Impact Factor ISSN: 1449-1907

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2020; 17(10):1375-1384. doi:10.7150/ijms.41793 This issue Cite
Research Paper
Cladribine Induces ATF4 Mediated Apoptosis and Synergizes with SAHA in Diffuse Large B-Cell Lymphoma Cells
Linyan Xu1,2,3#, Jun Jiao1,3#, Xiaoshen Sun1,3#, Wei Sang1,2,3, Xiang Gao1,3, Pu Yang4, Dongmei Yan2, Xuguang Song2, Cai Sun2, Mengdi Liu1,3, Yuanyuan Qin1,3, Yu Tian2, Feng Zhu1,2,3, Lingyu Zeng1,2,3, Zhenyu Li1,2,3, Kailin Xu1,2,3 ![]()
1. Blood Diseases Institute, Xuzhou Medical University, Xuzhou, Jiangsu, China.
2. Department of Hematology, the Affiliated Hospital of Xuzhou Medical University, Xuzhou, Jiangsu, China.
3. Key Laboratory of Bone Marrow Stem Cell, Xuzhou, Jiangsu, China.
4. Department of Hematology, Luoyang Central Hospital Affiliated to Zhengzhou University, Luoyang, Henan, China.
#These authors contributed equally to this work.
Received 2019-11-4; Accepted 2020-5-23; Published 2020-5-30
Abstract
Cladribine is a purine nucleoside analog used to treat B-cell chronic lymphocytic leukemia and hairy cell leukemia, also functions as an inhibitor of DNA synthesis to block the repair of the damaged DNA. The therapeutic role of cladribine against diffuse large B-cell lymphoma cells (DLBCL) is still undefined. In the present study, we demonstrated that cladribine inhibited cell proliferation and induced G1 phase arrest in human DLBCL cells. Furthermore, we showed that cladribine induced apoptosis by decreasing the expression of c-FLIPL and increasing the expression of DR4 and the cleaved form of caspase8. Cladribine also upregulated the expression of Bax, and downregulated the expression of Mcl-1 and Bcl-2 in a dose-dependent manner. It also activated endoplasmic reticulum (ER) stress, and ATF4 expression was required for cladribine induced apoptosis. Also, we showed that suberoylanilide hydroxamic acid (SAHA) enhanced the pro-apoptotic role of cladribine. Collectively, cladribine activated extrinsic and intrinsic apoptotic signaling pathways via stimulating ER stress signaling pathway and eliciting synergistic effect with SAHA in DLBCL cells.
Keywords: cladribine, ATF4, SAHA, apoptosis, DLBCL
Introduction
Diffuse large B-cell lymphoma (DLBCL) is a heterogeneous disease in molecular biology, histomorphology, gene expression, and clinical prognosis, and the incidence rate is increasing annually [1]. For most patients, DLBCL is readily curable with immunochemotherapy R-CHOP regimen; however, 30-40% of patients are relapsed, and about 10% of patients are primary refractory [2, 3]. The standard second-line of treatment for relapsed and refractory (R/R) DLBCL is autologous stem cell transplantation (ASCT) [2]. However, several patients who still cannot be cured succumb to the tumor [4]. Nowadays, an important objective is to develop competent and personalized therapeutic strategies.
Cladribine (2-chloro-2′-deoxyadenosine, 2-CdA), a purine analog, is an intermediate for the synthesis of 2-deoxyribonucleotide [5]. It is a prodrug and enters the cells through the nucleoside transporters [6]. This, in turn, results in the accumulation of active metabolite within the cell, resulting in disruption of cellular metabolism, DNA damage, and subsequent apoptosis [7,8]. Cladribine exhibits selective toxicity toward both proliferative and non-proliferative lymphocytes [9]. It can also decrease DNA methylation by indirectly inhibiting DNA methyltransferase and consuming methyl donors, thus promoting cell apoptosis [10, 11]. Cladribine has anti-lymphoma activity and has been widely used in the treatment of several hematologic malignancies, including hairy cell leukemia (HCL) and chronic lymphocytic leukemia (CLL) and achieved satisfactory results [12-14]. Cladribine is also an effective therapeutic agent in the management of mantle cell lymphoma (MCL), follicular lymphoma (FL), and extra nodal B-cell marginal zone lymphoma of the mucosa-associated lymphoid tissue (MALT) [12,15,16]. However, the role of cladribine in DLBCL is not well documented.
The combined application of drugs is also an effective strategy for the treatment of cancers [17]. The two important epigenetic modifications, methylation and acetylation, play significant roles in controlling gene expression. They have been implicated in aberrant oncogene expression in various cancers [18-20]. Recent reports have shown that both DNA methyltransferase and histone deacetylase (HDAC) inhibitors can synergistically cause global alterations in gene expression responsible for cell proliferation, differentiation, and death [21,22]. Cladribine can inhibit DNA methylation [10,11], while suberoylanilide hydroxamic acid (SAHA), which is also known as vorinostat, is a powerful HDAC inhibitor that arrests growth and causes apoptosis in many types of cancers [23]. Our previous study has shown that SAHA and cladribine synergistically induce apoptosis in natural killer-large granular lymphocyte leukemia (NK-LGLL) [24]. Cladribine and SAHA, in combination with gemcitabine and busulfan, also provided synergistic cytotoxicity in lymphoma cell lines [25]. Stephen E. Spurgeon showed a phase 1/2 study, which using SAHA, cladribine, and rituximab in relapsed B-cell non-Hodgkin lymphoma (NHL) and previously untreated MCL resulted in a high complete response rate [26]. In this study, we also tried to investigate whether combined SAHA and cladribine have a synergistic effect in DLBCL cells.
Apoptosis is recognized as an important programmed cell death; however, the apoptotic program is often decreased in hematologic malignancies. Several anti-cancer therapies and antineoplastic drugs induce apoptosis to eliminate tumor cells; hence, induction of apoptosis is considered one of the effective approaches of antitumor therapy [27-30]. Prolonged or severe endoplasmic reticulum (ER) stress impairs the normal functioning of the cells, and could induce cell apoptosis to remove damaged cells [31]. The activating transcription factor 4 (ATF4) is a stress-responsive protein that shows elevated expression in ER stress. Several stress signals, namely hypoxia, amino acid deficiency, and oxidative stress, can induce ER stress activation and elevate ATF4 expression [32,33]. There is an increased expression of ATF4 in cancer cells, and it promotes apoptosis induced by chemotherapeutic drugs by increasing the expression of DNA damage-inducible transcript 3 (DDIT3) [34,35].
In the present study, we investigated the effect of cladribine on proliferation and cell cycle in DLBCL cells and established the role of cladribine induced apoptosis both in intrinsic and extrinsic pathways. Meanwhile, we testified that ATF4 played an important role in cladribine mediated apoptosis.
Materials and methods
Cell culture
The human DLBCL cell lines U2932, OCI-LY10, SUDHL2, WSU-DLCL2, and DB were originally obtained from the American Type Culture Collection (Manassas, VA, USA) and grown in IMDM medium. HEK293T cells were cultured in DMEM medium. All cells were cultured with 10% fetal bovine serum at 37 °C in a humidified atmosphere consisting of 5% CO2.
Antibody and reagents
Compound cladribine was purchased from Sigma-Aldrich (Saint Louis, Missouri, US). The c-FLIP antibody was purchased from EZNO Life Sciences (Farmington, NY, USA). Cyclin D1, Cyclin E, p21, p27, DR4, caspase8, caspase3, PARP, Bax, Mcl-1, Bcl-2, ATF3, CHOP, ATF4, and β-actin antibodies were purchased from Cell Signaling Technology (Danvers, MA, USA).
Cell viability assay
Cells were seeded in 96-well plates with 2×104 cells per well and treated with cladribine with the indicated concentrations for indicated time. Then Cell Counting Kit-8 (CCK-8) (Dojindo Laboratories, Kumamoto, Japan) was used to measure cell viability according to the manufacturer's instruction.
Western blot
Human DLBCL cells were harvested, lysed, and detected protein concentrations by BCA assay. Then 50 µg protein samples were separated by SDS-PAGE electrophoresis, transferred to PVDF membranes, blocked with 5% skim milk and incubated with primary antibodies at 4 °C overnight. After washing the membranes and incubating with horseradish peroxidase-conjugated secondary antibodies, bounded antibodies were detected by enhanced chemiluminescence (ECL) kit (BD Biosciences, San Jose, CA, USA), and pictures were captured using GE Image Quant LAS4000 mini.
Apoptosis assay
Apoptosis was detected by Annexin V-PE/7-AAD apoptosis detection kit purchased from BD Biosciences following the manufacture's protocol. Briefly, the cells were seeded in 6-well plates with 2×106 cells per well and treated with cladribine for 24 h. Then harvest the cells and resuspend with binding buffer containing 5 µL Annexin V-PE and 5 µL 7-AAD (BD Biosciences, San Jose, CA, USA). At the same time, we conducted three controls used to set up compensation and quadrants: unstained cells, cells only stained with 5 µL Annexin V-PE, cells only stained with 5 µL 7-AAD. Samples were incubated in the dark for 15 minutes and detected by flow cytometer within 1 h. All data were analyzed using FlowJo software.
Cell cycle analysis
Cell cycle analysis was detected by flow cytometry. Cells were seeded in 6-well plates with 2×106 cells per well and treated with cladribine for 24 h. Then harvest the cells and fix with 70% ice-cold ethanol at -20 °C. The next day, the cells were washed and resuspended with PBS. Meanwhile, stain the cells with propidium iodide (contain RNase A: 50 µg/mL) for 30 min protection from light and then detect the cells using a flow cytometer. Measure the forward scatter (FS), side scatter (SS) to identify cells and use pulse area, pulse width to exclude cell debris, clumps, and doublets. Single cells were acquired with the maximum emission of PI less than 605 nm and analyzed the data by ModFit LT 3.3 software (Verity Software House Inc., Topsham, ME, USA).
RNA extraction and real-time qPCR
The cells were collected, and RNA was extracted with TRIzol™ reagent (Life Technologies, Gaithersburg, MD, USA) according to the manufacturer's instruction. The concentration was measured at 260 nm, and RNA was used to synthesize to cDNA using M-MLV reverse transcriptase (Life Technologies, Gaithersburg, MD, USA). Then q-PCR was performed according to the manufacturer's instruction using SYBR® Green Supermix (Roche, Indianapolis, IN, USA). Sequences of primers for RT-qPCR genes are listed in Table 1. The PCR amplification was conducted using the following procedure: 94 °C for 2 min, followed by 35 cycles of 94 °C for 30 s, 60 °C for 30 s, 72 °C for 35 s, and a final extension at 72 °C for 2 min. The threshold cycle number (Ct) was calculated, and the relative mRNA expression of target genes was calculated by 2-ΔΔCt method.
Sequences of primers for RT-qPCR
| Name | Sequences |
|---|---|
| Cyclin D1 | 5′-GCCCGAGGAGCTGCTGCAAA -3′(forward) 5′-GCAACGAAGGTCTGCGCGTG -3′(reverse) |
| Cyclin E | 5′-TTCTTGAGCAACACCCTCTTCTGCAGCC -3′(forward) 5′-TCGCCATATACCGGTCAAAGAAATCTTGTGCC -3′(reverse) |
| P21 | 5′-TGAGCCGCGACTGTGATG-3′(forward) 5′-GTCTCGGTGACAAAGTCGAAGTT-3′(reverse) |
| P27 | 5′- TGCAACCGACGATTCTTCTACTCAA-3′(forward) 5′- CAAGCAGTGATGTATCTGATAAACAAGGA -3′(reverse) |
| ATF4 | 5′-GCTAAGGCGGGCTCCTCCGA- 3′(forward) 5′-ACCCAACAGGGCATCCAAGTCG-3′(reverse) |
| ATF3 | 5′-TGATGCTTCAACACCCAGGCC-3′(forward) 5′-AGGGGACGATGGCAGAAGCA-3′(reverse) |
| CHOP | 5′-CATCACCACACCTGAAAGCA -3′(forward) 5′-TCAGCTGCCATCTCTGCA-3′(reverse) |
| β-Actin | 5′-CTCCATCCTGGCCTCGCTGT -3′(forward) 5′-GCTGTCACCTTCACCGTTCC- 3′(reverse) |
ShRNA oligonucleotide design and construction of stable cell lines
The ATF4 shRNA oligonucleotide sequences were designed as 5'-GCCTAGGTCTCTTAGATGATT-3' (1), 5'-GCCAAGCACTTCAAACCTCAT-3' (2). After cloning shRNA sequences into a pLVshRNA-eGFP vector, the desired plasmids were transfected into HEK-293T cells accompanied by packaging plasmid psPAX2 and envelope plasmid pMD2.G using transfection reagent Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA). Collect the lentivirus supernatant and concentrate the lentivirus for infection into DLBCL cells. The infected cells were then treated with puromycin for screening stable cell lines. Finally, the screened stable cells were identified by flow cytometry and western blot.
Statistical analysis
Data were shown as means ± standard deviation (SD). Two-tailed unpaired Student's t-test was used to analyze the statistical significance between two treatment groups. One-way ANOVAs was used to analyze the statistical significance between multiple treatment groups. Statistical significance was established as *P<0.05, **P<0.01, ***P<0.001. All statistical analyses were performed using the GraphPad Prism software ver. 6.0 (GraphPad Software Inc., La Jolla, CA, USA).
Results
Cladribine inhibits human DLBCL cells proliferation
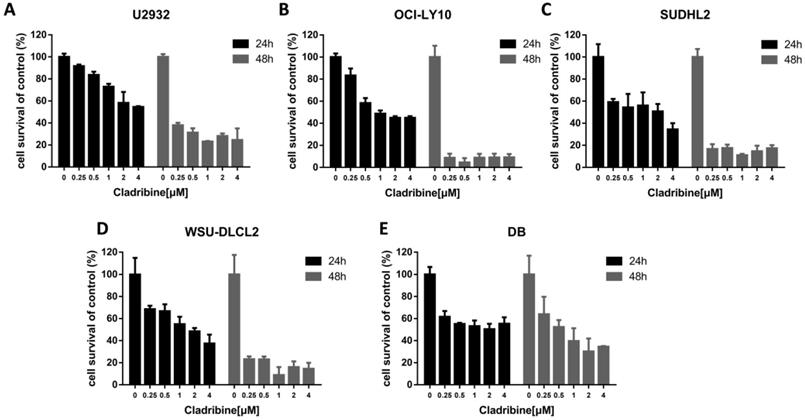
To determine the effect of cladribine on the growth of DLBCL cells, CCK-8 assay was performed to detect the viability. As shown in Figure 1A-E, cladribine exhibited notable suppression of cell proliferation in five DLBCL cells. Concurrently, it was observed that the inhibitory effect was more profound with the time elapse.
Cladribine inhibits human DLBCL cells proliferation. A-E. U2932 (A), OCI-LY10 (B), SUDHL2 (C), WSU-DLCL2 (D) and DB (E) cells were incubated with the indicated concentrations of cladribine for 24 h or 48 h, then CCK-8 assay was performed to detect the viability. Error bars, mean ± SD.
Cladribine induces G1 phase arrest in human DLBCL cells
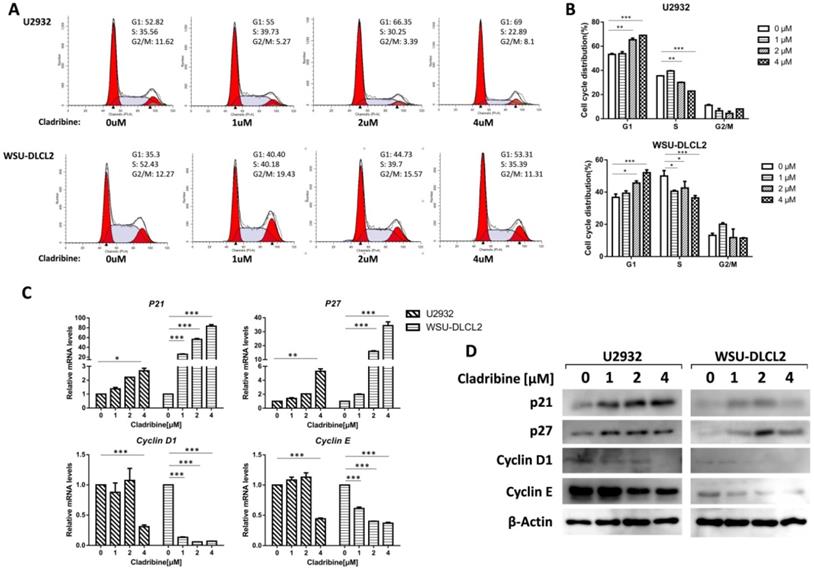
To further study the underlying mechanism of the inhibition of DLBCL cell proliferation by cladribine, flow cytometry analysis was carried out to investigate the cell cycle distribution after treating the cells with cladribine. The cells in the G1 phase were increased in a dose-dependent fashion both in U2932 and WSU-DLCL2 cells, while the population in the S phase was decreased from 35.56% to 22.89% in U2932 cells and from 52.43% to 35.39% in WSU-DLCL2 cells (Figure 2A and 2B). Moreover, when the mechanism of G1 phase arrest was studied, the mRNA levels of Cyclin D1 and Cyclin E were found to decrease, while those of P21 and P27 were increased (Figure 2C). Moreover, western blot showed the expressions of Cyclin D1 and Cyclin E were decreased, while there were elevated expressions of p21 and p27 in U2932 and WSU-DLCL2 cells (Figure 2D). Taken together, these results indicate that cladribine causes G1 phase arrest via decreasing the expressions of Cyclin D1 and Cyclin E, and increasing the expressions of p21 and p27 in DLBCL cells.
Cladribine induces G1 phase arrest in human DLBCL cells. A. U2932 and WSU-DLCL2 cells were incubated with the indicated concentrations of cladribine for 24 h. Then cells were harvested and prepared for cell cycle analysis. B. Percentages of the subpopulation of cells at different cell cycle phases were determined from three independent experiments. C. U2932 and WSU-DLCL2 cells were incubated with the indicated concentrations of cladribine for 24 h. The expressions of Cyclin D1, Cyclin E, P21, and P27 mRNA were assessed by real-time PCR. Error bars, mean ± SD. *P < 0.05; **P < 0.01; ***P < 0.001. D. U2932 and WSU-DLCL2 cells were incubated with the indicated concentrations of cladribine for 24 h. Then whole cells were harvested and subjected to western blot using Cyclin D1, Cyclin E, p21, and p27 antibodies.
Cladribine induces apoptosis and activates extrinsic and intrinsic signaling pathways in human DLBCL cells
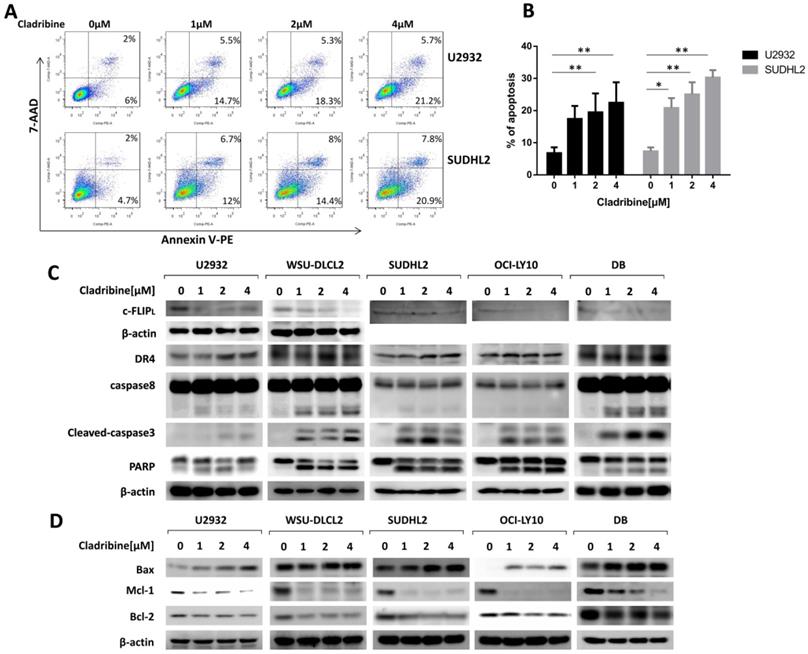
Furthermore, we performed a flow cytometric assay to elucidate the apoptotic effect and found that cladribine treatment induced apoptosis of U2932 and SUDHL2, and its percentage significantly increased with an increase in concentration (Figure 3A and 3B). The apoptotic signaling pathway was further activated. As shown by western blotting, the level of death receptor DR4 was upregulated in U2932, OCI-LY10, SUDHL2, WSU-DLCL2, and DB cells (Figure 3C). The expression of anti-apoptotic protein c-FLIP was decreased, and the cleavage of caspase8 was elevated in these cells (Figure 3C). Moreover, cladribine treatment increased the cleaved forms of caspase3 and PARP, indicating that it induces the extrinsic apoptotic pathway. Furthermore, we examined that cladribine raised the expression of pro-apoptotic protein Bax, and reduced the expression of anti-apoptotic proteins Mcl-1 and Bcl-2 in a dose-dependent manner (Figure 3D), suggesting the role of cladribine in inducing intrinsic apoptotic pathway. Taken together, these results indicate cladribine induces apoptosis and activates extrinsic and intrinsic signaling pathways in human DLBCL cells.
Cladribine induces apoptosis and activates exogenous and endogenous apoptotic signaling pathways in human DLBCL cells. A. U2932 and SUDHL2 cells were incubated with the indicated concentrations of cladribine for 24 h, and then cells were harvested and subsequently stained with Annexin-V-PE and 7-AAD and analyzed by flow cytometry for apoptosis. B. Percentages of apoptotic cells were determined from three independent experiments. Error bars, mean ± SD. *P < 0.05; **P < 0.01. C and D. U2932, WSU-DLCL2, SUDHL2, OCI-LY10, and DB cells were incubated with the indicated concentrations of cladribine for 24 h. Then whole cells were harvested and subjected to western blot using c-FLIP, DR4, caspase8, caspase3, PARP (C) and Bax, Mcl-1, Bcl-2 (D) antibodies.
Cladribine activates endoplasmic reticulum stress
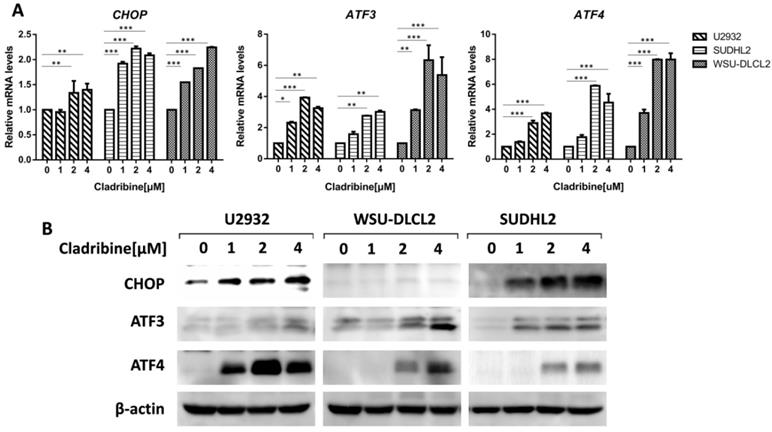
To elucidate the mechanism of cladribine-induced apoptosis in DLBCL cells, we examined the mRNA levels of CHOP, ATF3, and ATF4, which were considered as important markers of ER stress and found that their expressions were enhanced in a dose-dependent fashion (Figure 4A). Moreover, we confirmed that their protein levels were also increased (Figure 4B). Collectively, these results indicate that cladribine activates ER stress.
Cladribine activates ER stress. A-B. U2932, SUDHL2 and WSU-DLCL2 cells were incubated with the indicated concentrations of cladribine for 24 h, and then whole cells were harvested and subjected to real-time PCR assay (A) or western blot analysis using ATF3, CHOP, and ATF4 antibodies (B). Error bars, mean ± SD. *P < 0.05; **P < 0.01; ***P < 0.001.
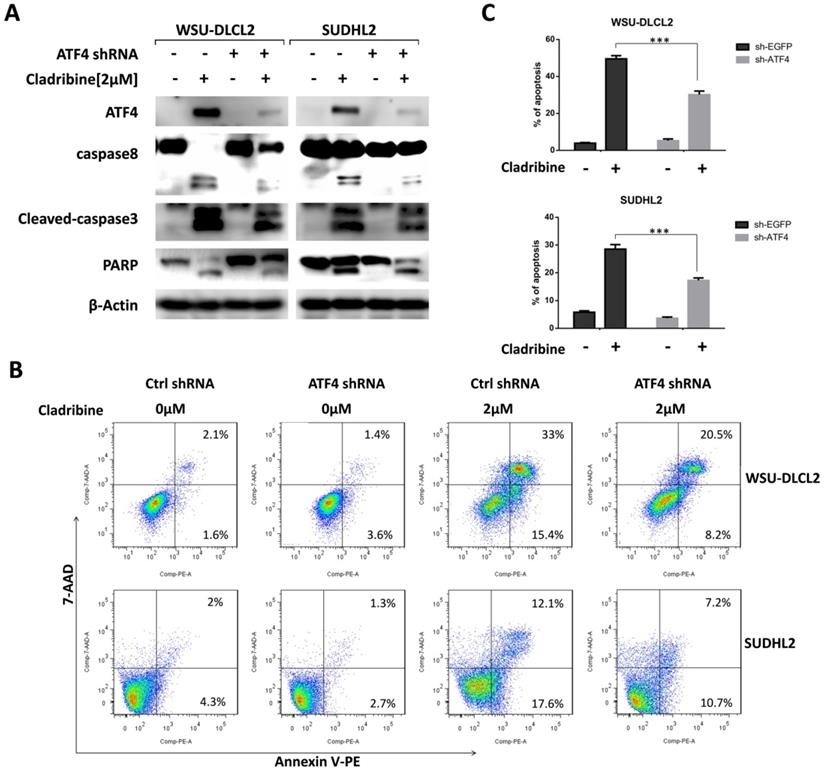
ATF4 expression is required for cladribine induced apoptosis
We then focused on the role of ATF4 by designing ATF4-shRNA to inhibit ATF4 expression. The inhibition of ATF4 up-regulation decreased the cleavage of caspase8, caspase3, and PARP in WSU-DLCL2 and SUDHL2 cells (Figure 5A). Consistently, the percentage of apoptotic cells in control shRNA-transfected cells was 48.4%, whereas it was only 28.7% in ATF4 shRNA-transfected WSU-DLCL2 when cells were exposed to cladribine. In SUDHL2 cells, the corresponding proportion was decreased from 29.7% to 17.9% (Figure 5B and 5C) indicating ATF4 contributes to cladribine induced apoptosis.
ATF4 expression is required for cladribine-induced apoptosis. A. WSU-DLCL2 and SUDHL2 cells were transduced with ATF4-shRNA and control-shRNA lentivirus and constructed stable knockdown cell lines. Then cells were incubated with cladribine for 24 h. Whole cells were harvested and subjected to western blot analysis using ATF4, caspase8, caspase3, and PAPR antibodies. B and C. WSU-DLCL2 and SUDHL2 stable cell lines with ATF4 knockdown were incubated with cladribine for 24 h, and then cells were detected with Annexin V/7-AAD by flow cytometry (B), and the percentages of cell apoptosis were determined from three independent experiments (C). Error bars, mean ± SD. ***P < 0.001.
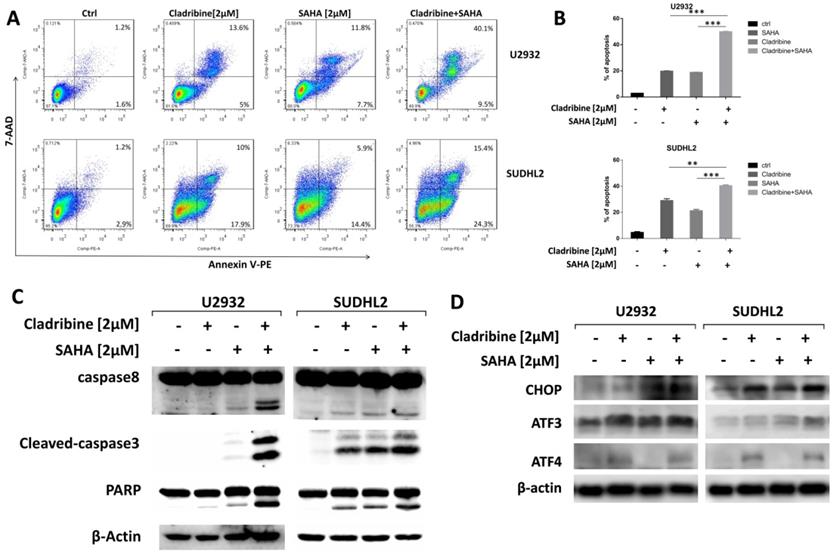
SAHA enhances the pro-apoptotic role of cladribine
To explore whether SAHA enhances the pro-apoptotic effect of cladribine in DLBCL cells, we treated U2932 and SUDHL2 cells with cladribine alone or in combination with SAHA. When cells were treated with a combination of cladribine and SAHA, an increase in apoptosis was detected using Annexin V staining-flow cytometry (Figures 6A and 6B). We confirmed the enhanced cleavage of caspase8, caspase3, and PARP using western blot test in the combination group compared to cladribine or SAHA alone (Figure 6C). In addition, we discovered that SAHA cannot alter the expression of ATF4, and combination of cladribine and SAHA also did not affect the expression of ATF4 (Figure 6D). SAHA slightly increased the expression of CHOP, ATF3, and combination of cladribine and SAHA also increased CHOP and ATF3 expression mildly (Figure 6D). Collectively, these results indicate that SAHA enhanced cladribine-mediated apoptosis, and partially enhanced ER stress.
SAHA enhanced the pro-apoptotic role of cladribine. A and B. U2932 and SUDHL2 cells were treated with cladribine and SAHA for 24 h. Then the whole cells were harvested and detected with Annexin V/7-AAD by flow cytometry (A), and percentages of cell apoptosis were determined from three independent experiments (B). Error bars, mean ± SD. **P < 0.01; ***P < 0.001. C and D. U2932 and SUDHL2 cells were treated with cladribine and SAHA for 24 h. Then the whole cells were harvested and subjected to western blot analysis using caspase8, caspase3, PAPR antibodies (C) and CHOP, ATF3, ATF4 antibodies (D).
Discussion
The prognosis of DLBCL has largely improved with the application of monoclonal anti-CD20 antibody rituximab; however, there is still a group of patients who cannot benefit from the standard treatment. So developing efficacious or specific therapeutic strategies continues to be an important objective [2-4]. Cladribine is a purine analog, which has been used in the treatment of B and T cell lymphoid malignancies, including multiple sclerosis, HCL, and CLL [5]. Meanwhile, cladribine also has an antitumor role in MCL, FL, and MALT [12-16]. In the study, we showed that cladribine inhibited cell proliferation and induced G1 phase arrest in human DLBCL cells. Furthermore, we showed that cladribine induced apoptosis and activated ER stress, and ATF4 expression was required for cladribine induced apoptosis. Also, we showed that SAHA enhanced the pro-apoptotic role of cladribine.
In the present study, we showed that cladribine induced apoptosis by decreasing the expression of c-FLIP and increasing the expression of DR4 and the cleaved form of caspase8, indicating that cladribine induced extrinsic apoptotic pathway. It is reported that cladribine induces human leukemia cell line apoptosis through the Fas/Fas ligand pathway [36], our results further established the role of cladribine in inducing extrinsic apoptotic pathway. Furthermore, the expressions of anti-apoptotic proteins Mcl-1 and Bcl-2 were decreased and the expression of pro-apoptotic protein Bax was increased, suggesting that cladribine also activated the intrinsic apoptotic pathway in DLBCL cells. These results were consistent with the earlier studies that cladribine induces apoptosis via activation of caspases 3, 6, 8, and 9 and decreasing the level of the anti-apoptotic protein Mcl-1 [37,38]. Also, studies have shown cladribine reduces the mitochondrial transmembrane potential, releases of cytochrome C and translocates AIF from mitochondria to the nucleus and causes chromatin condensation [37,39]. However, cladribine cannot change Bax and Bcl-2 expression in B-CLL cells in patients who express high levels of Bcl-2, Bax, and Bak proteins [37]. The study by V. Singh revealed a contrary view that cladribine induced caspase-independent apoptosis [39,40], so it induces apoptosis via various patterns that may be dependent on different cell types and gene expression. Whether cladribine would induce caspase-independent apoptosis in DLBCL cells remains to be further explored.
The mechanism of cladribine induced apoptosis has been extensively studied. It has been reported that cladribine activates the tumor suppressor p53, which further activates the intrinsic apoptotic pathway [37,41]. The intracellular calcium ion concentration, p38 mitogen-activated protein kinase, extracellular signal-regulated kinases 1 and 2 also probably play roles in the induction of apoptosis by cladribine [42,43]. In our study, we observed that cladribine elevated CHOP, ATF3, and ATF4 expression, which are important components of ER stress, indicating cladribine activated ER stress. Severe ER stress could induce apoptosis [44,45], and ATF4 promotes apoptosis induced by chemotherapeutic drugs such as bortezomib, cisplatin, and arsenic trioxide [34,46,47]. So we evaluated whether ER stress influenced cladribine induced apoptosis. We mainly focused on the important component ATF4 and found that the inhibition of ATF4 upregulation decreased the cleavage of caspases8, 3, and PARP induced by cladribine. Meanwhile, the percentage of apoptotic cells was also decreased in ATF4 shRNA-transfected cells when exposed to cladribine, suggesting that ATF4 participated in cladribine-induced apoptosis. Consistently, Swetlana Mactier reported ER stress pathway is involvement in cladribine induced apoptosis in Raji cells [48].
The combination therapy is gradually becoming a preferable practice in DLBCL treatment. The combination of different drugs might improve the prognosis of patients by suppressing cell proliferation, enhancing cell apoptosis, and surmounting drug resistance [49-51]. It has been found that combination of cladribine with other chemotherapy drugs shows good synergistic effects. Cladribine, in combination with bendamustine or STAT3 inhibitor, exhibited inhibitory activity in multiple myeloma cells [52,53]. A combination of cladribine with anthracyclines also induced apoptosis in lymphocytes in patients with B-cell chronic lymphocytic leukemia [54]. Our previous study also showed that SAHA and cladribine synergistically induced apoptosis in NK-LGL leukemia [24]. In the present study, we also investigated the combined synergistic effect of SAHA with cladribine in inducing apoptosis in DLBCL cells. The expressions of CHOP and ATF3 were slightly elevated with a combination of SAHA with cladribine, but the expression of ATF4 was not changed, so the detailed mechanism of the synergistic role needs to be further explored.
In summary, we showed that cladribine inhibited cell proliferation and induced G1 phase arrest and apoptosis in human DLBCL cells. We further demonstrated that cladribine activated ER stress, and ATF4 expression was required for cladribine induced apoptosis. Moreover, we showed that SAHA enhanced the pro-apoptotic role of cladribine and slightly enhanced ER stress. Collectively, cladribine activated extrinsic and intrinsic apoptotic signaling pathways by stimulating ATF4 expression and eliciting a synergistic effect with SAHA in DLBCL cells.
Abbreviations
2-CdA, 2-chloro-2′-deoxyadenosine; ASCT, autologous stem cell transplantation; ATF, activating transcription factor; CCK-8, cell counting kit-8; CHOP, C/EBP homologous protein; CLL, chronic lymphocytic leukemia; DDIT3, DNA damage-inducible transcript 3; DLBCL, diffuse large B-cell lymphoma; DR, death receptor; ECL, enhanced chemiluminescence; ER, endoplasmic reticulum; FS, forward scatter; FL, follicular lymphoma; HCL, hairy cell leukemia; HDAC, histone deacetylase; MALT, mucosa-associated lymphoid tissue; MCL, mantle cell lymphoma; NHL, non-Hodgkin lymphoma; NK-LGLL, natural killer-large granular lymphocyte leukemia; R-CHOP, rituximab, cyclophosphamide, doxorubicin, vincristine, and prednisone; R/R, relapsed and refractory; RT-qPCR, real-time quantitative polymerase chain reaction; SAHA, suberoylanilide hydroxamic acid; SD, standard deviation; SS, side scatter.
Acknowledgements
The research was supported by National Natural Science Foundation of China (81700199, 81500121); Natural Science Foundation of Jiangsu Province (BK20170257, BK20171181); Natural Science Foundation of the Jiangsu Higher Education Institutions of China (17KJB320019); Project funded by China Postdoctoral Science Foundation (2017M611917, 2016M601891) and Jiangsu Planned Projects for Postdoctoral Research Funds (1601099C, 1701134C).
Authors' contributions
LYX, JJ and XSS performed the experiments, WS and XG analyzed the results, PY and DMY made the figures, XGS and CS wrote the manuscript. MDL, YYQ and YT analyzed the results. FZ, LYZ and ZYL reviewed the manuscript, KLX designed the experiment. All authors read and approved the final manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Riedell PA, Smith SM. Double hit and double expressors in lymphoma: Definition and treatment. Cancer. 2018;124:4622-32
2. Gisselbrecht C, Van Den Neste E. How I manage patients with relapsed/refractory diffuse large B cell lymphoma. Br J Haematol. 2018;182:633-43
3. Zhang J, Medeiros LJ, Young KH. Cancer Immunotherapy in Diffuse Large B-Cell Lymphoma. Front Oncol. 2018;8:351
4. Sehn LH, Gascoyne RD. Diffuse large B-cell lymphoma: optimizing outcome in the context of clinical and biologic heterogeneity. Blood. 2015;125:22-32
5. Giovannoni G. Cladribine to Treat Relapsing Forms of Multiple Sclerosis. Neurotherapeutics. 2017;14:874-87
6. Owen RP, Badagnani I, Giacomini KM. Molecular determinants of specificity for synthetic nucleoside analogs in the concentrative nucleoside transporter, CNT2. J Biol Chem. 2006;281:26675-82
7. Beutler E. Cladribine (2-chlorodeoxyadenosine). Lancet. 1992;340:952-6
8. Comi G, Hartung HP, Kurukulasuriya NC. et al. Cladribine tablets for the treatment of relapsing-remitting multiple sclerosis. Expert opin pharmaco. 2013;14:123-36
9. Carson DA, Wasson DB, Taetle R. et al. Specific toxicity of 2-chlorodeoxyadenosine toward resting and proliferating human lymphocytes. Blood. 1983;62:737-43
10. Stephen S, Margaret Y, Phillips JD. et al. Cladribine: not just another purine analogue? Expert Opin Inv Drug. 2009;18:1169
11. Wyczechowska D, Fabianowska-Majewska K. The effects of cladribine and fludarabine on DNA methylation in K562 cells. Biochem Pharmacol. 2003;65:219-25
12. Sigal DS, Miller HJ, Schram ED. et al. Beyond hairy cell: the activity of cladribine in other hematologic malignancies. Blood. 2010;116:2884-96
13. Johnston JB. Mechanism of action of pentostatin and cladribine in hairy cell leukemia. Leukemi lymphoma. 2011;52(Suppl 2):43-5
14. Freyer CW, Gupta N, Wetzler M. et al. Revisiting the role of cladribine in acute myeloid leukemia: an improvement on past accomplishments or more old news? Am J Hematol. 2015;90:62-72
15. Spurgeon SE, Pindyck T, Okada C. et al. Cladribine plus rituximab is an effective therapy for newly diagnosed mantle cell lymphoma. Leukemia lymphoma. 2011;52:1488-94
16. Kiesewetter B, Dolak W, Simonitsch-Klupp I. et al. Long-term safety and activity of cladribine in patients with extranodal B-cell marginal zone lymphoma of the mucosa-associated lymphoid tissue (MALT) lymphoma. Hematol oncol. 2017;35:177-86
17. Eckschlager T, Plch J, Stiborova M. et al. Histone Deacetylase Inhibitors as Anticancer Drugs. Int J Mol Sci. 2017 18
18. Biswas S, Rao CM. Epigenetics in cancer: Fundamentals and Beyond. Pharmacol Therapeut. 2017;173:118-34
19. Dawson MA, Kouzarides T. Cancer epigenetics: from mechanism to therapy. Cell. 2012;150:12-27
20. Kanwal R, Gupta S. Epigenetic modifications in cancer. Clin Genet. 2012;81:303-11
21. Ken M, Eline M, Els VV. et al. Epigenetic modulating agents as a new therapeutic approach in multiple myeloma. Cancers. 2013;5:430-61
22. Griffiths EA, Gore SD. DNA methyltransferase and histone deacetylase inhibitors in the treatment of myelodysplastic syndromes. Semin Hematol. 2008;45:23-30
23. Chun P. Histone deacetylase inhibitors in hematological malignancies and solid tumors. Arch Pharm Res. 2015;38:933-49
24. Sun X, Hasanali ZS, Chen A. et al. Suberoylanilide hydroxamic acid (SAHA) and cladribine synergistically induce apoptosis in NK-LGL leukaemia. Br J Haematol. 2015;168:371-83
25. Ji J, Valdez BC, Li Y. et al. Cladribine, gemcitabine, busulfan, and SAHA combination as a potential pretransplant conditioning regimen for lymphomas: A preclinical study. Exp Hematol. 2016;44:458-65
26. Spurgeon SE, Sharma K, Claxton DF. et al. Phase 1-2 study of vorinostat (SAHA), cladribine and rituximab (SCR) in relapsed B-cell non-Hodgkin lymphoma and previously untreated mantle cell lymphoma. Br J Haematol. 2019;186:845-54
27. Mohammad RM, Muqbil I, Lowe L. et al. Broad targeting of resistance to apoptosis in cancer. Semin Cancer Biol. 2015;35(Suppl):S78-S103
28. Goldar S, Khaniani MS, Derakhshan SM. et al. Molecular mechanisms of apoptosis and roles in cancer development and treatment. Asian Pac J cancer P. 2015;16:2129-44
29. Kaczanowski S. Apoptosis: its origin, history, maintenance and the medical implications for cancer and aging. Phys Biol. 2016;13:031001
30. Zaman S, Wang R, Gandhi V. Targeting the apoptosis pathway in hematologic malignancies. Leukemia lymphoma. 2014;55:1980-92
31. Cubillos-Ruiz JR, Bettigole SE, Glimcher LH. Tumorigenic and Immunosuppressive Effects of Endoplasmic Reticulum Stress in Cancer. Cell. 2017;168:692-706
32. Rozpedek W, Pytel D, Mucha B. et al. The Role of the PERK/eIF2alpha/ATF4/CHOP Signaling Pathway in Tumor Progression During Endoplasmic Reticulum Stress. Curr Mol Med. 2016;16:533-44
33. Nakka VP, Prakash-babu P, Vemuganti R. Crosstalk Between Endoplasmic Reticulum Stress, Oxidative Stress, and Autophagy: Potential Therapeutic Targets for Acute CNS Injuries. Mol Neurobiol. 2016;53:532-44
34. Xu LY, Su L, Liu XG. PKC delta Regulates Death Receptor 5 Expression Induced by PS-341 through ATF4-ATF3/CHOP Axis in Human Lung Cancer Cells. Mol Cancer Ther. 2012;11:2174-82
35. Wang WA, Groenendyk J, Michalak M. Endoplasmic reticulum stress associated responses in cancer. Bba-Mol Cell Res. 2014;1843:2143-9
36. Nomura Y, Inanami O, Takahashi K. et al. 2-Chloro-2 '-deoxyadenosine induces apoptosis through the Fas/Fas ligand pathway in human leukemia cell line MOLT-4. Leukemia. 2000;14:299-306
37. Perez-Galan P, Marzo I, Giraldo P. et al. Role of caspases and apoptosis-inducing factor (AIF) in cladribine-induced apoptosis of B cell chronic lymphocytic leukemia. Leukemia. 2002;16:2106-14
38. Wawryk-Gawda E, Chylińska-Wrzos P, Lis-Sochocka M. et al. Intrinsic Apoptosis Pathway in Fallopian Tube Epithelial Cells Induced by Cladribine. The Scientific World J. 2014;2014:928036
39. Singh V, Prajeeth CK, Gudi V. et al. 2-Chlorodeoxyadenosine (cladribine) induces apoptosis in human monocyte-derived dendritic cells. Clin Exp Immunol. 2013;173:288-97
40. Marzo I, Perez-Galan P, Giraldo P. et al. Cladribine induces apoptosis in human leukaemia cells by caspase-dependent and -independent pathways acting on mitochondria. Biochem J. 2001;359:537-46
41. Johnston JB. Mechanism of Action of Pentostatin and Cladribine in Hairy Cell Leukemia. Leukemia lymphoma. 2011;52(Suppl 2):43-5
42. Smal C, Lisart S, Maerevoet M. et al. Pharmacological inhibition of the MAPK/ERK pathway increases sensitivity to 2-chloro-2 '-deoxyadenosine (CdA) in the B-cell leukemia cell line EHEB. Biochem Pharmaco. 2007;73:351-8
43. Conrad DM, Robichaud MRJ, Mader JS. et al. 2-Chloro-2 '-deoxyadenosine-induced apoptosis in T leukemia cells is mediated via a caspase-3-dependent mitochondrial feedback amplification loop. Int J Oncol. 2008;32:1325-33
44. Iurlaro R, Munoz-Pinedo C. Cell death induced by endoplasmic reticulum stress. Febs J. 2016;283:2640-52
45. Rozpedek W, Pytel D, Mucha B. et al. The Role of the PERK/eIF2 alpha/ATF4/CHOP Signaling Pathway in Tumor Progression During Endoplasmic Reticulum Stress. Curr Mol Med. 2016;16:533-44
46. Srivastava RK, Li CZ, Ahmad A. et al. ATF4 regulates arsenic trioxide-mediated NADPH oxidase, ER-mitochondrial crosstalk and apoptosis. Arch Biochem Biophys. 2016;609:39-50
47. Ju SM, Jo YS, Jeon YM. et al. Phosphorylation of eIF2 alpha suppresses cisplatin-induced p53 activation and apoptosis by attenuating oxidative stress via ATF4-mediated HO-1 expression in human renal proximal tubular cells. Int J Mol Med. 2017;40:1957-64
48. Mactier S, Henrich S, Che YP. et al. Comprehensive Proteomic Analysis of the Effects of Purine Analogs on Human Raji B-Cell Lymphoma. J Proteome Res. 2011;10:1030-42
49. Miao Y, Medeiros LJ, Xu-Monette ZY. et al. Dysregulation of Cell Survival in Diffuse Large B Cell Lymphoma: Mechanisms and Therapeutic Targets. Front Oncol. 2019;9:107
50. Herting F, Friess T, Umana P, Middleton S. et al. Chemotherapy-free, triple combination of obinutuzumab, venetoclax and idasanutlin: antitumor activity in xenograft models of non-Hodgkin lymphoma. Leukemia Lymphoma. 2018;59:1482-5
51. Kuo HP, Ezell SA, Schweighofer KJ. et al. Combination of Ibrutinib and ABT-199 in Diffuse Large B-Cell Lymphoma and Follicular Lymphoma. Mol Cancer Ther. 2017;16:1246-56
52. Cai B, Wang SL, Huang JC. et al. Cladribine and bendamustine exhibit inhibitory activity in dexamethasone-sensitive and -resistant multiple myeloma cells. Am J Transl Res. 2013;5:36-46
53. Ma J, Wang SL, Zhao M. et al. Therapeutic potential of cladribine in combination with STAT3 inhibitor against multiple myeloma. Bmc Cancer. 2011;11:255
54. Szmigielska-Kaplon A, Smolewski P, Najder M. et al. Evaluation of apoptosis induced in vitro by cladribine (2-CdA) combined with anthracyclines in lymphocytes from patients with B-cell chronic lymphocytic leukemia. Ann Hematol. 2002;81:508-13
Author contact
![]() Corresponding author: Kailin Xu, Blood Diseases Institute, Xuzhou Medical University, Xuzhou, 84 West Huaihai Road, Xuzhou, China. E-mail: lihmdcom; Tel: 86-516-85802382; Fax: 86-8580-1527.
Corresponding author: Kailin Xu, Blood Diseases Institute, Xuzhou Medical University, Xuzhou, 84 West Huaihai Road, Xuzhou, China. E-mail: lihmdcom; Tel: 86-516-85802382; Fax: 86-8580-1527.