Impact Factor ISSN: 1449-1907
- Issue 9; 2026
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Volume 23; 2026
- Past Issues
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Conclusions
Abbreviations
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2020; 17(4):525-535. doi:10.7150/ijms.39745 This issue Cite
Research Paper
Therapeutic Hypothermia Protects Against Heat Stroke-Induced Arterial Hypotension via Promoting Left Ventricular Performance in Rats
Wen-Ching Ko1,2, Cheng-Hsien Lin2, Jie-Jen Lee2, Ching-Ping Chang3 ![]() , Chien-Ming Chao4,5
, Chien-Ming Chao4,5 ![]()
1. Department of Surgery, Mackay Memorial Hospital, Taipei, Taiwan
2. Department of Medicine, Mackay Medical College, New Taipei, Taiwan
3. Department of Medical Research, Chi Mei Medical Center, Tainan, Taiwan
4. Department of Intensive Care Medicine, Chi Mei Medical Center, Liouying, Tainan 73657, Taiwan
5. Department of Nursing, Min-Hwei College of Health Care Management, Tainan, Taiwan
Received 2019-8-29; Accepted 2020-1-8; Published 2020-2-10
Abstract
We aimed to ascertain whether therapeutic hypothermia (TH) acts as cardioprotective management for heat stroke (HS). Adult male rats under general anesthesia were exposed to whole-body heating (43°C for 70 min) to induce HS. Rats with HS displayed hyperthermia (core body temperature 42°C vs. 36°C); hypotension (30 mmHg vs. 90 mmHg mean arterial blood pressure); suppressed left ventricular (LV) performance (stroke volume 52 μl/min vs. 125 μl/min), ejection fraction (0.29% vs. 0.69%), relaxation factor (72 ms vs. 12 ms), and arterial elastance (0.31 mmHg/ μl vs. 10 mmHg/ μl); increased myocardial injury markers (e.g., creatine kinase-MB: 86 U/L vs. 24 U/L, cardiac troponin I: 3.08 ng/ml vs. 0.57 ng/ml); increased myocardial oxidative stress markers (e.g., malondialdehyde: 6.52 nmol/mg vs. 1.06 nmol/mg, thiobarbituric acid-reactive substances: 29 nmol/g vs. 2 nmol/g); decreased myocardial antioxidants (e.g., superoxide dismutase: 6 unit/mg vs. 17 unit/mg, reduced glutathione: 0.64 nmol/mg vs. 2.53 nmol/mg); increased myocardial proinflammatory cytokines (e.g., tumor necrosis factor-α 3200 pg/ml vs. 1000 pg/ml, interleukin-6: 668 pg/ml vs. 102 pg/ml); and increased cardiac damage scores (2.2 vs. 0.3). TH therapy significantly reversed the following conditions: HS-induced hyperthermia (37.5°C core body temperature), hypotension (71 mmHg), suppressed LV performance (stroke volume: 97 μl/min, ejection fraction: 0.65%, relaxation factor: 39 ms, and arterial elastance: 0.99 mmHg/μl), increased myocardial injury markers (e.g., creatine kinase-MB: 37 U/L, cardiac troponin I: 1.06 ng/ml), increased myocardial oxidative stress markers (e.g., malondialdehyde: 2.68 nmol/mg, thiobarbituric acid-reactive substances: 12.3 nmol/g), decreased myocardial antioxidants (e.g., superoxide dismutase: 13.3 unit/mg, reduced glutathione: 2.71 mmol/mg), increased myocardial proinflammatory cytokines (e.g., tumor necrosis factor-α 1500 pg/ml, interleukin-6: 108 ng/ml); and increased cardiac damage scores (0.9). We thus conclude that TH protects against HS-induced arterial hypotension by promoting LV performance in rats. These results add to the literature regarding the use of TH as cardioprotective management for HS.
Keywords: myocardial injury, therapeutic hypothermia, heatstroke, cardiac mechanical efficiency, arterial elastance
Introduction
Temperature elevation (either spontaneous/ infection-related or induced by external warming) can exacerbate all types of neurologic injury (cardiac arrest, ischemic stroke, traumatic brain injury, and hemorrhagic stroke) [1]. Therapeutic hypothermia (TH) is an effective therapy for comatose patients, improving cardiac and neurologic outcomes after the return of spontaneous circulation following cardiac arrest [2-8]. After the return of spontaneous circulation following cardiac arrest, all comatose adult patients should receive target temperature management with a target temperature between 32°C and 36°C [9]. Patients with heat stroke (HS) present electrocardiographic abnormalities, including a predominant “S” wave and a depressed “ST” segment [10]. HS rats also display subcellular alterations (e.g., mitochondrial hypertrophy and intracellular edema) in their myocardial tissue [11]. In vitro studies have also demonstrated that heat stress causes time- and dose-dependent apoptosis, necrosis, and autophagy in rat cardiomyocytes [12]. TH is the current therapy of choice for HS because no pharmacologic agent is currently available [13-15]. Although the true mechanisms underlying the beneficial effect of TH on HS reactions remain unclear, evidence has accumulated to indicate that TH may improve outcomes of HS via reducing cardiac dysfunction [16, 17]. Indeed, our previous findings showed that after the onset of HS, animals displayed hypotension and altered cardiac protein profiles, which could be reversed by TH. This raises the possibility that cardiac dysfunction contributes to HS genesis, which can be ameliorated by TH [18].
In this study, to address this question, we used a rat model of HS [19] that exhibits hyperthermia, arterial hypotension, suppressed left ventricular (LV) performance and cardiac inflammatory and oxidative damage. We tested the hypothesis that TH protects against HS-induced arterial hypotension via promoting LV performance by pressure-volume loop analyses in HS rats. Based on the instructions provided by previous studies, our HS animals received target temperature management (32-36°C).
Materials and Methods
Ethics Statement
This study strictly adhered to the recommendations in the Guide for the Care and Use of Laboratory Animals of the Ministry of Science and Technology (MOST) of the Republic of China. The protocol was approved by the Chi Mei Medical Center Institutional Review Board for Animal Care and Use (Assurance Number: 106121110). All effects were made to minimize the suffering of the experimental rats.
Animals and Surgery
Sixty adult male Sprague-Dawley rats (250±12 g) obtained from BioLASCO Taiwan Co. and housed at an ambient temperature of 22±1°C with a 12-h light-dark cycle. Pellet rat chow and tap water were available ad libitum. Rats were anesthetized with an intraperitoneal (i.p.) dose of pentobarbital sodium (60 mg/kg). The femoral artery of anesthetized rats was cannulated with polyethylene tubing (PE-60) for measurement of mean arterial blood pressure (MABP) and HR. Both MABP and HR were monitored continuously using a pressure transducer (ADInstruments Inc., Springs, CO, USA) and a chart recorder (ADInstruments Inc.).
HS Induction
HS was induced by exposing anesthetized rats in a folded heating pad at 43°C controlled by 43°C water-circulating pad for 70 min as detailed previously [20]. Then, the heated rats were allowed to recover at room temperature of 26°C. Twenty minutes following the termination of the 70-min heat stress, all the heated rats displayed both excessive hyperthermia (~42.0°C) and arterial hypotension (~30mm Hg), indicating the occurrence of HS [21] (please see Figure 2).
TH
Before the start of the thermal experiments, the body core temperature of all anesthetized animals was maintained at 36.2°C using a water-circulating folded heating pad. Immediately after the termination of heat stress, all heated rats were subjected to TH (16°C water-circulating folded cooling pad for 30 min). Their body core temperatures were decreased to ~38°C and then maintained at 36°C by a folded heating pad at 26°C in a room at 26°C.
Experimental Groups and Experimental Procedures
The animals were assigned to one of the following 3 groups: the NC group (n=10): rats with body core temperature maintained at 36°C in a room at 26°C throughout the experiment; the HS+non-TH group (n=10): rats treated with HS (43°C for 70 min) followed by non-TH (26°C for 30 min); and the HS+TH group (n=10): rats treated with HS (43°C for 70 min) followed by TH (16°C for 30 min). The survival rate was obtained for each group. All 3 groups of rats were subjected to pressure-volume (P-V) loop analyses.
P-V Loop Analyses
A 1.2 F catheter-tip pressure transducer (Scisense, London, Ontario, Canada) was inserted into the right carotid artery to measure the baseline arterial pressure and then fed retrograde into the left ventricle to record baseline hemodynamics in the closed chest with the ADVantageTM (Scisense, London, Ontario, Canada). The signals of P and V were visually checked for quality and recorded for later analysis [22, 23]. The following hemodynamic parameters were calculated: HR, heart rate (beats/min); ESP, LV end-systolic pressure (mmHg); EDP, LV end-diastolic pressure (mmHg); SV, stroke volume (μl); ESV, LV end-systolic volume (μl); EDV, LV end-diastolic volume (μl); CO, cardiac output (μl/min); EF, ejection fraction (%); maximal dp/dt, (mmHg/s); Pmax, maximum LV pressure (mmHg); Vmax, maximum LV volume (μl/s); SW, stroke work (mmHg/ml); minimum dp/dt (mm Hg/s); Pmin, minimum LV pressure (mmHg); Tau (γ) Glantz time constant of ventricular relaxation (ms).
H&E Staining
At twenty minutes after the onset of HS, both the blood and heart tissue were collected from all groups of rats under anesthesia. The blood samples were centrifuged to obtain the serum for further biochemical assays. The heart tissue was immersed in 10% neutral formalin, dehydrated through graded alcohols and embedded in paraffin wax. Ten-micrometer thick sections were cut, stained with H&E and examined under a light microscope (Carl Zeiss GmbH, Göttingen, Germany). Myocardial damage was scored using published morphologic criteria [24]: 0, no damage; 1 (mild), interstitial edema and localized necrosis; 2 (moderate), widespread myocardial cell swelling and necrosis; 3 (severe), necrosis with contraction bands and compressed capillaries; or 4 (highly severe), diffuse necrosis with contraction bands, compressed capillaries, and hemorrhage. Histological sections were evaluated in a blinded manner by two examiners. The total cardiac damage score for each heart was calculated as an average of all the component injury scores.
Estimation of Myocardial Injury Markers
Myocardial injury was assessed in rats after 70 min of heat stress plus 20 min of room temperature recovery via estimating specific biomarkers, cardiac troponin I, lactate dehydrogenase, and creatine kinase-MB isoenzyme in serum samples using commercially available standard kits. All measurements were performed according to the manufacturer's instructions.
Estimation of Oxidative Stress and Antioxidant Defense System
The supernatant obtained from the 10% homogenate of heart tissue was used for the estimation of the end product of lipid peroxidation (malondialdehyde, MDA) reacted with thiobarbituric acid in acidic conditions and formed a pink colored chromophore measured at 532 nm using a microplate reader [25]. Thiobarbituric acid-reactive substances (TBARS) were expressed as nM/g tissue. Superoxide dismutase (SOD) activity in the heart tissue was measured using the SOD assay kit [26]. We used the method of Sinha [27] to measure the activity of catalase in the heart tissue. Additionally, 5,5'-dithiobis (2-nitrobenzoic acid) (DTNB) was used to estimate the levels of reduced glutathione (GSH) in the heart tissue [28].
Estimation of Two Pro-inflammatory Cytokines (TNF-α and IL-6) and One Anti-inflammatory Cytokine (IL-10)
We estimated the levels of two pro-inflammatory cytokines (TNF-α and IL-6) and one anti-inflammatory cytokine (IL-10) in the heart tissue homogenate of control and experimental groups by specific ELISA kits, according to the manufacturers' instructions.
Statistical Analysis
The results for multiple independent experiments are expressed as the mean±standard deviation (SD). Survival rates were compared using Kaplan-Meier analysis followed by the log-rank test. One-way analysis of variance followed by the Student-Newman-Keuls post hoc test was performed to analyze differences between multiple groups. P<0.05 was considered significant.
Results
Prolonging the Survival Rate and Reducing Excessive Hyperthermia and Arterial Hypotension in HS Rats by TH
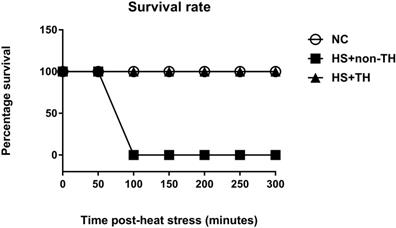
The potential reduction in the lethality of HS in rats was first analyzed by determining their survival rates. Heat stress (43°C for 70 min) significantly reduced the percent survival from 100% in the normothermic control (NC) group rats to 0% in the HS rats without TH (HS+non-TH) (P<0.001; Figure 1). In contrast, the HS+TH group rats exhibited a significant (P<0.001; Figure 2) increase in the percent survival (100%). In addition, the occurrence of excessive hyperthermia (~42°C vs. ~38°C colonic temperature), as well as arterial hypotension (~30 mmHg vs. 60 mmHg) at 20 min following the termination of HS (or 90 min after heat-stroke onset), was significantly reduced by TH, as demonstrated in the HS+TH group rats (P<0.01; Figure 2).
Kaplan-Meier analysis followed by log-rank tests were performed to determine the percent survival in NC rats (⚪), in heated rats without TH following HS (HS+non-TH) (◼) and in heated rats with TH (HS+TH) (▲). Data are expressed as the means±SD of 10 rats per group. *P<0.001, HS+non-TH vs. NC; +P<0.001, HS+TH vs HS+non-TH.
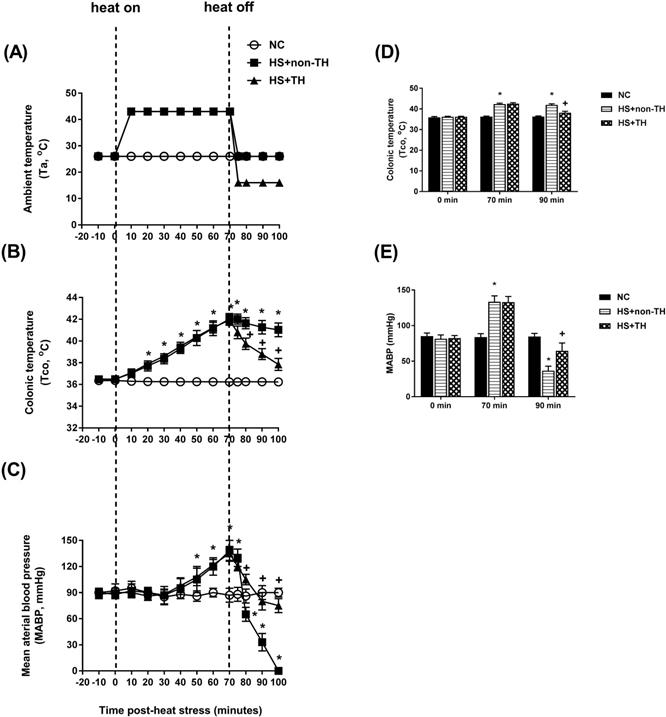
Time course of the change in ambient temperature (A, Ta), colonic temperature (B, Tco) and mean arterial blood pressure (C, MABP) in NC rats (⚪), in rats without TH following HS (HS+non-TH) (◼), and in rats receiving TH following HS (HS+TH) (▲). The Tco (D) and MABP (E) were obtained 0, 70, and 90 min after the initiation of heat exposure (ambient temperature in non-heated controls ) in heat stroke rats. All heated groups were exposed to heat (43 oC) for exactly 70 min and were then allowed to recover at room temperature (26 oC). Data are expressed as the means±SD of 10 rats per group. *P<0.01, HS+non-TH vs. NC; +P<0.05, HS+TH vs. HS+non-TH.
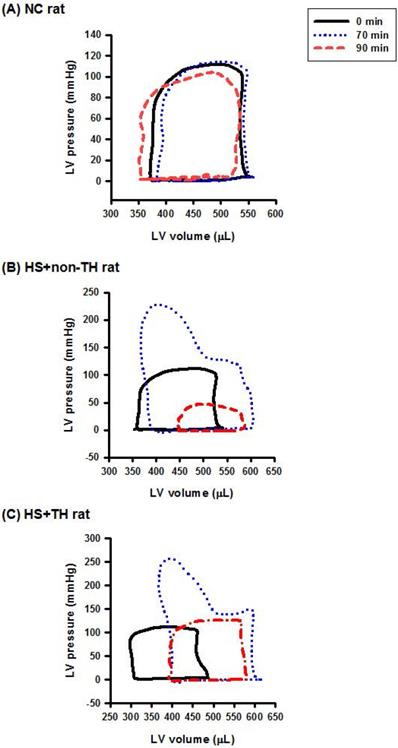
TH-induced Reversion of Suppressed LV Performance in HS Rats
As shown in both Figure 3 and Table 1, compared with the NC group rats, the HS+non-TH groups had significantly lower values of heart rate (HR) (53±4 beats/min vs. 349±5 beats/min), end-systolic pressure (ESP) (15±1 mmHg vs. 119±3 mmHg), CO (2982±22 μl/min vs. 44594±37 μl/min), EF (0.29±0.02% vs. 0.69±0.04%) and arterial elastance (Ea) (0.31±0.02 mmHg/μl vs. 10.1±0.3 mmHg/μl) but had significantly higher values of relaxation factor (γ) (72±1 ms vs. 12±0 ms). Notably, the suppressed left ventricle hemodynamics in the HS+non-TH group rats were significantly reversed by TH therapy in the HS+TH group rats: HR (512 ±12 beats/min), ESP (71±4 beats/min), CO (45740±35 μl/min), EF (0.65±0.03%), Ea (0.99±0.02 mmHg/μl), and γ (39±1 ms) (Table 1 and Figure 3).
Hemodynamic parameters and indexes of systolic and diastolic function derived from left pressure-volume relationships in NC, HS rats without TH (HS+non-TH), and HS rats with TH (HS+TH).
| Group of rats | NC rats | HS+non-TH | HS+TH | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Time points | 0 min | 90 min | 0 min | 90 min | 0 min | 90 min | |||
| Parameters | |||||||||
| HR, beats/min | 344±6 | 349±5 | 351±7 | 53±4* | 347±6 | 512±12+ | |||
| ESP, mmHg | 117±4 | 119±3 | 119±5 | 15±1 | 118±2 | 71±4+ | |||
| EDP, mmHg | 11±1 | 10±1 | 12±1 | 5±1* | 10±1 | 8±1+ | |||
| SV, μl | 122±5 | 125±4 | 124±3 | 52±4* | 123±2 | 97±5+ | |||
| ESV, μl | 298±6 | 303±5 | 302±4 | 418±9* | 299±5 | 305±6+ | |||
| EDV, μl | 409±5 | 416±7 | 401±6 | 459±5* | 404±5 | 402±6+ | |||
| CO, μl/min | 44,662±32 | 44,594±37 | 44,686±39 | 2,982±22* | 44,573± | 45,740±35+ | |||
| Systolic indexes | |||||||||
| EF, % | 0.65±0.03 | 0.69±0.04 | 0.66±0.04 | 0.29±0.02* | 0.64±0.04 | 0.65±0.03+ | |||
| Maximal dp/dt, mmHg/s | 9,982±8 | 9,990±12 | 9,988±18 | 1,492±37* | 9,991±12 | 6,176±98+ | |||
| Pmax, mmHg | 144±5 | 139±4 | 139±6 | 37±3* | 137±5 | 93±5+ | |||
| Vmax, μl/s | 408±7 | 416±7 | 414±6 | 514±7* | 409±6 | 428±5+ | |||
| SW, mmHg/μl | 13,395±81 | 13,406±83 | 13,416±74 | 1,258±64* | 13,395±77 | 9,121±107+ | |||
| Diastolic indexes | |||||||||
| Minimum do/dt, mmHg | -5,600±18 | -5,692±25 | -5,688±22 | -159±15* | -5,697±33 | -4,416±125+ | |||
| Pmin, mmHg | 293±6 | 288±9 | 301±8 | 404±9* | 288±7 | 313±7+ | |||
| Relaxation factor (γ) | |||||||||
| ms | 12±0 | 12±0 | 12±0 | 72±1* | 12±0 | 39±1+ | |||
| Aortic vascular index | |||||||||
| Ea, mmHg/μl | 0.99±0.02 | 10.1±0.3 | 0.98±0.02 | 0.31±0.02* | 0.97±0.02 | 0.99±0.02+ | |||
Values are expressed as means±SEM; n=10 for each group. HR, heart rate; ESP, end-systolic pressure; EDP, end-diastolic pressure; SV, stroke volume; ESV, LV end-systolic pressure; SV, stroke volume; ESV, LV end-systolic volume; Pmax, maximum LV pressure; Vmax, maximum dv/dt; Pmin, minimum LV pressure; γ, relaxation factor (Tau Glantz or time constant of ventricular relaxation); Ea, arterial elastance; and Vmin, minimum dp/dt. *P<0.05 vs NC. +P<0.05 vs HS+non-TH.
Representative LV pressure-volume loops obtained from the steady-state of (A) an NC rat, (B) an HS+non-TH rat, and (C) an HS+TH rat at time “0 min” (―), time “70 min” (…), and time “90 min” (-·-·) after onset of heat stress. As depicted in (B), the HS+non-TH rat displayed a characteristic right shift and declined in the amplitude of the pressure-volume signal in the pressure-volume loops at the time “90 min”. The abnormal amplitude of the pressure-volume signal in the pressure-volume loop at the time “90 min” (or the time point for the onset of HS) was significantly reversed by the TH, as demonstrated in HS+TH rats. The definitions of the group abbreviations are provided in the legend of Figure 1.
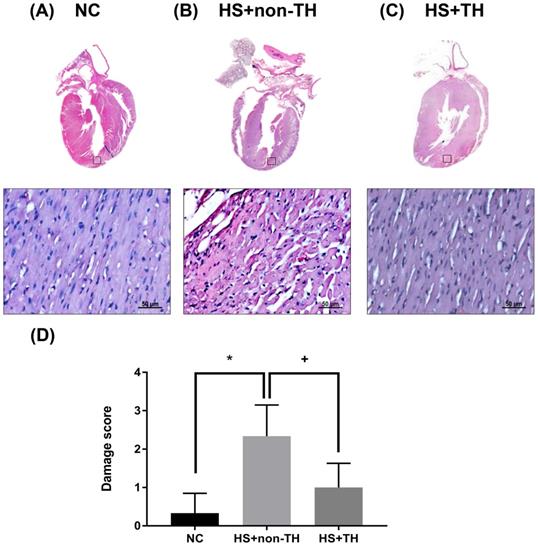
TH Attenuates HS-induced Pathological Changes in the Myocardial Tissues
The cardiac histology of the NC control rats revealed a normal appearance showing normal and centrally arranged nuclei, the connective tissue also appeared normal, and the cardiac muscle fibers were well arranged. When compared with the NC group (Figure 4 A), the HS+non-TH group of rats revealed significant pathological changes in the myocardial tissues (Figure 4 B). The changes observed in the HS+non-TH group included cellular edema or atrophy, inflammatory cell infiltration, coagulative necrosis, and liquefactive necrosis (Figure 4 B). In contrast, the myocardial tissue from the HS+TH group exhibited less cellular edema, mild leukocytic infiltration, and muscle necrosis (Figure 4 C). Semi-quantitative assessment of the histological lesions also confirmed that the HS+TH group had a significantly higher cardiac damage score than did the HS+non-TH group (Figure 4 D).
Histological analysis of myocardial tissues. Representative hematoxylin and eosin (H&E) staining results for myocardial sections in rats of the (A) NC, (B) HS+non-TH and (C) HS+TH groups. (D) Semiquantitative analysis of H&E staining in the rats (n=6 per group). *P<0.05, compared with NC, +P<0.05, compared with HS+non-TH. At twenty min after HS onset, gross observation, and 400x magnification with H&E staining revealed apparent differences in the degree of injury between the different experimental groups. Scale bars: 50 µm.
Reversing the Increased Concentrations of Myocardial Injury Markers in HS Rats by TH Therapy
Compared with the NC group rats, the HS+non-TH group rats had significantly increased serum levels of creatine kinase-MB (86±9 U/L vs. 24±4 U/L), lactate dehydrogenase (155±12 U/L vs. 51±8 U/L), and cardiac troponin I (3.08±0.72 ng/ml vs. 0.57±0.08 ng/ml) (Table 2). However, compared with the HS+non-TH group rats, the HS+TH group rats had significantly decreased serum levels of creatine kinase-MB (37±6 U/L), lactate dehydrogenase (76±9 U/L) and cardiac troponin I (1.06±0.11 ng/ml) (P<0.05; Table 2).
Levels of myocardial injury markers, myocardial antioxidant and oxidative stress markers, and myocardial inflammatory cytokines in NC, HS rats without TH (HS+non-TH), and HS rats with TH (HS+TH).
| Groups of rats | (NC) | (HS+non-TH) | (HS+TH) | |
|---|---|---|---|---|
| Parameters | ||||
| Myocardial injury markers | ||||
| Creatine kinase-MB (U/L) | 24±4 | 86±9* | 37±6+ | |
| Lactate dehydrogenase (U/L) | 51±8 | 155±12* | 76±9+ | |
| Cardiac troponin I (ng/ml) | 0.57±0.08 | 3.08±0.72* | 1.06±0.11+ | |
| Myocardial antioxidant and markers oxidative stress markers | ||||
| MDA (nmol/mg) | 1.06±0.19 | 6.52±1.02* | 2.68±0.63+ | |
| TBARS (nmol/g) | 2±1 | 29±4* | 12±3+ | |
| SOD (unit/mg) | 17±3 | 6±2* | 13±3+ | |
| Catalase (nM of H2O2- consumption/min/mg) | 9±2 | 5±2* | 10±3+ | |
| GSH (mmol/mg) | 2.53±0.24 | 0.64±0.32* | 2.71±0.26+ | |
| Inflammatory cytokines | ||||
| TNF-α (pg/ml) | 1000±380 | 3200±310* | 1500±320+ | |
| IL-6 (pg/ml) | 102±24 | 668±85* | 108±35+ | |
| IL-10 (pg/ml) | 3008±660 | 992±227* | 2955±441+ | |
Values are expressed as the means±SD; n=10 for each group. MDA, malondialdehyde; TBARS, thiobarbituric acid-reactive substances; SOD, superoxide dismutase; TNF-α, tumor necrosis factor-alpha; IL-6, interleukin-6; and IL-10, interleukin-10. *P<0.05 vs NC. +P<0.05 vs HS+TH.
Reversing the Increased Levels of Cardiac Oxidative Stress in HS Rats by TH
Compared with the NC group rats, rats in the HS+non-TH group had significantly increased cardiac levels of MDA (6.52±1.02 nmol/mg vs. 1.06±0.19 nmol/mg) and TBARS (29±4 nmol/g vs. 2±1 nmol/g) (Table 2; P<0.05). In contrast, compared with the NC group rats, the HS+non-TH group rats had significantly decreased cardiac levels of SOD (6±2 units/mg vs. 17±3 units/mg), catalase (5±2 nM H2O2 consumption/min/mg protein vs. 9±2 nM H2O2 consumption/min/mg protein) and GSH (0.64±0.32 mmol/mg protein vs. 2.53±0.24 mmol/mg protein) (Table 2, P<0.05). TH significantly reversed both the increased cardiac levels of both MDA (2.68±0.63 nmol/ng) and TBAS (12±3 nmol/g) and the decreased cardiac levels of SOD (13±3 unit/mg), catalase (10±3 nM), and GSH (2.71±0.26 mmol/mg) (Table 2).
Reversing the Increased Cardiac Levels of Inflammatory Status in Heatstroke Rats by TH
Compared with the NC group rats, the HS+non-TH group rats had a significant increase in the cardiac levels of both TNF-α (3200±310 pg/ml vs. 1000±380 pg/ml) and IL-6 (668±85 pg/ml vs. 102±24 pg/ml) as well as a significant decrease in the cardiac levels of the anti-inflammatory cytokine IL-10 (992±227 pg/ml vs. 3008±660 pg/ml) (Table 2). However, TH significantly reversed these alterations induced by HS (TNF-α, 1500±320 pg/ml vs. 3200±310 pg/ml; IL-6, 108±35 pg/ml vs. 668±85 pg/ml; IL-10, 2955±441 pg/ml vs. 992±227 pg/ml) (Table 2).
Discussion
In the present study, we subjected anesthetized rats to severe heat stress (43°C for 70 min) to induce HS [19]. The occurrence of body core temperature elevation (above 42°C) and decreased mean arterial pressure (below 35 mmHg) was taken as the time point of HS onset. At this time point, our heated rats displayed excessive hyperthermia, arterial hypotension, decreased stroke volume, decreased cardiac output, decreased ejection fraction, decreased stroke work, increased relaxation factor, and decreased arterial elastance. Additionally, HS rats had an increase in the serum levels of several cardiac damage markers, including cardiac troponin I, lactate dehydrogenase, and creatinine kinase-MB. The heated rats also had increased cardiac levels of pro-inflammatory cytokines such as TNF-α and IL-6 and decreased cardiac levels of the anti-inflammatory cytokine IL-10. Furthermore, HS rats displayed increased cardiac extents of lipid peroxide formation (revealed by increased cardiac levels of both MDA and TBARs [29] and decreased cardiac levels of SOD, catalase, and GSH (reflecting the decreased capability of antioxidant enzymes) [30-32]. Notably, decreasing the body core temperature from ~42.5°C to ~38°C by TH significantly reversed the hypotension and reduced the myocardial inflammatory and oxidative injury in HS rats.
TH (32 oC) reduced the inflammatory response following ischemia/reperfusion injury in rat hearts [33]. TH decreased the inflammatory cytokines in the risk zone of the heart, which included the IL-6 and TNF-α, inducible nitric oxide synthase, and others. Additionally, TH reduced levels of many inflammatory mediators including radical oxygen species (ROS) and pro-inflammatory cytokines (such as IL-1β, IL-6, and TNF-α) [34]. In our present study, we choose to measure two pro-inflammatory cytokines (IL-6 and TNF-α) as well as one anti-inflammatory cytokine (IL-10) for the elucidation of inflammatory status. At the same time, we choose to measure cardiac contents of MDA and TBARS for the elucidation of lipid peroxidation formation and cardiac contents of SOD, catalase, and GSH for the elucidation of the decreased capability of anti-oxidant enzymes.
As mentioned in the Introduction section, in patients who have been successfully resuscitated after cardiac arrest due to ventricular fibrillation, TH causes both a favorable neurologic outcome and reduced mortality [35]. Epidemiological studies of HS have also recommended that the goal of clinical therapies be to normalize the body (core and skin) temperature and the function of multiple vital organs as rapidly as possible [13-15]. Indeed, our present study confirmed that TH significantly attenuated myocardial injury, multiple organ damage, and lethality in heated rats. TH adopted immediately after HS onset significantly increased the percent survival from 0% to 100% in heated rats. Maintaining cardiac function by TH is vital for preserving adequate perfusion of vital organs and for decreasing multiple organ damage, especially brain damage, during HS. The goal of clinical therapy for heat stroke is to normalize body temperature, but approximately 30% of HS survivors experience disabilities as well as neurological dysfunction [13-15]. Multiple organ dysfunction syndromes continue to manifest in HS patients after TH [13-15]. Numerous preclinical studies of TH have suggested optimal cooling conditions, such as depth duration, and a temporal therapeutic window for effective protection [36]. To administer such TH in HS, more investigation is still needed to understand the optional clinical setting better. For example, the induction of TH of 32-34 oC at the onset of ischemia provides effective cardioprotection in male rodent models of acute myocardial infarction [37]. In the present study, we choose TH of 32 oC, without TH of a lower temperature for the treatments in male rats. This is a limitation of the present study. Additionally, this might be another limitation of the present study that we did not use female rats for the treatments.
Our present results are consistent with many previous findings. For example, in murine hemorrhagic shock, TH can modulate and release cardiac pro-inflammatory cytokines [38]. In a rat model with controlled hemorrhagic shock, TH can better preserve systolic and diastolic functions [39] and can attenuate myocardial apoptosis [20, 40]. Hypothermia treatment preserves mitochondrial integrity and viability of cardiomyocytes after ischemia-reperfusion injury [41]. TH treatment can ameliorate cardiac dysfunction and help preserve both mitochondrial integrity and electron transport activity for postcardiac arrest myocardial dysfunction [41]. Long noncoding RNA upregulated in hypothermia-treated cardiomyocytes protects against myocardial infarction by improving mitochondrial function [42]. Although percutaneous coronary intervention (PCI) reduces infarct size, PCI-mediated tissue reperfusion of ischemic tissue causes irreversible myocardial damage [43]. Therefore, new therapies for acute myocardial infarction should consider the reduction of both ischemia- and reperfusion-mediated tissue injuries [44, 45]. During acute myocardial ischemia, pharmacological compounds are unable to reach the ischemic tissues due to limited blood flow. Combination therapy with TH could provide a new strategy for early intervention. Indeed, in this study, we demonstrated that TH is cardioprotective during myocardial ischemia (due to hypotension) right at the onset of HS.
The induction of mild hypothermia of 32-34 oC at the onset of ischemia provides effective cardioprotection in an experimental model of acute myocardial infarction [46]. In contrast, induction of cooling at reperfusion alone does not reduce infarct size both in the experimental [46] and clinical settings [47]. In the present study, immediately after the termination of heat stress (or 70 min after the start of heat stress), all heated rats were subjected to TH (16 oC water-circulating folded cooling pad for 30 min)(Figure 2). In heated rats, the values of MABP started to fall at 70 min and downed to a value of ~30 mmHg from the control levels of ~96 mmHg at 90 min. Therefore, TH was performed in the present study at a suitable time for the treatment but not to be late.
The injured brain stimulates innate immune molecule production and these induced molecules, including radical oxygen species (ROS), protease, and pro-inflammatory cytokines (such as IL-1β, IL-6, and TNF-α) can activate more inflammatory cells, leading to a vicious cycle of death and inflammatory activation [48]. Indeed, TH lower numbers of infiltration neutrophils and activated macrophages in the ischemia and reduces levels of many inflammatory mediators including ROS [49], pro-inflammatory cytokines (such as IL-1β, IL-6, and TNF-α) [50] and others. In our present study, TH may offer the potential to interrupt the ischemic cascade (e.g., myocardial inflammation and oxidative stress), reduce myocardial injury, and improve functional independence. In the acute stage of ischemic cascade, the reduction in blood flow results in anaerobic metabolism and decreased molecular energy production. This, in turn, causes increased sodium influx and potassium efflux, disruption of ionic homeostasis and excitatory glutamate release, cellular edema, and secondary inflammation [34]. Uninhibited glutamate release leads to mitochondrial dysfunction, free radical formation, and expansion of the infarct. Hypothermia only induced in ischemia can improve cardiomyocyte contractility and mitochondrial respiratory function in the model of cultured cardiomyocyte [51]. Additionally, TH preserves mitochondrial integrity and viability of cardiomyocytes after ischemia-reperfusion injury [52]. Putting these observations together, it appears that TH can alleviate the deteriorations of oxidative stress and inflammation via preserving mitochondrial integrity and viability of cardiomyocytes after heat stroke in rats.
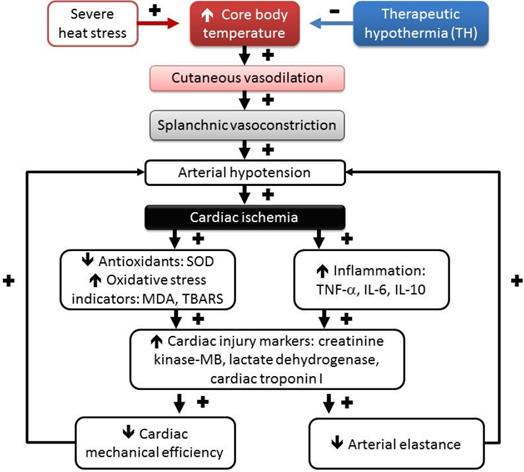
As depicted in Figure 5, environmental heat stress increases body core temperature, cutaneous blood flow, and metabolism and progressively decreases splanchnic blood flow [21], which results in arterial hypotension and myocardial ischemia. In heated rats, myocardial ischemia, inflammatory, and oxidative damage might cause cardiac cell death by apoptosis and autophagy [40, 53]. Myocardial injury caused by severe heat ed rats, the stress might cause further arterial hypotension by decreasing both cardiac mechanical efficiency and arterial elastance and result in vital organ ischemia [19]. Decreasing the whole-body temperature with TH (~32°C) significantly attenuates cardiac ischemic, inflammatory and oxidative damage, arterial hypotension (by decreasing cardiac mechanical efficiency and arterial elastance), and multiple organ ischemia and injury (Figure 5). Thus, we conclude that TH reverses the suppressed LV performance and subsequently maintains normal levels of arterial blood pressure in rats. Finally, it should be stressed that the effect of TH on the survival of HS rats may be related to its systemic, noncardiovascular effects (for instance, neurologic protection). The major limitation of the present study is that we focused mainly on cardiac protection.
Temporal associations are driving the HS reactions, including hyperthermia, hypotension, splanchnic vasoconstriction, cardiac oxidative stress, cardiac inflammation, and lethality following heat stress, which can be affected by therapeutic hypothermia (TH) treatment. (+), aggravation, and (-), mitigation. Environmental heat stress increases cutaneous blood flow and metabolism and progressively decreases splanchnic blood flow (including heart, intestine, brain, and others). Decreasing mean arterial pressure by heat stress causes myocardial ischemia. Myocardial ischemia causes myocardial oxidative stress and inflammation and results in a reduction in both cardiac mechanical efficiency and arterial elastance, causing further hypotension. SOD, superoxide dismutase; MDA, malondialdehyde; TBARS, thiobarbituric acid reactive substances; TNF-α, tumor necrosis factor-alpha; IL-6, interleukin-6; IL-10, interleukin-10.
Conclusions
In summary, severe heat stress (43°C for 70 min) caused hyperthermia (~42°C), hypotension (~30 mmHg), and suppressed LV performance (due to myocardial inflammatory and oxidative injury) in heated rats. In addition, the percent survival was decreased from a value of 100% in the normothermic controls to a new value of 0% in the heated rats without TH. However, when applied during myocardial ischemia (due to hypotension), TH significantly reversed the myocardial injury and dysfunction, preserved an adequate blood supply to vital organs and subsequently improved survival in heated rats.
Abbreviations
TH: Therapeutic hypothermia; HR: Heart rate; ESP: Left ventricular (LV) end-systolic pressure; EDP: LV end-diastolic pressure; SV: Stroke volume; ESV: LV end-systolic pressure; EDV: LV end-diastolic volume; CO: Cardiac output; EF: Ejection fraction; Pmax: Maximal LV pressure; Vmax: Maximal LV volume; SW: Stroke work; Pmin: Minimum LV pressure; Tau: Glantz time constant of ventricular relaxation; MDA: Malondialdehyde; TBARS: Thiobarbituric acid-reactive substances; SOD: Superoxide dismutase; DTNB: Dithionitrobenzoic acid; GSH: Reduced glutathione; TNF-α: Tumor necrosis factor-α; IL-1β: Interleukin-1β.
Acknowledgements
The authors appreciate Miss Mon-Tsung Ho for her excellent technical and executive assistance.
Funding
This research was funded by the Ministry of Sciences and Technology (Taiwan) under grant nos. MOST 108-2314-B-715-001 (C.H.L.) and MOST 107-2314-B-384-007-MY3 (C.P.C.), by the Chi Mei Medical Center (Taiwan) under grant no. CLFHR10825 (C.-M.C.), by the Mackay Memorial Hospital under grant no. MMH-MM-10807 (C.-H. L.) and by the Mackay Medical College under grant no. 1081A01 (C.-H. L.).
Author Contributions
Conceptualization, W.C.K. and C.H.L.; methodology, C.H.L. and C.P.C.; formal analysis, W.C.K.; investigation, C.H.L. and C.P.C.; writing, W.C.K., C.P.C. and C.M.C.; data curation, C.H.L. and W.C.K.; supervision, J. J. L. and C.P.C.; projection administration, C.M.C.; funding acquisition, C.H.L., C.P.C., and C.M.C.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Polderman KH. Cooling after cardiac arrest-the longer the better? J Thorac Dis. 2018;10:S3300-S04
2. Group THaCAS. Mild Therapeutic Hypothermia to Improve the Neurologic Outcome after Cardiac Arrest. New England Journal of Medicine. 2002;346:549-56
3. Nolan JP, Morley PT, Hoek TL. et al. Therapeutic hypothermia after cardiac arrest. An advisory statement by the Advancement Life support Task Force of the International Liaison committee on Resuscitation. Resuscitation. 2003;57:231-5
4. Donnino MW, Andersen LW, Berg KM. et al. Temperature Management After Cardiac Arrest: An Advisory Statement by the Advanced Life Support Task Force of the International Liaison Committee on Resuscitation and the American Heart Association Emergency Cardiovascular Care Committee and the Council on Cardiopulmonary, Critical Care, Perioperative and Resuscitation. Circulation. 2015;132:2448-56
5. Nolan JP, Soar J. Postresuscitation care: entering a new era. Curr Opin Crit Care. 2010;16:216-22
6. Bernard SA, Gray TW, Buist MD. et al. Treatment of Comatose Survivors of Out-of-Hospital Cardiac Arrest with Induced Hypothermia. New England Journal of Medicine. 2002;346:557-63
7. Peberdy MA, Callaway CW, Neumar RW. et al. Part 9: post-cardiac arrest care: 2010 American Heart Association Guidelines for Cardiopulmonary Resuscitation and Emergency Cardiovascular Care. Circulation. 2010;122:S768-86
8. Geocadin RG, Wijdicks E, Armstrong MJ. et al. Practice guideline summary: Reducing brain injury following cardiopulmonary resuscitation: Report of the Guideline Development, Dissemination, and Implementation Subcommittee of the American Academy of Neurology. Neurology. 2017;88:2141-49
9. Neumar RW, Otto CW, Link MS. et al. Part 8: adult advanced cardiovascular life support: 2010 American Heart Association Guidelines for Cardiopulmonary Resuscitation and Emergency Cardiovascular Care. Circulation. 2010;122:S729-67
10. Akhtar MJ, al-Nozha M, al-Harthi S. et al. Electrocardiographic abnormalities in patients with heat stroke. Chest. 1993;104:411-4
11. Lin MT, Chai CY, Sun SC. et al. Myocardial lesions produced by external heat or cold exposure in rats. Chin J Physiol. 1977;22:115-25
12. Qian L, Song X, Ren H. et al. Mitochondrial mechanism of heat stress-induced injury in rat cardiomyocyte. Cell Stress Chaperones. 2004;9:281-93
13. Argaud L, Ferry T, Le QH. et al. Short- and long-term outcomes of heatstroke following the 2003 heat wave in Lyon, France. Arch Intern Med. 2007;167:2177-83
14. Dematte JE, O'Mara K, Buescher J. et al. Near-fatal heat stroke during the 1995 heat wave in Chicago. Ann Intern Med. 1998;129:173-81
15. Wallace RF, Kriebel D, Punnett L. et al. Prior heat illness hospitalization and risk of early death. Environ Res. 2007;104:290-5
16. Chang CK, Chang CP, Chiu WT. et al. Prevention and repair of circulatory shock and cerebral ischemia/injury by various agents in experimental heatstroke. Curr Med Chem. 2006;13:3145-54
17. Chauhan NR, Kapoor M, Prabha Singh L. et al. Heat stress-induced neuroinflammation and aberration in monoamine levels in hypothalamus are associated with temperature dysregulation. Neuroscience. 2017;358:79-92
18. Cheng BC, Chang CP, Tsay YG. et al. Body cooling causes normalization of cardiac protein expression and function in a rat heatstroke model. J Proteome Res. 2008;7:4935-45
19. Hsu SF, Chao CM, Chang CP. et al. Heat shock protein 72 may improve hypotension by increasing cardiac mechanical efficiency and arterial elastance in heatstroke rats. Int J Cardiol. 2016;219:63-9
20. Li J, Beiser DG, Wang H. et al. Therapeutic hypothermia cardioprotection in murine hemorrhagic shock/resuscitation differentially affects p38alpha/p38gamma, Akt, and HspB1. J Trauma. 2011;71:1262-70
21. Chen SH, Lin MT, Chang CP. Ischemic and oxidative damage to the hypothalamus may be responsible for heat stroke. Curr Neuropharmacol. 2013;11:129-40
22. Cheng CP, Suzuki M, Ohte N. et al. Altered ventricular and myocyte response to angiotensin II in pacing-induced heart failure. Circ Res. 1996;78:880-92
23. Cheng CP, Ukai T, Onishi K. et al. The role of ANG II and endothelin-1 in exercise-induced diastolic dysfunction in heart failure. Am J Physiol Heart Circ Physiol. 2001;280:H1853-60
24. Qin-Wei Z, Yong-Guang LI. Berberine attenuates myocardial ischemia reperfusion injury by suppressing the activation of PI3K/AKT signaling. Exp Ther Med. 2016;11:978-84
25. Ohkawa H, Ohishi N, Yagi K. Assay for lipid peroxides in animal tissues by thiobarbituric acid reaction. Anal Biochem. 1979;95:351-8
26. Kumar M, Kasala ER, Bodduluru LN. et al. Baicalein protects isoproterenol induced myocardial ischemic injury in male Wistar rats by mitigating oxidative stress and inflammation. Inflamm Res. 2016;65:613-22
27. Sinha AK. Colorimetric assay of catalase. Anal Biochem. 1972;47:389-94
28. Ellman GL. Tissue sulfhydryl groups. Arch Biochem Biophys. 1959;82:70-7
29. Monje ML, Toda H, Palmer TD. Inflammatory blockade restores adult hippocampal neurogenesis. Science. 2003;302:1760-5
30. Inci S, Ozcan OE, Kilinc K. Time-level relationship for lipid peroxidation and the protective effect of alpha-tocopherol in experimental mild and severe brain injury. Neurosurgery. 1998;43:330-5 discussion 35-6
31. Imam SZ, Ali SF. Selenium, an antioxidant, attenuates methamphetamine-induced dopaminergic toxicity and peroxynitrite generation. Brain Res. 2000;855:186-91
32. Serarslan Y, Yonden Z, Ozgiray E. et al. Protective effects of tadalafil on experimental spinal cord injury in rats. J Clin Neurosci. 2010;17:349-52
33. Shi J, Dai W, Kloner RA. Therapeutic Hypothermia Reduces the Inflammatory Response Following Ischemia/Reperfusion Injury in Rat Hearts. Therapeutic hypothermia and temperature management. 2017;7:162-70
34. Kuczynski AM, Demchuk AM, Almekhlafi MA. Therapeutic hypothermia: Applications in adults with acute ischemic stroke. Brain circulation. 2019;5:43-54
35. Hypothermia after Cardiac Arrest Study Group. Mild therpeutic hypothermia to improve the neurological outcome after cardiac arrest. N Engl J Med. 2002;346:549-56
36. Kurisu K, Yenari MA. Therapeutic hypothermia for ischemic stroke; pathophysiology and future promise. Neuropharmacology. 2018;134:302-09
37. Kohlhauer M, Pell VR, Burger N. et al. Protection against cardiac ischemia-reperfusion injury by hypothermia and by inhibition of succinate accumulation and oxidation is additive. Basic research in cardiology. 2019;114:18-18
38. Beiser DG, Wang H, Li J. et al. Plasma and myocardial visfatin expression changes are associated with therapeutic hypothermia protection during murine hemorrhagic shock/resuscitation. Resuscitation. 2010;81:742-8
39. Li T, Lin X, Zhu Y. et al. Short-term, mild hypothermia can increase the beneficial effect of permissive hypotension on uncontrolled hemorrhagic shock in rats. Anesthesiology. 2012;116:1288-98
40. Lin CH, Wu WS, Lin MT. et al. Attenuating ischemia-induced H9c2 myoblasts apoptosis by therapeutic hypothermia. Am J Med Sci. 2010;339:258-65
41. Huang CH, Chiang CY, Pen RH. et al. Hypothermia treatment preserves mitochondrial integrity and viability of cardiomyocytes after ischaemic reperfusion injury. Injury. 2015;46:233-9
42. Zhang J, Yu L, Xu Y. et al. Long noncoding RNA upregulated in hypothermia treated cardiomyocytes protects against myocardial infarction through improving mitochondrial function. Int J Cardiol. 2018;266:213-17
43. Hausenloy DJ, Yellon DM. Myocardial ischemia-reperfusion injury: a neglected therapeutic target. J Clin Invest. 2013;123:92-100
44. Kohlhauer M, Pell VR, Burger N. et al. Protection against cardiac ischemia-reperfusion injury by hypothermia and by inhibition of succinate accumulation and oxidation is additive. Basic Res Cardiol. 2019;114:18
45. Heusch G. Cardioprotection research must leave its comfort zone. Eur Heart J. 2018;39:3393-95
46. Tissier R, Hamanaka K, Kuno A. et al. Total liquid ventilation provides ultra-fast cardioprotective cooling. Journal of the American College of Cardiology. 2007;49:601-05
47. Erlinge D, Götberg M, Lang I. et al. Rapid endovascular catheter core cooling combined with cold saline as an adjunct to percutaneous coronary intervention for the treatment of acute myocardial infarction. The CHILL-MI trial: a randomized controlled study of the use of central venous catheter core cooling combined with cold saline as an adjunct to percutaneous coronary intervention for the treatment of acute myocardial infarction. Journal of the American College of Cardiology. 2014;63:1857-65
48. Yenari MA, Han HS. Neuroprotective mechanisms of hypothermia in brain ischaemia. Nature reviews. Neuroscience. 2012;13:267-78
49. Perrone S, Szabó M, Bellieni CV. et al. Whole body hypothermia and oxidative stress in babies with hypoxic-ischemic brain injury. Pediatric neurology. 2010;43:236-40
50. Wang Q, Tang XN, Yenari MA. The inflammatory response in stroke. Journal of neuroimmunology. 2007;184:53-68
51. Li H, Fang X, Yang Z. et al. Ischemia hypothermia improved contractility under normothermia reperfusion in the model of cultured cardiomyocyte. In vitro cellular & developmental biology. Animal. 2012;48:284-92
52. Huang C-H, Chiang C-Y, Pen R-H. et al. Hypothermia treatment preserves mitochondrial integrity and viability of cardiomyocytes after ischaemic reperfusion injury. Injury. 2015;46:233-39
53. Cheng BC, Huang HS, Chao CM. et al. Hypothermia may attenuate ischemia/reperfusion-induced cardiomyocyte death by reducing autophagy. Int J Cardiol. 2013;168:2064-9
Author contact
![]() Corresponding authors: Ching-Ping Chang, Ph.D., Department of Medical Research, Chi Mei Medical Center, Tainan, Taiwan. Mailing address: No. 901, Zhonghua Rd., Yongkang District, Tainan City 710, Taiwan. Phone: +886-6-2812811; Fax: +886-6-2832639; Email: jessica.cpchangcom and Chien-Ming Chao, MD, Department of Intensive Care Medicine, Chi Mei Medical Center, Liouying, Tainan 73657, Taiwan. Mailing address: No.201, Taikang Taikang Vil., Liuying Dist., Tainan City 73657 Taiwan. Email: ccm870958com.tw; Phone:+886-6- 622-6999.
Corresponding authors: Ching-Ping Chang, Ph.D., Department of Medical Research, Chi Mei Medical Center, Tainan, Taiwan. Mailing address: No. 901, Zhonghua Rd., Yongkang District, Tainan City 710, Taiwan. Phone: +886-6-2812811; Fax: +886-6-2832639; Email: jessica.cpchangcom and Chien-Ming Chao, MD, Department of Intensive Care Medicine, Chi Mei Medical Center, Liouying, Tainan 73657, Taiwan. Mailing address: No.201, Taikang Taikang Vil., Liuying Dist., Tainan City 73657 Taiwan. Email: ccm870958com.tw; Phone:+886-6- 622-6999.