNA response elements (MREs) to induce gene silencing. More and more researches demonstrated that ceRNA network plays crucial roles in numerous cancers [17-18].
Targeting protein for Xenopus kinesin-like protein 2 (TPX2) is a microtubule-associated protein, which was involved in mitotic spindle formation [19-20]. Growing evidence has proved that increased TPX2 contribute to the poor prognosis by triggering spindle dysfunctions, subsequent chromosomal instability or abnormal DNA damage response in tumor development [21]. TPX2 overexpression is positively correlated with lymphatic metastasis, tumor grade and stage, negatively with the survival rate [22-24]. However, there are no systematic studies to investigate the prognostic value of TPX2 in LUAD. This study aims to construct a critical TPX2-related ceRNA network in LUAD.
Material and Methods
Data collection and significant differentially expressed RNAs screening
The gene expression profile of GSE32863 based on the GPL6884 platform, was selected from Gene Expression Omnibus (GEO) (http://www.ncbi.nlm.nih.gov/geo/) [25]. GSE32863 included 58 pairs of LUAD and normal lung tissues, normalized microarray data was reanalyzed using the GEO2R online tools (https://www.ncbi.nlm.nih.gov/geo/geo2r/) [26]. The Cancer Genome Atlas (TCGA) (http://cancergenome.nih.gov/) contains expression profiles and prognostic information, so we gathered RNA-sequencing data of LUAD as well. Then “limma” package in R was performed to identify differentially expressed lncRNAs (DElncRNAs), differentially expressed miRNAs (DEmiRNAs) and differentially expressed genes (DEGs). Volcano plot was generated using the “ggplot2” R package, and the “Venn Diagram” package was performed to overlap DEGs. Down-regulated gene was defined as logFC<0, on the contrary, up-regulated was defined as log FC >0. The significant differentially expressed RNAs were defined as p< 0.01 and |logFC| > 1, statistically.
The functional enrichment and pathway analysis
Gene ontology (GO) analysis is a common method to identify functional enrichment of gene data. And the function and pathway analysis were constructed through Database for Annotation, Visualization and Integrated Discovery (DAVID) (https://david.ncifcrf.gov/). Then functional enrichment of candidate genes were visualized using “GOplot” R package. As for signaling pathway analysis, “cluster profile” R package was used to discover significant pathways. p < 0.05 was used to significant terms statistically. To identify the potential function of TPX2, Gene set enrichment analysis (GSEA) (http://software.broadinstitute.org/gsea/index.jsp) was conducted between the low-expression and high-expression groups [27]. And p < 0.05 was regarded as statistically significant.
Candidate gene selection
Search Tool for the Retrieval of Interacting Genes (STRING) (http://string-db.org/) was used to obtain the predicted interactions for overlapping DEGs with confidence score >0.4 [28], protein-protein interaction (PPI) network of overlapping DEGs was visualized by Cytoscape [29]. The CytoHubba plugin gave us a novel insight into the importance of networks by ranking nodes [30], according to which we found hub genes. The Molecular Complex Detection (MCODE) plugin was performed to select linked genes with degree cutoff = 2 and k-core = 2.
Construction of a TPX2-related ceRNA network
LncRNA and mRNA have the same miRNA response elements (MREs) by competitively binding to miRNA to regulate each other's expression. In this study, a ceRNA network of lncRNAs, miRNAs, and TPX2 was constructed to explore their functions. The relationship between TPX2 and target miRNAs was analyzed by using three databases miRTarBase (http://mirtarbase.mbc.nctu.edu.tw/), TargetScan (http://www.targetscan.org/) and StarBase (http://starbase.sysu.edu.cn/). The unions of the prediction results of three databases were used to perform further analysis. LncRNAs targeted by the miRNAs were retrieved from the StarBase.
Statistical analysis
Statistical analysis was performed using SPSS and R 3.6.2 software. The differences of TPX2 expression levels between the LUAD and normal lung groups were determined by Student's t-test. The Chi-square test was used to analyze the associations between TPX2 expression and clinicopathological factors of LUAD patients. The survival R packages were performed for survival analysis of TPX2 and survival curve was displayed by Kaplan-Meier plotter and examined by log-rank test. Univariate and multivariate Cox analysis were used to evaluate the prognostic value between TPX2 and other clinicopathological fators. Lung cancer explorer (LCE) database (http://lce.biohpc.swmed.edu/lungcancer/index.php#page-top) includes gene expression and clinical data from more than 6,700 lung cancer patients, which provides forest plots to summarize tumor-normal standardized mean difference and hazard ratios [31]. The p < 0.05 was considered to be statistically significant.
Results
Differentially expressed RNAs in LUAD
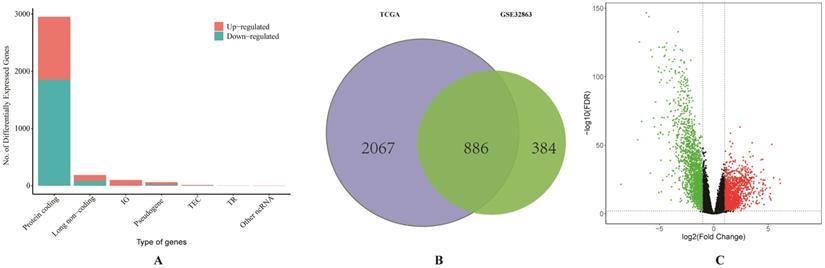
We firstly obtained the gene expression profile of 58 pairs of LUAD tissues and their matched normal lung tissues from GSE32863. A total of 1270 DEGs were identified by using GEO2R. Then the RNA-sequencing data and clinical data of LUAD were downloaded from TCGA. Based on the differentially expressed RNAs analysis by limma R package, a total of 186 DElncRNAs, 150 DEmiRNAs and 2953 DEGs were identified, and the proportion of all differentially expressed RNAs from TCGA was visualized (Figure 1A). As it shown in Figure 1C, the result of volcano plot displayed the distribution of DEGs in the dimensions of -log (FDR) and logFC. On the basis of p <0.01 and |Log2(FC)|>1, a total of 886 overlapping DEGs, including 279 up-regulated and 607 down-regulated genes, were screened from two studies (Figure 1B).
Comprehensive analysis of differentially expressed RNAs. (A) The bar graph of the proportion of all differentially expressed RNAs from TCGA database. (B) Identification of differential expressed genes from TCGA and GSE32863. (C) The volcano plot of differentially expressed genes from TCGA database.
Enrichment analysis of DEGs in LUAD
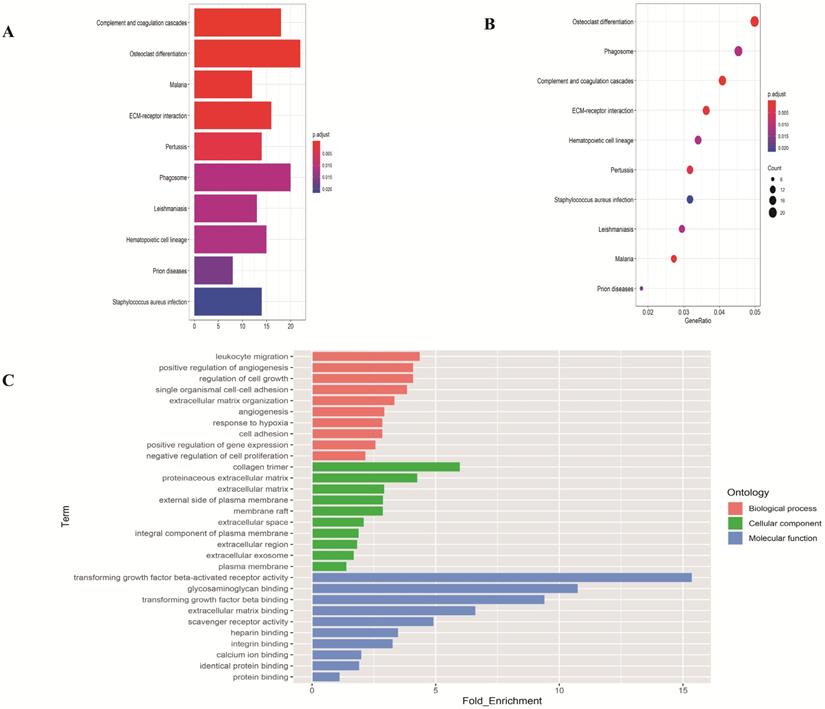
To investigate the function of the DEGs, GO and KEGG analysis were performed using the web-based tool DAVID. Enriched terms with p <0.05 were displayed (Figure 2). The genes were mainly enriched in leukocyte migration and positive regulation of angiogenesis in the biological process; collagen trimer and proteinaceous extracellular matrix in the cellular component; transforming growth factor beta-activated receptor activity and glycosaminoglycan binding in the molecular function.
Functional enrichment analysis of overlapping differentially expressed genes in LUAD. (A-B) The top ten pathways of overlapping differentially expressed genes in barplot and dotplot. (C) Gene Ontology terms of overlapping differentially expressed genes.
Construction of the PPI network and screening of modules and hub genes
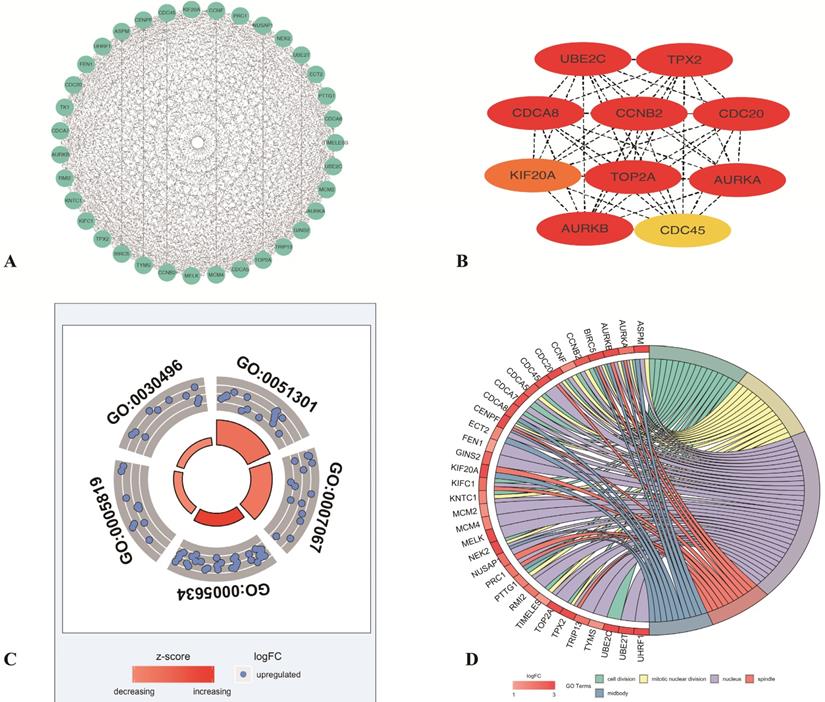
To identify the most important proteins and biological modules which may be involved in the progression of LUAD the PPI network was generated by overlapping genes using STRING and the interactors of top 10 hub genes were visualized using Cytoscape software. Subsequently, to explore the significance of these DEGs, the top 1 module, which displayed the most densely connected region, was selected (Figure 3A). The result showed the top 10 hub nodes (TPX2, TOP2A, AURKB, CCNB2, CDC20, CDCA8, AURKA, UBE2C, KIF20A, CDC45) with the highest degrees in LUAD (Figure 3B). And to explore the possible function of these 35 selected DEGs, the functional annotation of 35 genes (PTTG1, TPX2, ASPM, NUSAP1, TOP2A, TYMS, TK1, CDCA7, AURKB, CCNB2, TRIP13, CDC20, NEK2, UHRF1, CDCA8, FEN1, MCM2, MCM4, KNTC1, MELK, BIRC5, CENPF, KIFC1, AURKA, CCNF, TIMELESS, UBE2C, PRC1, UBE2T, KIF20A, CDC45, GINS2, CDCA5, RMI2, ECT2) made by M-code showed that 5 GO terms were statistically significant (p <0.05). The result showed the interaction between 35 DEmRNAs and their associated GO terms, in which the highest GO term was cell division (Figure 3C-D), and 16 DEGs (KIFC1, CDCA8, CCNB2, CDCA7, TIMELESS, NEK2, CCNF, TPX2, KNTC1, CENPF, BIRC5, AURKA, CDC20, PTTG1, UBE2C, CDCA5) were contained in this term (Table 1). Above all, top 10 hub genes, which were contained in these 16 genes, indicated that these proteins may serve crucial roles in the whole protein interaction network. Then in consideration of the enrichment results of highest GO term, TPX2 was selected for further analysis.
The top5 GO terms of 35 differentially expressed genes
| Category | ID | Term | Genes | adj_pval |
|---|---|---|---|---|
| BP | GO:0051301 | cell division | KIFC1, CDCA8, CCNB2, CDCA7, TIMELESS, NEK2, CCNF, TPX2, KNTC1, CENPF, BIRC5, AURKA, CDC20, PTTG1, UBE2C, CDCA5 | 1.44E-13 |
| BP | GO:0007067 | mitotic nuclear division | CCNB2, TIMELESS, NEK2, CCNF, KNTC1, TPX2, CENPF, AURKA, BIRC5, CDC20, PTTG1, AURKB, CDCA5, ASPM | 1.03E-12 |
| CC | GO:0005634 | nucleus | KIFC1, PRC1, NEK2, KNTC1, AURKA, PTTG1, AURKB, TYMS, CDC45, CDCA8, CDCA7, TOP2A, CDCA5, ASPM, FEN1, TRIP13, GINS2, CCNF, TPX2, NUSAP1, CENPF, RMI2, BIRC5, CDC20, MCM2, ECT2, MCM4, UHRF1, CCNB2, TIMELESS, MELK, UBE2T | 1.01E-10 |
| CC | GO:0005819 | spindle | KIFC1, PRC1, TPX2, NUSAP1, CENPF, AURKA, BIRC5, CDC20, AURKB, KIF20A | 9.10E-10 |
| CC | GO:0030496 | midbody | CDCA8, PRC1, NEK2, CENPF, AURKA, BIRC5, AURKB, ECT2, ASPM, KIF20A | 1.63E-09 |
Screening the key genes and further GO analysis from PPI network. (A-B) Hub genes selected using MCODE and CytoHubba. (C) The outer circle represents the expression (logFC) of 35-differentially expressed genes in each enriched GO (gene ontology) terms. The inner circle indicates the significance of GO terms (log10-adjusted p values). Red points indicate upregulated gene and blue points indicate downregulated gene. (D) The circle indicates the correlation between 35 differentially expressed genes and their gene ontology terms.
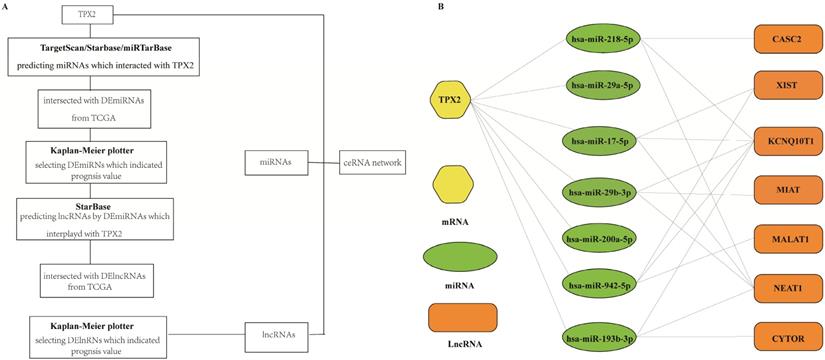
Construction of TPX2-related ceRNA network
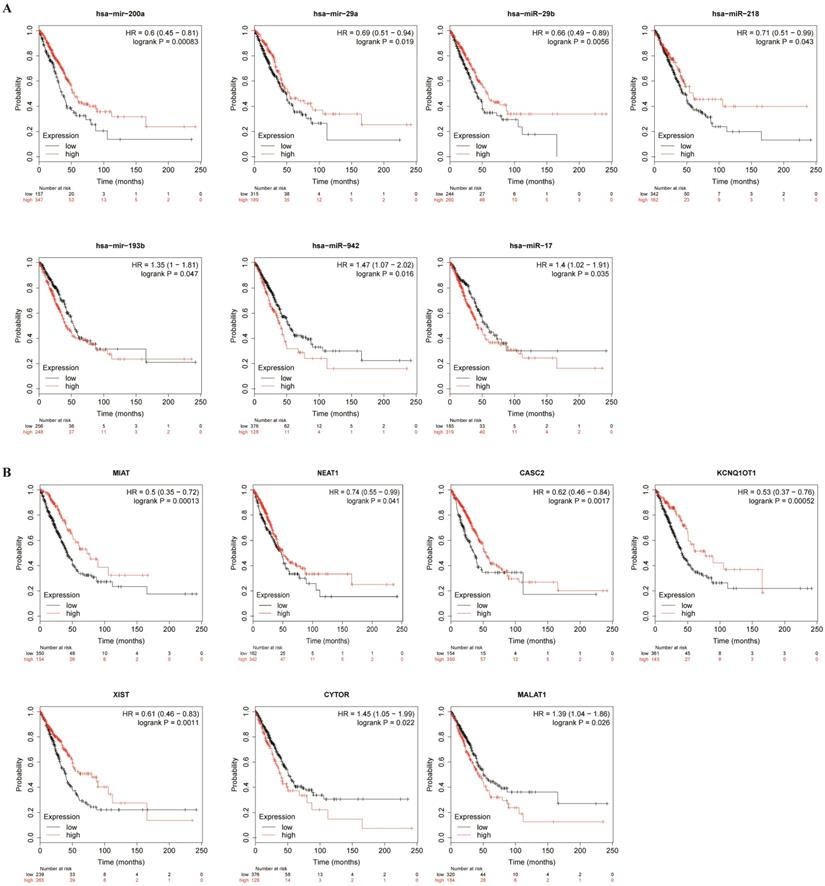
To better predict the upstream regulation mechanism of TPX2, we constructed a ceRNA network. At the beginning, the miRNAs targeted by TPX2 were predicted via StarBase, miRTarBase and TargetScan. We identified a total of 35 DEmiRNAs from TCGA, and we further assessed prognostic values of these 35 predicted DEmiRNAs using Kaplan-Meier plotter database. The results showed that 7 of 35 DEmiRNAs (miR-942-5p, miR-193b-3p, miR-29b-3p, miR-17-5p, miR-218-5p, miR-29a-5p and miR-200a-5p) were significantly associated with overall survival (all p <0.05) (Figure 4A). Subsequently, the lncRNAs targeted by 7 DEmiRNAs were predicted via StarBase. A total of 30 DElncRNAs were predicted, and only 7 of 30 DElncRNAs (KCNQ1OT1, XIST, MALAT1, CYTOR, NEAT1, CASC2, MIAT) have prognostic values (all p <0.05) (Figure 4B). Ultimately, we constructed the TPX2-related ceRNA network of LUAD, including 7 DElncRNAs, 7 DEmiRNAs as well as TPX2, which functioned as prognostic biomarkers for LUAD patients (Figure 5A-B).
Kaplan-Meier curve analysis of TPX2-related DElncRNAs and DEmiRNAs in LUAD. (A) Prognostic values of DEmiRNAs in LUAD. (B) Prognostic values of DElncRNAs in LUAD.
(A) A flow diagram of TPX2-related ceRNA network. (B) Construction of lncRNA-miRNA-TPX2 network in LUAD.
TPX2 expression and correlation with clinical characteristics
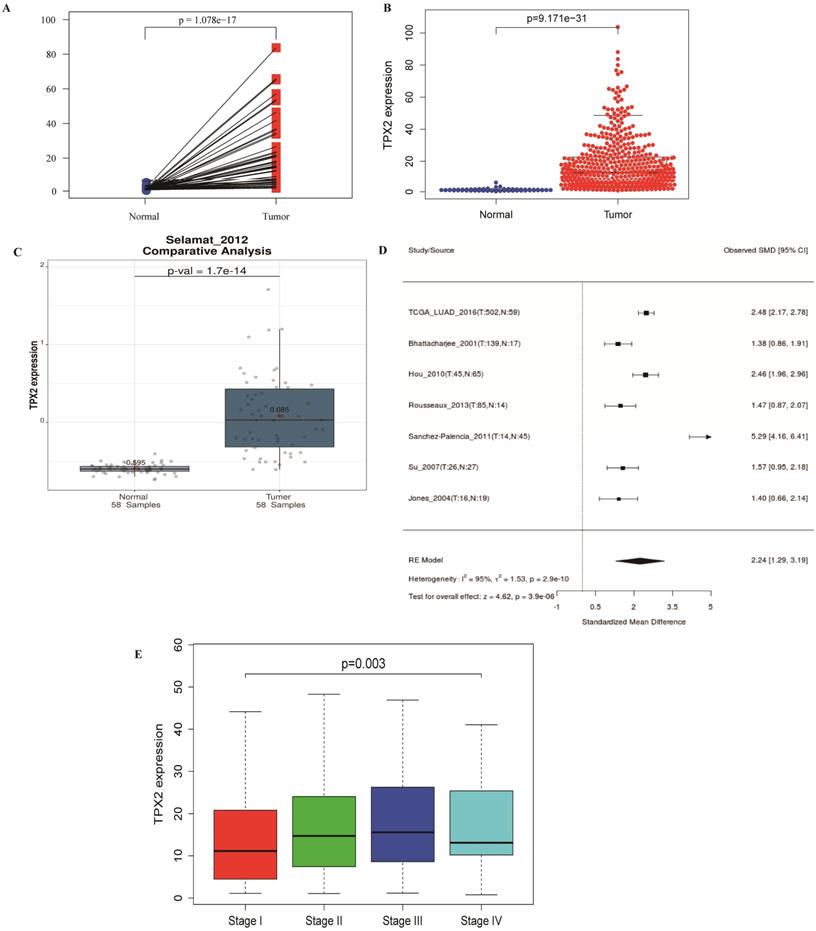
486 LUAD and normal lung samples from the TCGA database were enrolled, and a simple summary of the data was showed in Table 2. The expression of TPX2 in LUAD tissues was significantly up-regulated compared with normal tissues (p <0.001), which is consistent with GEO database (Figure 6A-C). The expression of TPX2 in the 7 independent datasets was pooled in a forest plot (Figure 6D). The SMD was 1.40 (95% CI: 1.29-3.19; I2 =95%; p = 2.0e-10), which suggested that the occurrence of LUAD is related to expression levels of TPX2. To determine whether TPX2 expression level was related to LUAD progression, expression level of TPX2 from 458 patients was analyzed. Clinical characteristics and their relations with TPX2 were performed, and the results showed TPX2 expression level was significantly associated with gender (p=0.005), stage (p=0.001), pathological T stage (p=0.018) and pathological N stage (p=0.004). However, no significant difference was found in age (p=0.133) and pathological M stage (p=0.703) (Table 3). Figure 6E showed that the expression level of TPX2 displayed strong correlation with the tumor stage in patients with LUAD (p <0.05).
Clinical characteristics of LUAD patients obtained from TCGA database
| Clinical characteristics | Variable | Patients, n (%) |
|---|---|---|
| Age | >65 | 240 (49) |
| ≤65 | 227 (47) | |
| unknown | 19 (4) | |
| Gender | Male | 222 (46) |
| Female | 264 (54) | |
| Pathology stage | I | 262 (54) |
| II | 112 (23) | |
| III | 79 (16) | |
| IV | 25 (5) | |
| unknown | 8 (2) | |
| Pathology T stage | T1 | 163 (34) |
| T2 | 260 (53) | |
| T3 | 41 (8) | |
| T4 | 19 (4) | |
| unknown | 3 (1) | |
| Pathology M stage | M0 | 333 (69) |
| M1 | 24 (5) | |
| Mx | 129 (26) | |
| Pathology N stage | N0 | 312 (64) |
| N1 | 90 (19) | |
| N2 | 70 (14) | |
| N3 | 2 (1) | |
| Nx | 12 (2) |
Association between TPX2 expression levels and clinicopathological characteristics of LUAD patients
| Characteristic | n=458 | TPX2 | ||
|---|---|---|---|---|
| Low (n=229) | High (n=229) | p value | ||
| Age (years) | 0.133 | |||
| <65 | 206 | 95 | 111 | |
| ≥65 | 252 | 134 | 118 | |
| Gender | 0.005 | |||
| Male | 208 | 89 | 119 | |
| Female | 250 | 140 | 110 | |
| Stage | 0.001 | |||
| I | 246 | 143 | 103 | |
| II | 105 | 39 | 66 | |
| III | 74 | 30 | 44 | |
| IV | 25 | 11 | 14 | |
| unknown | 8 | 6 | 2 | |
| Pathological T stage | 0.018 | |||
| T1 | 158 | 96 | 62 | |
| T2 | 239 | 105 | 134 | |
| T3 | 39 | 17 | 22 | |
| T4 | 19 | 9 | 10 | |
| unknown | 3 | 2 | 1 | |
| Pathological N stage | 0.004 | |||
| N0 | 295 | 159 | 136 | |
| N1 | 84 | 34 | 50 | |
| N2+3 | 67 | 26 | 41 | |
| Nx | 12 | 10 | 2 | |
| Pathological M stage | 0.703 | |||
| M0 | 305 | 154 | 151 | |
| M1 | 24 | 10 | 14 | |
| unknown | 129 | 65 | 64 | |
The relationship between TPX2 expression and clinical characteristics in LUAD. (A-C) Expression of TPX2 in LUAD patients based on TCGA and GSE32863. (D) Meta-analysis for the expression of TPX2 in LUAD and normal lung tissues. (E) The relation between TPX2 expression and tumor stage in LUAD patients.
Prognostic significance of TPX2 in LUAD
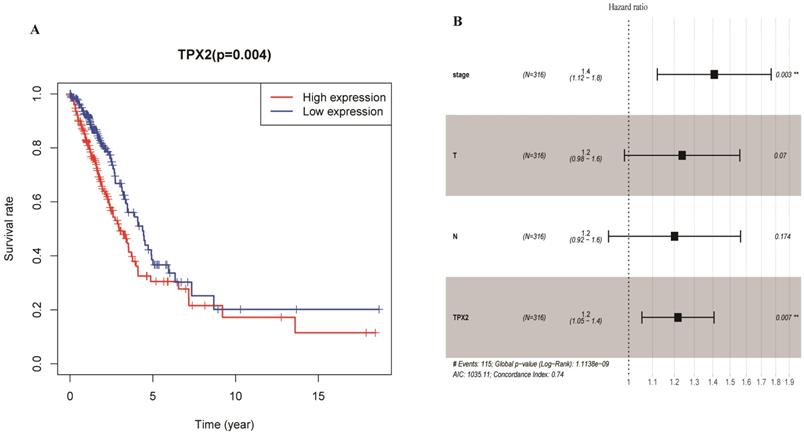
To elucidate the prognostic values of TPX2, we assessed the correlation between TPX2 expression level and overall survival rate in LUAD. On the basis of the median risk scores, a total of 458 patients were divided into a high risk group and a low risk group. The results showed that high expression of TPX2 was associated with worse overall survival for LUAD patients (p=0.004) (Figure 7A). Moreover, we used univariate and multivariate analysis to distinguish the risk factors correlated with the prognosis of LUAD patients by using Cox regression model. Univariate independent prognostic analysis showed that clinical stage, pathological T stage, pathological N stage and expression levels of TPX2 were statistically significant with overall survival (p <0.05). Multivariate independent prognostic analysis showed that clinical stage and the TPX2 expression can act as independent prognostic factors (p <0.05) (Figure 7B, Table 4).
Univariate and multivariate analysis of prognostic factors in LUAD
| Variables | Univariate analysis | Multivariate analysis | ||
|---|---|---|---|---|
| Hazard ratio (95% CI) | p value | Hazard ratio (95%CI) | p value | |
| Age | 1.002 (0.983-1.021) | 0.843 | ||
| Gender | 1.035 (0.717-1.495) | 0.852 | ||
| Stage | 1.654 (1.401-1.951) | <0.01 | 1.408 (1.121-1.768) | 0.003 |
| T | 1.632 (1.315-2.024) | <0.01 | 1.238 (0.983-1.560) | 0.070 |
| M | 1.757 (0.964-3.203) | 0.066 | ||
| N | 1.790 (1.459-2.196) | <0.01 | 1.201 (0.922-1.564) | 0.174 |
| TPX2 | 1.010 (1.003-1.017) | 0.003 | 1.218 (1.054-1.4077) | 0.007 |
CI: confidence interval.
Expression of TPX2 in LUAD was correlated with prognosis. (A) Overall survival analysis of LUAD patients with different TPX2 expression. (B) Forest plot of multivariate cox analysis in LUAD.
Gene Set Enrichment Analysis of TPX2
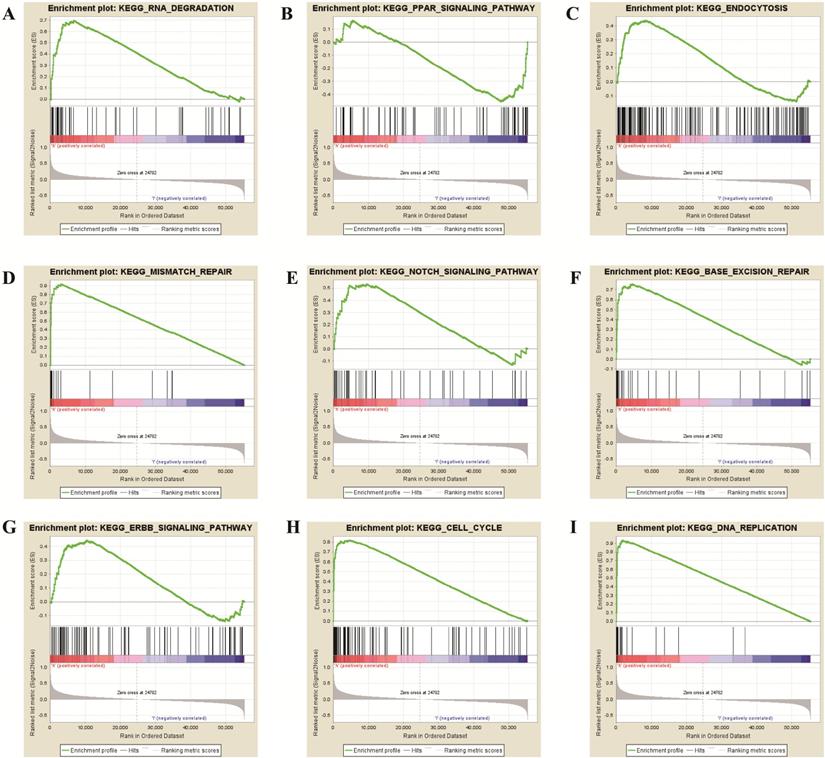
To further verify the potential function of TPX2 gene, GSEA was used to obtain the biological process. Ultimately, TPX2 was associated with cancer-related signaling pathways including “cell cycle”, “RNA degradation”, “mismatch repair”, “DNA replication”, “base excision repair”, “endocytosis”, “Notch signaling pathway” “PPAR signaling pathway” and “ERBB signaling pathway” (all p <0.05) (Figure 8, Table 5). Enrichment plots of GSEA demonstrated that the gene signatures of “Notch signaling pathway” (NES = 1.76) with higher TPX2 expression were more active than lower TPX2 expression (p <0.05, FDR <0.05). All these results suggested that TPX2 may facilitate tumorigenesis by taking part in several cancer-related signaling pathways in LUAD.
The significant enriched signaling pathways from GSEA results (p < 0.05)
| NAME | ES | NES | NOM p-val | FDR q-val |
|---|---|---|---|---|
| CELL CYCLE | 0.818 | 2.690 | 0.000 | 0.000 |
| OOCYTE MEIOSIS | 0.649 | 2.499 | 0.000 | 0.000 |
| HOMOLOGOUS RECOMBINATION | 0.897 | 2.417 | 0.000 | 0.000 |
| UBIQUITIN MEDIATED PROTEOLYSIS | 0.591 | 2.397 | 0.000 | 0.000 |
| SPLICEOSOME | 0.788 | 2.390 | 0.000 | 0.000 |
| NUCLEOTIDE EXCISION REPAIR | 0.765 | 2.370 | 0.000 | 0.000 |
| RNA DEGRADATION | 0.696 | 2.364 | 0.000 | 0.000 |
| P53 SIGNALING PATHWAY | 0.604 | 2.315 | 0.000 | 0.000 |
| MISMATCH REPAIR | 0.917 | 2.313 | 0.000 | 0.000 |
| BASAL TRANSCRIPTION FACTORS | 0.714 | 2.257 | 0.000 | 0.001 |
| PYRIMIDINE METABOLISM | 0.642 | 2.254 | 0.000 | 0.001 |
| DNA REPLICATION | 0.933 | 2.206 | 0.000 | 0.001 |
| PENTOSE PHOSPHATE PATHWAY | 0.732 | 2.145 | 0.000 | 0.002 |
| PROTEASOME | 0.859 | 2.129 | 0.000 | 0.002 |
| BASE EXCISION REPAIR | 0.759 | 2.099 | 0.000 | 0.003 |
| PATHWAYS IN CANCER | 0.467 | 2.034 | 0.002 | 0.006 |
| FC GAMMA R MEDIATED PHAGOCYTOSIS | 0.501 | 1.921 | 0.002 | 0.015 |
| PROSTATE CANCER | 0.478 | 1.846 | 0.008 | 0.024 |
| REGULATION OF ACTIN CYTOSKELETON | 0.452 | 1.829 | 0.008 | 0.027 |
| RNA POLYMERASE | 0.629 | 1.810 | 0.012 | 0.030 |
| ENDOCYTOSIS | 0.437 | 1.793 | 0.006 | 0.032 |
| NOTCH SIGNALING PATHWAY | 0.534 | 1.759 | 0.014 | 0.041 |
| GAP JUNCTION | 0.455 | 1.749 | 0.011 | 0.042 |
| PROTEIN EXPORT | 0.637 | 1.730 | 0.031 | 0.046 |
| ERBB SIGNALING PATHWAY | 0.445 | 1.724 | 0.014 | 0.046 |
| PRION DISEASES | 0.545 | 1.722 | 0.021 | 0.046 |
| ASTHMA | -0.729 | -1.765 | 0.046 | 0.120 |
| PPAR SIGNALING PATHWAY | -0.455 | -1.642 | 0.008 | 0.151 |
GSEA revealed that TPX2 was enriched in pathways of (A) RNA Degradation, (B) PPAR signaling pathway, (C) Endocytosis, (D) Mismatch Repair, (E) Notch signaling pathway, (F) Base Excision Repair, (G) ERBB signaling pathway, (H) Cell cycle, (I) DNA Replication.
Discussion
Lung cancer is the chief cause of cancer death (18.4% of the 9.6 million cancer death) [32], and LUAD is the most common subtype of lung cancer. Despite advances in the diagnosis and treatment, the overall survival rates remain poor in LUAD [33]. Recent studies have identified novel molecular biomarkers may provide a prognosis value in combination with patients' clinical parameters [34-35], and in which non-coding RNAs (ncRNAs) play vital roles in cancer initiation and progression [36]. For example, LncRNA DGCR5 promotes LUAD progression [37], miR-1323 promotes cell migration in LUAD [38]. With the increasing understanding of ceRNA, more and more people have studied the mechanism of ceRNAs in cancer. Yang et al. indicated that lncRNA LCAT1 regulates RAC1 in lung cancer [39]. And lncRNA WDFY3-AS2 promotes LUAD progression via targeting miR-491-5p/ZNF703 axis [40]. In this present study, we firstly constructed a new mRNA-miRNA-lncRNA regulatory network using RNA sequencing data and microchip data, in which each RNA in the network has important implications for the prognosis of LUAD.
A total of 886 significant DEGs were identified, which mainly enriched in cell growth, cell adhesion and metabolic pathways. Considering the results of functional enrichment analysis, these significant DEGs may regulate growth of LUAD. We obtained 35 core genes from PPI network and these genes were re-analyzed for functional enrichment, in which cell division were chosen for further study because it is the most significant GO terms (p=1.44E-13). Finally, 16 DEGs were significantly enriched in the cell division which is closely related to tumorigenesis. Considering the highest term of candidate genes in PPI, we identified TPX2 for further study. Besides, we discovered that most of these genes have been well studied except TPX2. For example, AURKA promotes tumor proliferation and was associated with worse prognosis in bladder cancer and gastrointestinal cancer [41-42]. Increased AURKB have a poor prognosis in NSCLC [43]. CDCA8 participated in lung carcinogenesis [44]. KIF20A promoted cell proliferation and inhibit apoptosis in LUAD [45]. And UBE2C was found to enhance lung cancer growth [46].
Subsequently, we predicted 30 DElncRNAs and 35 DEmiRNAs, in which 7 DEmiRNAs and 7 DElncRNAs functioned as prognostic biomarkers in LUAD. High expression levels of 4 DEmiRNAs (miR-218-5p, miR-29b-3p, miR-200a-5p, miR-29a-5p) have a better prognosis and high expression levels of 3 DEmiRNAs (miR-17-5p, miR-942-5p, miR-193b-3p) were associated with worse prognosis in LUAD. In the past studies, miR-218-5p inhibited proliferation and migration of NSCLC [47]. LncRNA H19 was found to promote epithelial-mesenchymal transition of LUAD by targeting miR-29b-3p and modifying STAT3 [48]. Wang A et al. proved that miR-29a-5p regulated TETs in colorectal cancer [49]. MiRNA-17-5p inhibited triggers apoptosis in NSCLC [50]. Then we predicted 30 DElncRNAs of 7 DEmiRNAs, and only 7 DElncRNAs were significantly associated with overall survival. Low expression levels of 2 DElncRNAs (MALAT1 and CYTOR) increased survival times. High expression levels of 5 DElncRNAs (KCNQ1OT1, XIST, NEAT1, CASC2, MIAT) have a better prognosis. In previous studies, KCNQ1OT1 indicated a better prognosis in lung cancer [51]. MALAT1 promoted the proliferation and invasion of NSCLC [52]. Finally, a prognosis-associated TPX2-DEmiRNA-DElncRNA network in LUAD was established.
Then our findings revealed that overexpressed TPX2 is correlated with poor prognosis in LUAD. Moreover, univariate and multivariate cox model proved that TPX2 expression can act as an independent factor for prognisis. For example, TPX2 is an independent prognostic factor in esophageal cancer [53-54], digestive system cancer [55] and clear cell renal cell carcinoma [56]. Furthermore, our analysis indicated TPX2 was mostly concentrated in tumor-related pathways. Among results of pathway, we ultimately focused on the Notch pathway, which is connected with cell proliferation, differentiation and survival. And Notch pathway played a crucial role in the development of multiple cancers [57-58]. Previous study showed that activation of the Notch pathway can lead to a malignant phenotype of lung cancer [59]. It has been noted that Notch1 signaling was reported to be activated in NSCLC [60]. Another study proved that Notch2 has a tumor suppressive effect in NSCLC [61]. Hassan et al. presented that Notch3 behaves as a tumor promoter pathway in NSCLC [62].
In conclusion, we constructed a novel TPX2-related ceRNA regulatory network by comprehensive bioinformatics analysis, in which all RNAs could be prognostic biomarkers of LUAD. However, these biomarkers need to be further validated by molecular biology experiments.
Abbreviations
DAVID: The Database for Annotation, Visualization and Integrated Discovery; GO: Gene ontology; KEGG: Kyoto Encyclopedia of Genes and Genomes; NSCLC: Non-small-cell lung cancer; LUAD: Lung adenocarcinoma; TPX2: Targeting protein for Xenopus kinesin-like protein 2; FDR: False discovery rate; ceRNA: Competing endogenous RNA; TCGA: The Cancer Genome Atlas Project; GEO: The Gene Expression Omnibus database; LCE: Lung cancer explorer; DElncRNAs: differentially expressed lncRNAs; DEmiRNAs: differentially expressed miRNAs; DEGs: differentially expressed genes; ncRNAs: non-coding RNAs.
Acknowledgements
This work was supported by grants from Major Scientific and Technological Innovation Project of Shandong Province [2018CXGC1212], Science and Technology Foundation of Shandong Province [2014GSF118084, 2016GSF121043], Medical and Health Technology Innovation Plan of Jinan City [201805002].
Ethics approval and consent to participate
Consent for participation for all patients was obtained through The Cancer Genome Atlas Project.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Fitzmaurice C, Dicker D, Pain A, Hamavid H, Moradi-Lakeh M, MacIntyre MF. et al. The Global Burden of Cancer 2013. JAMA oncology. 2015;1:505-27
2. Travis WD, Brambilla E, Noguchi M, Nicholson AG, Geisinger KR, Yatabe Y. et al. International association for the study of lung cancer/american thoracic society/european respiratory society international multidisciplinary classification of lung adenocarcinoma. Journal of thoracic oncology: official publication of the International Association for the Study of Lung Cancer. 2011;6:244-85
3. Latimer KM, Mott TF. Lung cancer: diagnosis, treatment principles, and screening. American family physician. 2015;91:250-6
4. Travis WD. Lung Cancer Pathology: Current Concepts. Clinics in chest medicine. 2020;41:67-85
5. Testa U, Castelli G, Pelosi E. Lung Cancers: Molecular Characterization, Clonal Heterogeneity and Evolution, and Cancer Stem Cells. Cancers. 2018 10
6. Schiller JH, Harrington D, Belani CP, Langer C, Sandler A, Krook J. et al. Comparison of four chemotherapy regimens for advanced non-small-cell lung cancer. The New England journal of medicine. 2002;346:92-8
7. Yu X, Zhang Y. Identification of a long non-coding RNA signature for predicting prognosis and biomarkers in lung adenocarcinoma. Oncology letters. 2020;19:2793-800
8. Ling H, Vincent K, Pichler M, Fodde R, Berindan-Neagoe I, Slack FJ. et al. Junk DNA and the long non-coding RNA twist in cancer genetics. Oncogene. 2015;34:5003-11
9. Kung JT, Colognori D, Lee JT. Long noncoding RNAs: past, present, and future. Genetics. 2013;193:651-69
10. Prensner JR, Iyer MK, Balbin OA, Dhanasekaran SM, Cao Q, Brenner JC. et al. Transcriptome sequencing across a prostate cancer cohort identifies PCAT-1, an unannotated lincRNA implicated in disease progression. Nature biotechnology. 2011;29:742-9
11. Ling H, Spizzo R, Atlasi Y, Nicoloso M, Shimizu M, Redis RS. et al. CCAT2, a novel noncoding RNA mapping to 8q24, underlies metastatic progression and chromosomal instability in colon cancer. Genome research. 2013;23:1446-61
12. Yin Z, Zhou Y, Ma T, Chen S, Shi N, Zou Y. et al. Down-regulated lncRNA SBF2-AS1 in M2 macrophage-derived exosomes elevates miR-122-5p to restrict XIAP, thereby limiting pancreatic cancer development. Journal of cellular and molecular medicine. 2020
13. Zhang C, Peng G. Non-coding RNAs: an emerging player in DNA damage response. Mutation research Reviews in mutation research. 2015;763:202-11
14. Mikell I, Crawford LB, Hancock MH, Mitchell J, Buehler J, Goodrum F. et al. HCMV miR-US22 down-regulation of EGR-1 regulates CD34+ hematopoietic progenitor cell proliferation and viral reactivation. PLoS pathogens. 2019;15:e1007854
15. Hu F, Yang J, Chen X, Shen Y, Chen K, Fu X. et al. LncRNA 1700020I14Rik/miR-297a/CGRP axis suppresses myocardial cell apoptosis in myocardial ischemia-reperfusion injury. Molecular immunology. 2020;122:54-61
16. Salmena L, Poliseno L, Tay Y, Kats L, Pandolfi PP. A ceRNA hypothesis: the Rosetta Stone of a hidden RNA language? Cell. 2011;146:353-8
17. Wang L, Cho KB, Li Y, Tao G, Xie Z, Guo B. Long Noncoding RNA (lncRNA)-Mediated Competing Endogenous RNA Networks Provide Novel Potential Biomarkers and Therapeutic Targets for Colorectal Cancer. International journal of molecular sciences. 2019 20
18. Qi X, Zhang DH, Wu N, Xiao JH, Wang X, Ma W. ceRNA in cancer: possible functions and clinical implications. Journal of medical genetics. 2015;52:710-8
19. Wittmann T, Boleti H, Antony C, Karsenti E, Vernos I. Localization of the kinesin-like protein Xklp2 to spindle poles requires a leucine zipper, a microtubule-associated protein, and dynein. The Journal of cell biology. 1998;143:673-85
20. Wittmann T, Wilm M, Karsenti E, Vernos I. TPX2, A novel xenopus MAP involved in spindle pole organization. The Journal of cell biology. 2000;149:1405-18
21. Neumayer G, Belzil C, Gruss OJ, Nguyen MD. TPX2: of spindle assembly, DNA damage response, and cancer. Cellular and molecular life sciences: CMLS. 2014;71:3027-47
22. Ma Y, Lin D, Sun W, Xiao T, Yuan J, Han N. et al. Expression of targeting protein for xklp2 associated with both malignant transformation of respiratory epithelium and progression of squamous cell lung cancer. Clinical cancer research: an official journal of the American Association for Cancer Research. 2006;12:1121-7
23. Shigeishi H, Fujimoto S, Hiraoka M, Ono S, Taki M, Ohta K. et al. Overexpression of the receptor for hyaluronan-mediated motility, correlates with expression of microtubule-associated protein in human oral squamous cell carcinomas. International journal of oncology. 2009;34:1565-71
24. Li B, Qi XQ, Chen X, Huang X, Liu GY, Chen HR. et al. Expression of targeting protein for Xenopus kinesin-like protein 2 is associated with progression of human malignant astrocytoma. Brain research. 2010;1352:200-7
25. Selamat SA, Chung BS, Girard L, Zhang W, Zhang Y, Campan M. et al. Genome-scale analysis of DNA methylation in lung adenocarcinoma and integration with mRNA expression. Genome research. 2012;22:1197-211
26. Davis S, Meltzer PS. GEOquery: a bridge between the Gene Expression Omnibus (GEO) and BioConductor. Bioinformatics (Oxford, England). 2007;23:1846-7
27. Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA. et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:15545-50
28. Szklarczyk D, Franceschini A, Wyder S, Forslund K, Heller D, Huerta-Cepas J. et al. STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic acids research. 2015;43:D447-52
29. Saito R, Smoot ME, Ono K, Ruscheinski J, Wang PL, Lotia S. et al. A travel guide to Cytoscape plugins. Nature methods. 2012;9:1069-76
30. Chin CH, Chen SH, Wu HH, Ho CW, Ko MT, Lin CY. cytoHubba: identifying hub objects and sub-networks from complex interactome. BMC systems biology. 2014;8(Suppl 4):S11
31. Cai L, Lin S, Girard L, Zhou Y, Yang L, Ci B. et al. LCE: an open web portal to explore gene expression and clinical associations in lung cancer. Oncogene. 2019;38:2551-64
32. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA: a cancer journal for clinicians. 2018;68:394-424
33. Misono S, Seki N, Mizuno K, Yamada Y, Uchida A, Sanada H. et al. Molecular Pathogenesis of Gene Regulation by the miR-150 Duplex: miR-150-3p Regulates TNS4 in Lung Adenocarcinoma. Cancers. 2019 11
34. Shi X, Tan H, Le X, Xian H, Li X, Huang K. et al. An expression signature model to predict lung adenocarcinoma-specific survival. Cancer management and research. 2018;10:3717-32
35. Sui J, Yang S, Liu T, Wu W, Xu S, Yin L. et al. Molecular characterization of lung adenocarcinoma: A potential four-long noncoding RNA prognostic signature. Journal of cellular biochemistry. 2019;120:705-14
36. Chan JJ, Tay Y. Noncoding RNA:RNA Regulatory Networks in Cancer. International journal of molecular sciences. 2018 19
37. Dong HX, Wang R, Jin XY, Zeng J, Pan J. LncRNA DGCR5 promotes lung adenocarcinoma (LUAD) progression via inhibiting hsa-mir-22-3p. Journal of cellular physiology. 2018;233:4126-36
38. Zhao H, Zheng C, Wang Y, Hou K, Yang X, Cheng Y. et al. miR-1323 Promotes Cell Migration in Lung Adenocarcinoma by Targeting Cbl-b and Is an Early Prognostic Biomarker. Frontiers in oncology. 2020;10:181
39. Yang J, Qiu Q, Qian X, Yi J, Jiao Y, Yu M. et al. Long noncoding RNA LCAT1 functions as a ceRNA to regulate RAC1 function by sponging miR-4715-5p in lung cancer. Molecular cancer. 2019;18:171
40. Ren P, Hong X, Chang L, Xing L, Zhang H. USF1-induced overexpression of long noncoding RNA WDFY3-AS2 promotes lung adenocarcinoma progression via targeting miR-491-5p/ZNF703 axis. Molecular carcinogenesis. 2020
41. Guo M, Lu S, Huang H, Wang Y, Yang MQ, Yang Y. et al. Increased AURKA promotes cell proliferation and predicts poor prognosis in bladder cancer. BMC systems biology. 2018;12:118
42. Wang-Bishop L, Chen Z, Gomaa A, Lockhart AC, Salaria S, Wang J. et al. Inhibition of AURKA Reduces Proliferation and Survival of Gastrointestinal Cancer Cells With Activated KRAS by Preventing Activation of RPS6KB1. Gastroenterology. 2019;156:662-75.e7
43. Takeshita M, Koga T, Takayama K, Ijichi K, Yano T, Maehara Y. et al. Aurora-B overexpression is correlated with aneuploidy and poor prognosis in non-small cell lung cancer. Lung cancer (Amsterdam, Netherlands). 2013;80:85-90
44. Hayama S, Daigo Y, Yamabuki T, Hirata D, Kato T, Miyamoto M. et al. Phosphorylation and activation of cell division cycle associated 8 by aurora kinase B plays a significant role in human lung carcinogenesis. Cancer research. 2007;67:4113-22
45. Zhao X, Zhou LL, Li X, Ni J, Chen P, Ma R. et al. Overexpression of KIF20A confers malignant phenotype of lung adenocarcinoma by promoting cell proliferation and inhibiting apoptosis. Cancer medicine. 2018;7:4678-89
46. Tang XK, Wang KJ, Tang YK, Chen L. Effects of ubiquitin-conjugating enzyme 2C on invasion, proliferation and cell cycling of lung cancer cells. Asian Pacific journal of cancer prevention: APJCP. 2014;15:3005-9
47. Zhu K, Ding H, Wang W, Liao Z, Fu Z, Hong Y. et al. Tumor-suppressive miR-218-5p inhibits cancer cell proliferation and migration via EGFR in non-small cell lung cancer. Oncotarget. 2016;7:28075-85
48. Liu L, Liu L, Lu S. lncRNA H19 promotes viability and epithelial-mesenchymal transition of lung adenocarcinoma cells by targeting miR-29b-3p and modifying STAT3. International journal of oncology. 2019;54:929-41
49. Wang A, Deng S, Chen X, Yu C, Du Q, Wu Y. et al. miR-29a-5p/STAT3 Positive Feedback Loop Regulates TETs in Colitis-Associated Colorectal Cancer. Inflammatory bowel diseases. 2020;26:524-33
50. Li H, Zhou H, Luo J, Huang J. MicroRNA-17-5p inhibits proliferation and triggers apoptosis in non-small cell lung cancer by targeting transforming growth factor beta receptor 2. Experimental and therapeutic medicine. 2017;13:2715-22
51. Sun X, Xin Y, Wang M, Li S, Miao S, Xuan Y. et al. Overexpression of long non-coding RNA KCNQ1OT1 is related to good prognosis via inhibiting cell proliferation in non-small cell lung cancer. Thoracic cancer. 2018;9:523-31
52. Liu C, Li H, Jia J, Ruan X, Liu Y, Zhang X. High Metastasis-Associated Lung Adenocarcinoma Transcript 1 (MALAT1) Expression Promotes Proliferation, Migration, and Invasion of Non-Small Cell Lung Cancer via ERK/Mitogen-Activated Protein Kinase (MAPK) Signaling Pathway. Medical science monitor: international medical journal of experimental and clinical research. 2019;25:5143-9
53. Hsu PK, Chen HY, Yeh YC, Yen CC, Wu YC, Hsu CP. et al. TPX2 expression is associated with cell proliferation and patient outcome in esophageal squamous cell carcinoma. Journal of gastroenterology. 2014;49:1231-40
54. Sui C, Song Z, Yu H, Wang H. Prognostic significance of TPX2 and NIBP in esophageal cancer. Oncology letters. 2019;18:4221-9
55. Wang G, Wang Q, Li Z, Liu C, He X. Clinical value of Xenopus kinesin-like protein 2 as a prognostic marker in patients with digestive system cancers: a systematic review and meta-analysis. OncoTargets and therapy. 2018;11:1229-43
56. Glaser ZA, Love HD, Guo S, Gellert L, Chang SS, Herrell SD. et al. TPX2 as a prognostic indicator and potential therapeutic target in clear cell renal cell carcinoma. Urologic oncology. 2017;35:286-93
57. Yuan X, Wu H, Xu H, Xiong H, Chu Q, Yu S. et al. Notch signaling: an emerging therapeutic target for cancer treatment. Cancer letters. 2015;369:20-7
58. Alketbi A, Attoub S. Notch Signaling in Cancer: Rationale and Strategies for Targeting. Current cancer drug targets. 2015;15:364-74
59. Dang TP. Notch, apoptosis and cancer. Advances in experimental medicine and biology. 2012;727:199-209
60. Licciulli S, Avila JL, Hanlon L, Troutman S, Cesaroni M, Kota S. et al. Notch1 is required for Kras-induced lung adenocarcinoma and controls tumor cell survival via p53. Cancer research. 2013;73:5974-84
61. Baumgart A, Mazur PK, Anton M, Rudelius M, Schwamborn K, Feuchtinger A. et al. Opposing role of Notch1 and Notch2 in a Kras(G12D)-driven murine non-small cell lung cancer model. Oncogene. 2015;34:578-88
62. Hassan WA, Yoshida R, Kudoh S, Motooka Y, Ito T. Evaluation of role of Notch3 signaling pathway in human lung cancer cells. Journal of cancer research and clinical oncology. 2016;142:981-93
Author contact
![]() Corresponding author: Prof. Yi-Qing Qu, MD, PhD, Department of Pulmonary and Critical Care Medicine, Qilu Hospital of Shandong University, Wenhuaxi Road 107#, Jinan 250012, China. Tel: +86 531 8216 9335; Fax: +86 531 8296 7544; E-mail: quyiqingedu.cn.
Corresponding author: Prof. Yi-Qing Qu, MD, PhD, Department of Pulmonary and Critical Care Medicine, Qilu Hospital of Shandong University, Wenhuaxi Road 107#, Jinan 250012, China. Tel: +86 531 8216 9335; Fax: +86 531 8296 7544; E-mail: quyiqingedu.cn.