Impact Factor ISSN: 1449-1907
- Issue 9; 2026
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Volume 23; 2026
- Past Issues
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Material and Methods
Methods
Results
Discussion
Conclusion
Acknowledgements
Abbreviations
Authors' contributions
References

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2020; 17(6):834-843. doi:10.7150/ijms.40959 This issue Cite
Research Paper
Action of low doses of Aspirin in Inflammation and Oxidative Stress induced by aβ1-42 on Astrocytes in primary culture
Adrian Jorda1,2*, Martin Aldasoro1*, Constanza Aldasoro1, Sol Guerra-Ojeda1, Antonio Iradi1, Jose Mª Vila1, Juan Campos-Campos1,2, Soraya L. Valles1 ![]()
1. Department of Physiology, School of Medicine, University of Valencia, Spain.
2. Faculty of Nursing and Podiatry, University of Valencia, Spain.
*These authors contributed equally to this work.
Received 2019-10-7; Accepted 2019-12-23; Published 2020-3-13
Abstract
Aspirin has been used as anti-inflammatory and anti-aggregate for decades but the precise mechanism(s) of action after the presence of the toxic peptide Aβ1-42 in cultured astrocytes remains poorly resolved. Here we use low-doses of aspirin (10-7 M) in astrocytes in primary culture in presence or absence of Aβ1-42 toxic peptide. We noted an increase of cell viability and proliferation with or without Aβ1-42 peptide presence in aspirin treated cells. In addition, a decrease in apoptosis, determined by Caspase 3 activity and the expression of Cyt c and Smac/Diablo, were detected. Also, aspirin diminished necrosis process (LDH levels), pro-inflammatory mediators (IL-β and TNF-α) and NF-ᴋB protein expression, increasing anti-inflammatory PPAR-γ protein expression, preventing Aβ1-42 toxic effects. Aspirin inhibited COX-2 and iNOS without changes in COX-1 expression, increasing anti-oxidant protein (Cu/Zn-SOD and Mn-SOD) expression in presence or absence of Aβ1-42. Taken together, our results show that aspirin, at low doses increases cell viability by decreasing inflammation and oxidative stress, preventing the deleterious effects of the Aβ1-42 peptide on astrocytes in primary culture. The use of low doses of aspirin may be more suitable for Alzheimer's disease.
Keywords: Amyloid-β, aspirin, inflammation, oxidative stress, Alzheimer's disease
Introduction
Alzheimer's disease (AD) causes decline in memory and is the most common neurodegenerative disease implicated in the aging process [1]. The prominent features of AD include amyloid plaques, intraneuronal tangles, cell death, inflammatory changes and oxidative stress [2,3,4].
Astroglia are the most prevalent cell type in the brain [5]. These cells have roles in the brain protecting against CNS injury and repairing nervous tissue after injury [6]. Data from our laboratory demonstrated that astrocytes increase neuronal viability and mitochondrial biogenesis, protecting from oxidative stress and inflammation induced by toxic amyloid peptide [7,8]. Also, astrocytes act in neuronal synapses, regulate the blood-brain barrier, providing nutrients to the nervous system and maintaining ion and metabolite balance, and also propagate calcium currents, release gliotransmitters, growth factors and inflammatory mediators [9,10,11]. Astrogliosis is produced by a reaction from astrocytes to inflammation, oxidative stress and cell death producing toxic products and inflammatory agents and oxidative stress mediators [3,12]. In astrocytes, complex changes and specific conflicts occur in different brain regions during the development of AD. The number of reactive astrocytes increases in AD, phagocytizing and reducing amyloid β (Aβ) deposition because these cells surround amyloid plaques and secrete proinflammatory factors [13,14].
Acetylsalicylic acid (aspirin) is used frequently as a member of the nonsteroidal anti-inflammatory drugs (NSAIDs) group [15]. Aspirin induces its effects by inhibiting cyclooxygenase (COX) and suppresses prostaglandins [16,17]. Two main isoforms of COX exist, COX-1 and COX-2. COX-1 is involved in the synthesis of thromboxane A2 (TXA2) [18] and COX-2 in prostacyclin biosynthesis [19,20]. In epidemiological studies of AD, a high dose of aspirin produces lower prevalence of AD [21] (Nilsson et al. 2003). Other authors using low-dose aspirin treatment indicated promising results for aspirin [22]. In this study, we were interested in exploring the action of aspirin in both inflammatory and ROS (reactive oxygen species) events associated with Alzheimer's disease (AD) by using the amyloid β1-42 in astrocytes in primary culture. The accumulation and precipitation of Aβ1-42 peptide has a neuropathological role associated with AD. However the Aβ40-1 peptide has non-toxic effects and is used as control.
Material and Methods
Materials
This study was approved by the Bioethics Committee of the School of Medicine of the University of Valencia, and the local Government of Valencia, Spain (2016/VSC/PEA/00220). All animals (Wilson rats from Charles River laboratory) were handled according to the recommendations of the Committee and with access to food and water. Animals were sacrificed with pentobarbital by the veterinary personnel. No randomization was performed to allocate subjects in the study. Rats (6 months old) were housed alone for 21 days during pregnancy. Female rat fetuses were obtained at day 21 of pregnancy prior to delivery. The fetal cortex was obtained to create a culture of astrocytes. Every assay was performed 3, 4 or 5 times, from different mother rats. No blinding procedures for each culture were made. Female rats were excluded if they were pregnant before the study. Rats were also excluded if the delivery of rats was 21 days before pregnancy.
Dulbecco's modified Eagle's medium (DMEM) and fetal bovine serum (FBS) were obtained from Gibco (Gibco Invitrogen Corporation, Barcelona, Spain). The oligomers Aβ (40-1 and 1-42), were prepared following manufacturer's instructions (Sigma-Aldrich biotechnology). Briefly, the peptides were dissolved in 100 µM phosphate buffered saline (PBS) and preparations were heated for 24 h at 37°C for assembly of the oligomers. Aspirin was obtained from Sigma-Aldrich biotechnology and dissolved in Krebs solution to the proper final concentration (10-7 M). 3-(4,5-dimethyl-2-thiazolyl)-2,5-dipheniyl-2H-tetrazolium bromide (MTT) was purchased from Sigma Chemical Co. (St Louis, MO). Enzyme-linked immunosorbent assay (ELISA) kits for IL-1β (Interleukin 1-β) and TNF-α (Tumor necrosis-α) from Pierce Biotechnology, Inc. (Rockford, USA). Western Blot Chemiluminescent Detection System (ECL) was from Amersham (Amersham Biosciences, Barcelona, Spain). Monoclonal anti-cytochrome C (Cyt c) antibody (1:500), monoclonal anti-Smac/Diablo antibody (Smac/Diablo) (1:500), monoclonal anti-nuclear factor κB antibody (NF-κB) (1:1000), monoclonal anti-Mn superoxide dismutase antibody (Mn-SOD) (1:500), monoclonal anti-cyclooxygenase 1 antibody (COX-1) (1:500), monoclonal anti-cyclooxygenase 2 antibody (COX-2) (1:500), monoclonal anti-inducible nitric oxide synthase antibody (iNOS) (1:500) from Santa Cruz Biotechnology (Madrid, Spain). Monoclonal anti-peroxisome proliferator-activated receptor antibody (PPAR-γ) (1:500) from Sigma Aldrich (Madrid, Spain). Polyclonal anti-Cu/Zn superoxide dismutase antibody (Cu/Zn-SOD) (1:500) from Assay Designs (Madrid, Spain). Monoclonal anti-tubulin antibody (1:1000) from Cell Signaling (Beverly, MA, USA). All other reagents were analytical or culture grade purity.
Methods
Primary Culture of Cortical Astrocytes
Cerebral cortical astrocytes were isolated from rat fetuses of 21 days gestation. Fetuses were obtained by caesarean section and decapitated. Cerebral cortices were removed and triturated 10-15 times through a Pasteur pipette. The cell suspension was filtered through nylon mesh with a pore size of 90 μm and diluted in DMEM containing 20% fetal bovine serum (FBS) supplemented with L-glutamine (1%), HEPES (10 mM), fungizone (1%), and antibiotics (1%). Cells were plated on T75 culture flask pretreated with poli-L-lysine. Cultures were maintained in a humidified atmosphere of 5% CO2/95% air at 37°C during 20 days. After 1 week of culture, the FBS content was reduced to 10%, and the medium was changed twice a week. By immunocytochemistry, 97% of cells are GFAP positive (data not shown).
Four groups were used. Group A, control received Aβ40-1 peptide, Group B, Aβ40-1 peptide + aspirin, Group C, Aβ1-42 toxic peptide and Group D, Aβ1-42 toxic peptide + aspirin. Initially, we used Aβ42-1 but due to its high cost, we assayed Aβ40-1 as we have used before [7].
MTT assay
Cell viability of the cultures was determined by the MTT assay. Cells were plated in 96 well culture plate and incubated with Asp during 24 h at 10-11 M, 10-9 M, 10-7 M, 10-5 M, Aβ1-42 15 µM or with Aβ1-42 15 µM + 10-7 M Asp. After cell treatments, the medium was removed and the cortical cells were incubated with red free medium and MTT solution [0.5 mg/ml, prepared in phosphate buffer saline (PBS) solution] for 4 h at 37ºC. Finally the medium was removed and formazan particles were dissolved in dimethyl sulfoxide (DMSO). Cell viability, defined as the relative amount of MTT reduction, was determined by spectrophotometry at 570 nm.
Trypan Blue Assay
Trypan blue exclusion assay was used to count the living cells and monitor cell proliferation. Astrocytes were isolated and seeded at 7x104 cells/35 mm dish. After 5 days of culture, cells were incubated without (control, C), with Asp (10-7 M), Aβ1-42 15 µM or with Aβ1-42 15 µM + 10-7 M Asp for 24 h. 1.5% trypan blue solution was applied to astrocytes cultures at room temperature for 3 min.
Lactate Dehydrogenase (LDH) Assay
To evaluate plasma membrane integrity, LDH release was determined by monitoring the leakage of the cytosolic LDH to the extracellular medium. LDH was measured spectrophotometrically at 340 nm, following the rate of conversion of reduced nicotinamide adenine dinucleotide to oxidized nicotinamide adenine dinucleotide.
Caspase 3 Activity Assay
Caspase 3 activity was measured in cytosolic fractions by using a highly sensitive colorimetric substrate, N-acetyl-Asp-Glu-Val-Asp p-nitroanilide (Ac-DEVD-pNA) following manufacturer´s instructions (CalBiochem, La Jolla, CA). Enzyme activity was calculated using manufacturer´s formulae, as pmol/min.
Cytokine Determination, IL-1β and TNFα
Cells were seeded, and at time of assay, the red phenol medium was removed and replaced by PBS containing 1 mg/ml bovine serum albumin (BSA), either in the presence or absence of Asp (10-7 M), with Aβ1-42 15 µM or Aβ1-42 15 µM + 10-7 M Asp). IL-1β and TNF-α concentration (pg/ml) were ascertained using ELISA kits (Pierce Biotechnology, Inc.).
Western Blot Analysis
Cultured cells were treated with lysis buffer and then mechanically degraded to release the proteins. Protein concentration was determined using modified Lowry method. Loading buffer (0.125 M Tris-HCl, pH 6.8, 2% SDS, 0.5% (v/v) 2-mercaptoethanol, 1% bromophenolblue and 19% glycerol) was added to protein sample and heated for 5 min at 95ºC. Proteins (20 µg) were separated on SDS-PAGE gels and transferred to nitrocellulose membranes in a humid environment using a transfer buffer (25 mM Tris, 190 mM glycine, and 20% methanol). Membranes were blocked with 5% milk in TBS-T (0.05% Tween-20) and incubated with primary antibodies overnight at 4ºC. Membranes were washed 3 times with wash buffer TBS-T (TBS, 0.2% Tween-20) and incubated with a secondary anti-rabbit IgG or anti-mouse IgG antibody conjugated to the enzyme horseradish peroxidase (HRP) for 1 h. Membranes were washed three times and proteins were detected using the ECL method as specified by the manufacturer. Autoradiography signals were assessed using digital image system ImageQuant LAS 4000 (GE Healthcare). Densitometry is the quantitative measurement of optical density in a photographic paper or photographic film, due to exposure to light. Concentration of protein was determined by densitometry analysis, expressed as arbitrary units relative to tubulin.
Statistical Analyses
All values are expressed as mean ± S.D. The differences between groups were determined with unpaired Student´s t-test. All statistical analyses were performed using the GraphPad Prism software (GrapshPad Software Inc., San Diego, CA, USA). Statistical significance was accepted at p ≤ 0.05.
Results
Asp and Cell Viability
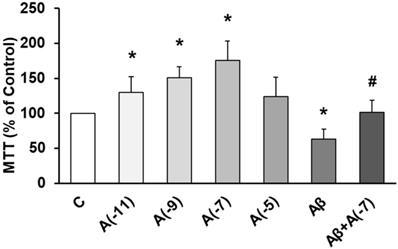
The role of Asp on cell viability was studied using MTT conversion assay. Fig. 1 shows that incubation with Asp at 10-11 M, 10-9 M, and 10-7 M significantly increased astrocyte viability vs control. On the other hand, Aβ1-42 significantly decreased cell viability (30%) compared to control cells. After incubation with Aβ1-42 + 10-7 M Asp, no significant changes were detected compared to control astrocytes and contrarily, an increase in cell viability was detected compared to cells with Aβ1-42 peptide alone.
Cell viability was determined by MTT assay in cells treated during 24 h. Astrocytes were incubated without Asp (control, C), with Asp at different concentrations (10-11, 10-9, 10-7 and 10-5 M), with Aβ1-42 (15 µM) or Aβ1-42 (15 µM) + Asp (10-7 M) for 24 h. Data are means ± SD of four independent experiments (three different rats). *p < 0.05 vs. control. # p < 0.05 vs Aβ1-42 treated cells.
Trypan blue exclusion assay was used to count the living cells and monitor cell proliferation. Astrocytes were isolated and seeded at 7x104 cells/35 mm dish. After 5 days of culture, cells were incubated without (control, C) or with Asp 10-7 M, Aβ1-42 15 µM or Aβ1-42 15 µM + Asp 10-7 M for 24 h. In control conditions proliferation was 0.93%, and previous incubation with Asp (10-7 M) increased proliferation by 9.53%. On the other hand, in presence of Aβ1-42 proliferation decreased 12.96% and with Aβ1-42 + Asp 10-7 M only decreased 5.37% (Table 1).
Astrocytes proliferation and counting living cells
| Seeding cells | 5 days of culture | After 24 h of incubation | % Proliferation | |
|---|---|---|---|---|
| C | 7 | 12.72±0.19 | 12.83±0.20 | +0.93 |
| Asp (10-7 M) | 7 | 12.70±0.11 | 13.91±0.16* | +9.53 |
| Aβ | 7 | 12.65±0.23 | 11.01±0.32* | -12.96 |
| Aβ + Asp (10-7 M) | 7 | 12.75±0.24 | 12.10±0.14* | -5.37 |
Astrocytes were isolated and seeded at 7x104 cells/35 mm dish during 5 days. At this time, cells were incubated without Asp (control, C), with Asp (10-7 M), Amyloid β1-42 (15 µM) or Amyloid β1-42 (15 µM) + Asp (10-7 M) for 24 h. Trypan blue exclusion was used to count the living cells and monitor cell proliferation. Data are mean ± SD of five independent experiments (four different rats). *p < 0.05 vs. control.
LDH and Caspase 3
Incubation of the astrocytes with Asp 10-7 M for 24 h decreased significantly LDH values (21%) compared with control cells. With Aβ1-42 (15 µM) an increase of LDH release (55%) was detected compared with control cells and this data was reversed with Asp (10-7 M) to control values (Fig. 2A).
Lactate dehydrogenase and caspase 3 activity. Astrocytes were incubated without Asp (control, C), with Asp (10-7 M), Aβ1-42 (15 µM) or Aβ1-42 (15 µM) + Asp (10-7 M) for 24 h. Panel A: Lactate dehydrogenase from supernatants of astrocytes. Panel B: Caspase 3 activity. Data are means ± SD of four independent experiments (four different rats). *p < 0.05 vs. control cells. #p < 0.05 vs. Aβ1-42 treated cells.
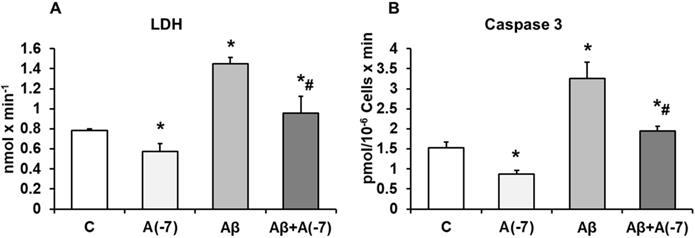
Incubation with Asp 10-7 M for 24 h, decreased Caspase 3 activity significantly (45%) compared to control cells, whereas Aβ1-42 (15 µM) activity was increased in presence of Asp (10-7 M), and prevented the toxic peptide effect (Fig 2B), indicating reduction of apoptosis when Asp is present on the culture with Aβ1-42.
Cytochrome c and Smac/DIABLO Expression
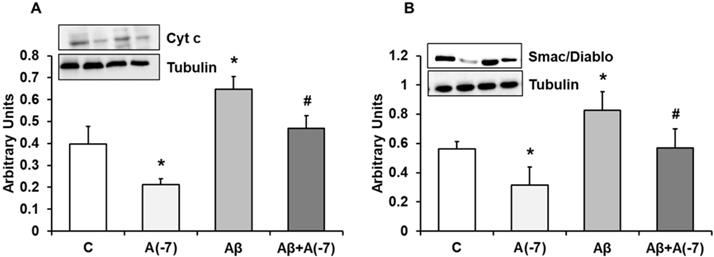
Fig. 3A shows cytochrome c expression in astrocytes in primary culture. Asp decreased cytochrome c expression by 2.2-fold at 10-7 M. Aβ1-42 increased cytochrome c expression compared to control cells. On the contrary, Asp (10-7 M) significantly reversed the cytochrome c increase expression induced by Aβ1-42. Fig. 3B shows Smac/Diablo expression in astrocytes in primary culture. Asp significantly decreased Smac/Diablo protein expression compared to control cells. Addition of Aβ1-42 significantly increased Smac/Diablo expression (1.3-fold) compared with control cells. Furthermore, Asp 10-7 M decreased Smac/Diablo expression induced by Aβ1-42 to control values. Consequently Asp addition produced a protective effect against the amyloid toxic peptide (Fig. 3).
Cytocrhome c and Smac/Diablo expression. Astrocytes were incubated without Asp (control, C), with Asp (10-7 M), Aβ1-42 (15 µM) or Aβ1-42 (15 µM) + Asp (10-7 M) for 24 h and collected to determine Cytocrhome c (panel A) or Smac/Diablo (panel B) protein expression by Western-blot. A representative immunoblot is shown in the top panel. Data are means ± SD of four independent experiments (four different rats). *p < 0.05 vs. control cells. #p < 0.05 vs. Aβ1-42 treated cells.
IL-1β and TNF-α Pro-inflammatory Cytokines
Secretion of the pro-inflammatory mediators, IL-1β and TNF-α, were detected by ELISA. Fig 4 shows that, in astrocytes, Asp (10-7 M) decreased 1.6-fold IL-1β release compared with control values (Fig 4A). Conversely, Aβ1-42 addition increased 2.5-fold IL-1β secretion compared with control cells. Aβ1-42 + Asp 10-7 M decreased IL-1β liberation to control values.
IL-1β and TNF-α determination. Astrocytes were incubated without Asp (control, C), with Asp (10-7 M), Aβ1-42 (15 µM) or Aβ1-42 (15 µM) + Asp (10-7 M). Cell culture supernatants were harvested and IL-1β (Panel A) and TNF-α (Panel B) were determined by ELISA. Values are means ± SD from four independent experiments (four different rats). *p < 0.05 vs control. #p < 0.05 vs. Aβ treated cells.
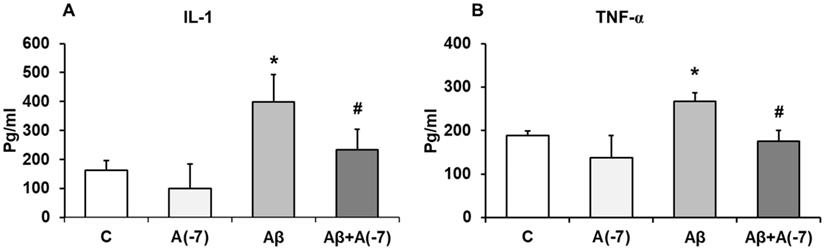
Fig. 4B shows that Asp (10-7 M) produced a significant decrease of TNF-α secretion (3.1-fold) compared to control cells. Aβ1-42 addition increased TNF-α liberation 1.5-fold. Moreover, Asp (10-7 M) prevented this toxic effect produced by Aβ1-42 peptide.
NF-κB and PPAR-γ Expression
NF-κB is a transcription factor that regulates positively gene expression of pro-inflammatory proteins. Fig. 5A shows that Asp decreased this protein expression about 1.25-fold compared to control astrocytes. When Aβ1-42 was added an increase (1.5-fold) of this transcription factor was detected compared to control results. Asp (10-7 M) addition returned NF-κB to control values.
NF-ᴋB and PPAR-γ expression. Astrocytes were incubated without Asp (control, C), with Asp (10-7 M), Aβ1-42 (15 µM) or Aβ1-42 (15 µM) + Asp (10-7 M) for 24 h and collected to determine NF-ᴋB (panel A) or PPAR-γ (panel B) protein expression by Western-blot. A representative immunoblot is shown in the top panel. Data are means ± SD of four independent experiments (four different rats). *p < 0.05 vs. control cells. #p < 0.05 vs. Aβ1-42 treated cells.
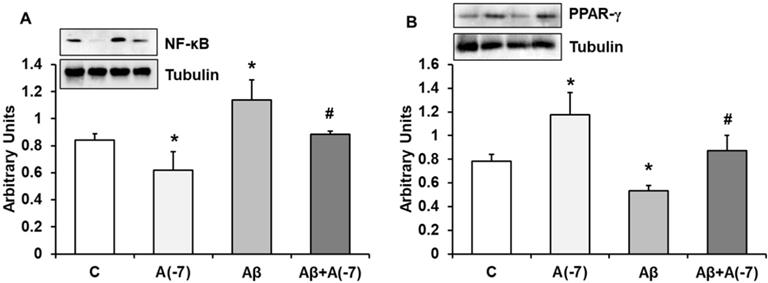
PPARs family regulates negatively gene expression of pro-inflammatory proteins. Fig. 5B shows PPAR-γ expression in astrocytes in culture. Asp increased PPAR-γ expression 1.5-fold at 10-7 M. Addition of Aβ1-42 decreased 1.6-fold this transcription factor compared to control results. Asp (10-7 M) addition returned PPAR-γ expression to control values.
COX-2 and iNOS Expression
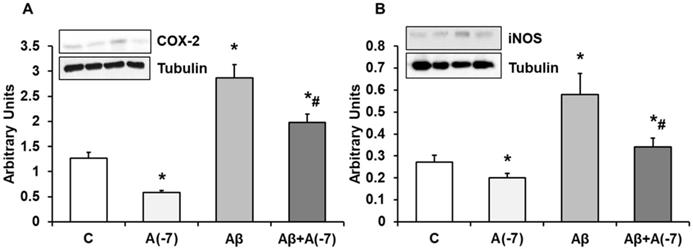
In Fig. 6, we detected a reduction of COX-2 (panel A) expression after addition of Asp (10-7 M) compared with control values. The presence of the toxic peptide Aβ1-42 increased the expression of COX-2 that was reversed by the incubation with Asp 10-7 M to control values. On the contrary, no changes were detected in COX-1 expression at any experimental condition (Data not shown). Fig. 6 (panel B) demonstrates a significand decrease in iNOS protein expression after Asp 10-7 M addition compared to control. Aβ1-42 produced a significant increase of iNOS expression. On the other hand, the presence of both the toxic peptide and Asp 10-7 M reduced significantly iNOS expression compared to Aβ1-42.
COX-2 and iNOS expression. Astrocytes were incubated without Asp (control, C), with Asp (10-7 M), Aβ1-42 (15 µM) or Aβ1-42 (15 µM) + Asp (10-7 M) for 24 h and collected to determine COX-2 (panel A) or iNOS (panel B) protein expression by Western-blot. A representative immunoblot is shown in the top panel. Data are means ± SD of four independent experiments (four different rats). *p < 0.05 vs. control cells. #p < 0.05 vs. Aβ1-42 treated cells.
Expression of CU/ZN-SOD and Mn-SOD Proteins
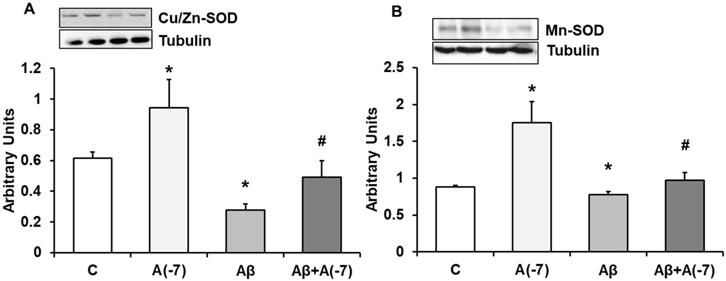
In astrocytes, Asp 10-7 M increased Cu/Zn-SOD (panel A) and Mn-SOD (panel B) expression compared to control cells (Fig. 7). In the presence of the Aβ1-42 toxic peptide, a decrease of both proteins compared with control values was reversed by Asp.
Cu/Zn-SOD and Mn-SOD expression. Astrocytes were incubated without Asp (control, C), with Asp (10-7 M), Aβ1-42 (15 µM) or Aβ1-42 (15 µM) + Asp (10-7 M) for 24 h and collected to determine Cu/Zn-SOD (panel A) or Mn-SOD (panel B) protein expression by Western-blot. A representative immunoblot is shown in the top panel. Data are means ± SD of four independent experiments (four different rats). *p < 0.05 vs. control cells. #p < 0.05 vs. Aβ1-42 treated cells.
Discussion
In this study we show that aspirin, at low-doses, protects from Aβ1-42 toxic peptide actions in astrocytes in primary culture, indicant the convenience to use low doses to obtain better benefices of aspirin. The aspirin increases cell viability and proliferation, decreases apoptosis (Caspase 3, Cyt c and Smac/Diablo) and necrosis (LDH), in the presence or absence of Aβ1-42 peptide. Moreover, the aspirin decreases pro-inflammatory mediators (IL-β and TNF-α) and NF-κB expression and increases anti-inflammatory PPAR-γ protein after addition of Aβ1-42. As also inhibits COX-2 and iNOS without changes in COX-1 expression and increases anti-oxidant proteins (Cu/Zn-SOD and Mn-SOD) expression in the presence or absence of Aβ1-42.
The role of astrocytes in the brain has been reviewed [23,24,25]. It is reported that astrocytes protect neurons against Aβ-amyloid peptide, decreasing inflammation, and oxidative stress, and increasing cell viability and mitochondrial biogenesis [7,8,26].
Low-dose of aspirin has been reported to reduce the incidence of Alzheimer's disease [27] and also the donation of its acetyl group to molecules that suppress protein aggregation in neurodegenerative diseases, including AD [28]. This mechanism could be implicated in the astrocytic protection observed in our study. Aβ1-42 peptide induces reduction in cell viability and increases apoptosis by cytochrome c and Smac/Diablo pathways [7,29]. However, simultaneous treatment with aspirin of Aβ1-42 treated cells protects them from neurotoxicity [29]. Reactive gliosis increases glial fibrillary acidic protein (GFAP) in AD where it is highly expressed and remarkably, aspirin produces a reduction in GFAP synthesis by blocking NF-κB in astrocytes culture [30]. It is reported that high doses aspirin can help reduce chronic AD inflammation [30]. However, our results show that aspirin, at low concentration (10-7 M), increases cell viability, diminishing necrosis and apoptosis. Burke et al. (2006) [31] reported that human daily ingestion of aspirin, 80 to 350 mg, produces plasma salicylate levels of 2.4 to 9.7 µg/ml, corresponding to 1.3 × 10-6 to 5.4 x 10-5 M of aspirin in the culture medium [31]. According to these results, the concentrations used in our experiments correspond to doses lower than 70 mg/day, considered as low doses of aspirin [32]. However, aspirin increases apoptosis in gastric mucosal cell line in a caspase-dependent manner through Smac/Diablo pathway [33]. Our results differ due to aspirin concentration used. Redlak et al. (2005) [33] used a concentration of 4 × 10-2 M whereas in our study we used 10-7 M.
Our results indicate that low concentrations of aspirin may decrease apoptosis as opposed to high doses, protecting without increasing the adverse mechanisms observed in other paper.
After Aβ1-42 addition, we detected an increase in COX-2 without changes in COX-1 expression. Both COX proteins could lead to adverse cellular effects derived from TXA2 and prostacyclin biosynthesis. Using LPS (bacterial lipopolysaccharide) to determine COX isoform expression in astrocytes, Font-Nieves et al. (2012) [34] showed a strong induction of COX-2 through an NF-κB-dependent mechanism [34]. In chronic inflammation and in the progression of neurodegenerative diseases, NF-κB activation plays an important role in astrocytes in primary culture [35], according to previous data obtained in our laboratory indicating that Aβ addition is also associated with an increase in NF-κB activity in astrocytes [7]. Lee et al. (2018) [36], have indicated that downregulation of inflammatory molecules such as iNOS and different cytokines (TNF-α, IL-1β and IL-6) could be a consequence of the NF-κB and MAP kinases inhibition in astrocytes [36]. Furthermore, NF-κB binding sites are present in the promoters of iNOS and COX-2 and are essential to modulate their transcription [37,38]. Results of Yao et al. (2014) [39] demonstrated that aspirin normalizes COX-2 and iNOS over-expression in astrocytes in part through inhibition of the NF-κB pathway [39]. As these authors indicated, we show a reduction in the expression of iNOS, COX-2 and NF-κB in the presence of aspirin.
Aspirin has additional targets in humans, besides the cyclooxygenases. Salicylic acid (SA), the primary metabolite of aspirin, binds human glyceraldehyde 3-phosphate dehydrogenase (GAPDH) and suppresses nuclear translocation and cell death [40]. It is known that GAPDH plays an important role in neurodegenerative diseases, including AD [41].
In neurodegenerative diseases such as AD, treatment with non-steroidal anti-inflammatory drugs (NSAIDs) has been reported [42] and COX-1 and COX-2 are the targets of those drugs. Furthermore both COX isoforms regulate PPARγ activity through prostaglandin synthesis [43]. Also, PPARγ has been shown to inhibit the expression of proinflammatory genes, such as iNOS [44,35] and has several inhibitory effects on inflammation, including reduction of NF-κB transcriptional activities and promotes anti-inflammatory mediators [43]. Potent synthetic antidiabetics such as rosiglitazone are agonists for PPAR-γ [45,46] and many NSAIDs are also PPAR-γ agonists [47]. Furthermore, reduction of Aβ induced by indomethacin or naproxen by inhibiting BACE1 (beta-site APP-cleaving enzyme 1) activity occurs in a PPAR-γ dependent manner [48]. In our study, we detected a decrease of PPAR-γ expression after Aβ addition that was reversed by treatment with aspirin. It is possible that aspirin could inhibit Aβ synthesis through the increment of PPAR-γ.
In this study we also investigated the possible antioxidant properties of aspirin using Aβ1-42 toxic peptide as a model. We showed an increase expression of Mn-SOD and Cu/Zn-SOD proteins after aspirin addition in culture of astrocytes with and without the toxic peptide. In agreement with our results, Dairam et al. (2006) [49] have demonstrated the antioxidant and neuroprotective effects of NSADs in an AD rat model [49]. Furthermore, aspirin-elicited lipoxin A4 inhibited ROS synthesis induced by LPS in microglial cells [50]. Also, aspirin minimizes the effect of free radicals induced by LPS in rat dopaminergic neurons [51]. Liu et al. (2017) [52] indicated that aspirin at doses of 75 and 100 mg/day, similar to that used in our study, stimulates the levels of superoxide dismutase (SOD). On the other hand, production of L-NMMA in microcirculation is inhibited by aspirin, due to cyclooxygenase inhibition or SOD increase [53,54].
Also, aspirin exerts pro-oxidant effects on Mn-SOD-deficient yeast cells causing apoptosis with mitochondrial involvement [55]. Furthermore, aspirin on focal cerebral ischemia-reperfusion rats reduced MDA content [56].
Conclusion
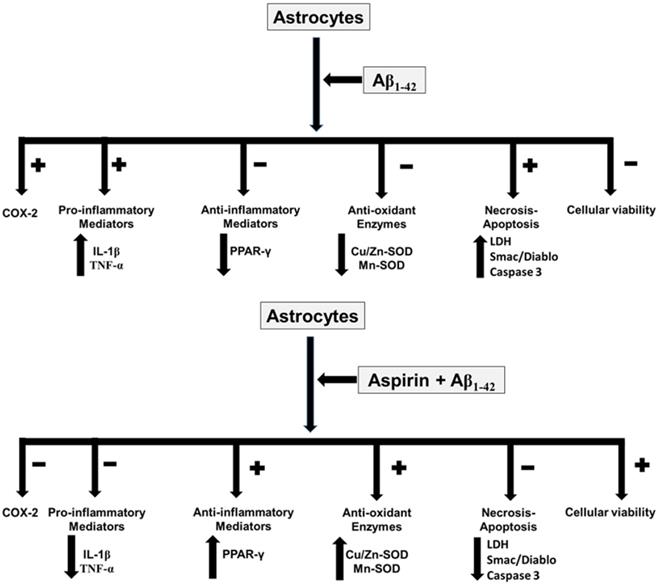
In conclusion, our results indicate that aspirin, at low doses, prevents toxic effects induced by Aβ1-42 peptide in astrocytes in primary culture, increasing cell viability and proliferation while decreasing apoptosis and necrosis. The key finding of our study is that aspirin at low doses prevents oxidative stress and inflammation induced by Aβ1-42 toxic peptide. So, the administration of low doses of aspirin could be useful in AD patients (Fig. 8).
Aspirin effects in astrocytes in primary culture. Asp increases cell viability, anti-inflammatory response and anti-oxidant proteins. On the other hand, Asp decreases pro-inflammatory mediators, necrosis and apoptosis.
Acknowledgements
Funding sources
This work was supported in part by grant from local government of Spain: Gvcs2007-AP-001.
Ethics approval consent
All animal procedures were carried out in accordance with the European legislation on the use and care of laboratory animals (CEE 86/609). Experimental research on mice was performed with the approval of the ethics committee on animal research of the University of Valencia (Spain) and Generalitat Valenciana local government (2016/VSC/PEA/00220). All procedures were performed in accordance with the 1964 Helsinki declaration and its later amendments.
Abbreviations
MTT: 3-(4,5-dimethyl-2-thiazolyl)-2,5-dipheniyl-2H-tetrazolium bromide; Asp: Acetylsalicylic acid: aspirin; AD: Alzheimer's disease; Aβ: Amyloid β; Cu/Zn: superoxide dismutase; COX: Cyclooxygenase; Cyt c: Cytochrome c; GFAP: Glial fibrillary protein; NF-κB: Nuclear factor κB; PPAR-γ: Peroxisome proliferator-activated receptor.
Authors' contributions
AJ designed the study, carried out the LDH and Caspase 3 experiments, contributed to wester-blot, and wrote the manuscript. MA designed the study, carried out the ELISA assays and helps to write the manuscript. CA, SGO and JCC contributed to western-blot experiments. AI contributed to statistics analysis. JV contributed to the look for references and statistics analysis. SLV conceived the study, participated in its design and coordination, and helped to draft the manuscript. All authors read and approved the final manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Lane CA, Hardy J, Schott JM. Alzheimer's disease. Eur J Neurol. 2018;25:59-70
2. Perry G, Cash AD, Smith M. Alzheimer disease and oxidative stress. J Biomed Biotechnol. 2002;2:120-123
3. Holtzman DM, Mandelkow E, Selkoe DJ, Alzheimer disease in 2020, Cold Spring Harb Perspect. Med. 2011. doi:10.1101/cshperspect.a011585.
4. Selkoe DJ, Hardy J. The amyloid hypothesis of Alzheimer's disease at 25 years. EMBO Mol Med. 2016;8:595-608
5. Figley CR, Stroman PW. The role(s) of astrocytes and astrocyte activity in neurometabolism, neurovascular coupling, and the production of functional neuroimaging signals. Eur J Neurosci. 2011;33:577-588
6. Frost GR, Li YM. The role of astrocytes in amyloid production and Alzheimer's disease. Open Biol. 2017:7 doi: 10.1098/rsob.170228
7. Aguirre-Rueda D, Guerra-Ojeda S, Aldasoro M, Iradi A, Obrador E, Ortega A. et al. Astrocytes protect neurons from Aβ1-42 peptide-induced neurotoxicity increasing TFAM and PGC-1 and decreasing PPAR-γ and SIRT-1. Int J Med Sci. 2015 a;12:48-56
8. Aguirre-Rueda D, Guerra-Ojeda S, Aldasoro M, Iradi A, Obrador E, Mauricio MD. et al. WIN 55,212-2, agonist of cannabinoid receptors, prevents amyloid β1-42 effects on astrocytes in primary culture. PLoS One. 2015 b. doi: 10.1371/journal.pone.0122843
9. Nedergaard M, Ransom B, Goldman SA. New roles for astrocytes: redefining the functional architecture of the brain. Trends Neurosci. 2003;26:523-530
10. Malarkey EB, Parpura V. Mechanisms of glutamate release from astrocytes. Neurochem Int. 2018;52:142-154
11. Verkhratsky A, Olabarria M, Noristani HN, Yeh CY, Rodriguez JJ. Astrocytes in Alzheimer's disease. Neurotherapeutics. 2010;7:399-412
12. Giunta B, Fernandez F, Nikolic WC, Obrego E, Rrapo E, Town T. et al. Inflammaging as a prodrome to Alzheimer's disease. J Neuroinflammation. 2008;5:51-65
13. Hou L, Liu Y, Wang X, Ma H, He J, Zhang Y. et al. The effects of amyloid-β42 oligomer on the proliferation and activation of astrocytes in vitro. Vitr Cell Dev Biol Anim. 2011;47:573-580
14. Farina C, Aloisi F, Meinl E. Astrocytes are active players in cerebral innate immunity. Trends Immunol. 2017;28:138-145
15. Green GA. Understanding NSAIDs: from aspirin to COX-2, Clin Cornerstone. 2001;3:50-60.
16. Vane JR, Botting R. The mechanism of action of aspirin. Thromb Res. 2003;110:255-258
17. Ertmer C, Rehberg S, Westphal M. Cyclooxygenase-2 inhibition and increased arterial vasoconstriction to vasopressin: what is the link? Crit Care Med. 2008;36:353-354
18. Caughey GE, Cleland LG, Penglis PS, Gamble JR, James MJ. Roles of cyclooxygenase (COX)-1 and COX-2 in prostanoid production by human endothelial cells: selective up-regulation of prostacyclin synthesis by COX-2. J Immunol. 2001;167:2831-2838
19. Aldasoro M, Mauricio MD, Serna E, Cortina B, Segarra G, Medina P. et al. Effects of aspirin, nimesulide, and SC-560 on vasopressin-induced contraction of human gastroepiploic artery and saphenous vein. Crit Care Med. 2008;36:193-197
20. McAdam BF, Catella-Lawson F, Mardini IA, Kapoor S, Lawson JA, FitzGerald GA. Systemic biosynthesis of prostacyclin by cyclooxygenase (COX)-2: the human pharmacology of a selective inhibitor of COX-2, Proc Nat Acad Sci USA. 1999; 96:272-277. Erratum in: Proc Nat Acad Sci USA. 1999;96:5890
21. Nilsson SE, Johansson B, Takkinen S, Berg S, Zarit S, McClearn G. Does aspirin protect against Alzheimer's dementia? A study in a swedish population-based sample aged or 80 years. Eur J Clin Pharmacol. 2003;59:313-319
22. Kern S, Skoog I, Ostling S, Kern J, Börjesson-Hanson A. Does low-dose acetylsalicylic acid prevent cognitive decline in women with high cardiovascular risk? A 5-year follow-up of a non-demented population-based cohort of Swedishelderly women. BMJ Open. 2012 doi:10.1136/bmjopen-2012-001288
23. Heneka MT, Carson MJ, El Khoury J, Landreth GE, Brosseron F, Feinstein DL. et al. Neuroinflammation in Alzheimer's disease. Lancet Neurol. 2015;14:388-405
24. Nirzhor SSR, Khan RI, Neelotpol S. The Biology of Glial Cells and Their Complex Roles in Alzheimer's Disease: New Opportunities in Therapy. Biomolecules. 2018:10 doi: 10.3390/biom8030093
25. Jorda A, Cauli O, Santonja JM, Aldasoro M, Aldasoro C, Obrador E. et al. Changes in Chemokines and Chemokine Receptors Expression in a Mouse Model of Alzheimer's Disease. Int J Biol Sci. 2019;15:453-463
26. Yao Y, Huang JZ, Chen Y, Hu HJ, Tang X, Li X. Effects and mechanism of amyloid β1-42 on mitochondria in astrocytes. Mol Med Rep. 2018;17:6997-7004
27. Pomponi MF, Gambassi G, Pomponi M, Masullo C. Alzheimer's Diseases fatty acids we eat may be linked to a specific protection via low-dose aspirin. Aging Dis. 2010;1:37-59
28. Ayyadevara S, Balasubramaniam M, Kakraba S, Alla R, Mehta JL, Shmookler-Reis RJ. Aspirin-Mediated Acetylation Protects Against Multiple Neurodegenerative Pathologies by Impeding Protein Aggregation. Antioxid Redox Signal. 2017;27:1383-1396
29. Parmar HS, Houdek Z, Pesta M, Vaclava C, Dvorak P, Hatina J. Protective Effect of Aspirin against Oligomeric Aβ42 Induced Mitochondrial Alterations and Neurotoxicity in Differentiated EC P19 Neuronal Cells. Curr Alzheimer Res. 2017;14:810-819
30. Bae MK, Kim SR, Lee HJ, Wee HJ, Yoo MA, Ock Oh S. et al. Aspirin-induced blockade of NF-kappaB activity restrains up-regulation of glial fibrillary acidic protein in human astroglial cells. Biochim Biophys Acta. 2006;1763:282-289
31. Burke A, Smyth E, Fitzgerald G, Goodman, Gilmman's the Pharmacological Basis of Therapeutics, 11th ed. McGraw-Hill, New York. 2006.
32. He J, Whelton PK, Vu B, Klag MJ. Aspirin and risk of hemorrhagic stroke: a meta-analysis of randomized controlled trials. JAMA. 1998;280:1930-1935
33. Redlak MJ, Power JJ, Miller TA. Role of mitochondria in aspirin-induced apoptosis in human gastric epithelial cells. Am J Physiol Gastrointest Liver Physiol. 2005;289:G731-8
34. Font-Nieves M, Sans-Fons MG, Gorina R, Bonfill-Teixidor E, Salas-Pérdomo A, Márquez-Kisinousky L. et al. Induction of COX-2 enzyme and down-regulation of COX-1 expression by lipopolysaccharide (LPS) control prostaglandin E2 production in astrocytes. J Biol Chem. 2012;287:6454-6468
35. Newcombe EA, Camats-Perna J, Silva ML, Valmas N, Huat TJ, Medeiros R. Inflammation: the link between comorbidities, genetics, and Alzheimer's disease. J Neuroinflammation. 2018;24:276
36. Lee JE, Park JS, Lee YY, Kim DY, Kang JL, Kim HS. Anti-inflammatory and anti-oxidant mechanisms of an MMP-8 inhibitor in lipoteichoic acid-stimulated rat primary astrocytes: involvement of NF-κB, Nrf2, and PPAR-γ signaling pathways. J Neuroinflammation. 2018;23:326
37. Chen CH, Sheu MT, Chen TF, Wang YC, Hou WC, Liu DZ. et al. Suppression of endotoxin-induced proinflammatory responses by citrus pectin through blocking LPS signaling pathways. Biochem Pharmacol. 2006;72:1001-1009
38. Wu KK. Control of cyclooxygenase-2 transcriptional activation by pro-inflammatory mediators, Prostaglandins Leukot Essent Fatty Acids. 2005;72:89-93.
39. Yao C, Yang D, Wan Z, Wang Z, Liu R, Wu Y. et al. Aspirin-triggered lipoxin A4 attenuates lipopolysaccharide induced inflammatory response in primary astrocytes. Int Immunopharmacol. 2014;18:85-89
40. Choi HW, Tian M, Manohar M, Harraz MM, Park SW, Schroeder FC. et al. Human GAPDH Is a Target of Aspirin's Primary Metabolite Salicylic Acid and Its Derivatives. PLoS One. 2015 doi:10.1371/journal.pone.0143447
41. El Kadmiri N, El Khachibi M, Slassi I, El Moutawakil B, Nadifi S, Soukri A. Assessment of GAPDH expression by quantitative real time PCR in blood of Moroccan AD cases. J Clin Neurosci. 2017;40:24-26
42. Lleo A, Galea E, Sastre M. Molecular targets of non-steroidal anti-inflammatory drugs in neurodegenerative diseases. Cell Mol Life Sci. 2007;64:1403-1408
43. Wahli W, Michalik L. PPARs at the crossroads of lipid signaling and inflammation. Trends Endocrinol Metab. 2012;23:351-363
44. Iglesias J, Morales L, Barreto GE. Metabolic and Inflammatory Adaptation of Reactive Astrocytes: Role of PPARs. Mol Neurobiol. 2017;54:2518-2538
45. Li AC, Glass CK. PPAR- and LXR-dependent pathways controlling lipid metabolism and the development of atherosclerosis. J Lipid Res. 2004;45:2161-2173
46. Sastre M, Klockgether T, Heneka MT. Contribution of inflammatory processes to Alzheimer's disease: molecular mechanisms. Int J Dev Neurosci. 2006;24:167-176
47. Lehmann JM, Lenhard JM, Oliver BB, Ringold GM, Kliewer SA. Peroxisome proliferator-activated receptors alpha and gamma are activated by indomethacin and other non-steroidal anti-inflammatory drugs. J Biol Chem. 1997;272:3406-3410
48. Sastre M, Dewachter I, Landreth GE, Willson TM, Klockgether T, van Leuven F. et al. Nonsteroidal anti-inflammatory drugs and peroxisome proliferator-activated receptor-gamma agonists modulate immunostimulated processing of amyloid precursor protein through regulation of beta-secretase. J Neurosci. 2003;23:9796-97804
49. Dairam A, Chetty P, Daya S. Non-steroidal anti-inflammatory agents, tolmetin and sulindac, attenuate oxidative stress in rat brain homogenate and reduce quinolinic acid-induced neurodegeneration in rat hippocampal neurons. Metab Brain Dis. 2006;21:221-233
50. Wu Y, Zhai H, Wang Y, Li L, Wu J, Wang F. et al. Aspirin-triggered lipoxin A₄ attenuates lipopolysaccharide-induced intracellular ROS in BV2 microglia cells by inhibiting the function of NADPH oxidase. Neurochem Res. 2012;37:1690-1696
51. Wang F, Zhai Z, Huang L, Li H, Xu Y, Qiao X. et al. Aspirin protects dopaminergic neurons against lipopolysaccharide-induced neurotoxicity in primary midbrain cultures. J Mol Neurosci. 2012;46:153-161
52. Liu F, Yang H, Li G, Zou K, Chen Y. Effect of a small dose of aspirin on quantitative test of 24-h urinary protein in patients with hypertension in pregnancy. Exp Ther Med. 2017;13:37-40
53. Rosenblum WI, Nishimura H, Nelson GH. L-NMMA in brain microcirculation of mice is inhibited by blockade of cyclooxygenase and by superoxide dismutase. Am J Physiol. 1992;262:H1343-1349
54. Grosser N, Schröder H. Aspirin protects endothelial cells from oxidant damage via the nitric oxide-cGMP pathway. Arterioscler Thromb Vasc Biol. 2003;23:1345-1351
55. Farrugia G, Bannister WH, Vassallo N, Balzan R. Aspirin-induced apoptosis of yeast cells is associated with mitochondrial superoxide radical accumulation and NAD(P)H oxidation. FEMS Yeast Res. 2013;13:755-768
56. Qiu LY, Yu J, Zhou Y, Chen CH. Protective effects and mechanism of action of aspirin on focal cerebral ischemia-reperfusion in rats. Yao Xue Xue Bao. 2003;38:561-564
Author contact
![]() Corresponding author: Dr. Soraya L. Valles. Department of Physiology. School of Medicine, University of Valencia. email: lilian.valleses Phone: 0034-963983813.
Corresponding author: Dr. Soraya L. Valles. Department of Physiology. School of Medicine, University of Valencia. email: lilian.valleses Phone: 0034-963983813.