Impact Factor ISSN: 1449-1907
- Issue 9; 2026
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Volume 23; 2026
- Past Issues
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
1. Introduction
2. Pathophysiology of chronic...
3. Chronic respiratory failure...
4. Management and Treatment
Conclusions
Abbreviations
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2019; 16(7):967-980. doi:10.7150/ijms.32752 This issue Cite
Review
Management of Chronic Respiratory Failure in Interstitial Lung Diseases: Overview and Clinical Insights
Paola Faverio1, Federica De Giacomi1, Giulia Bonaiti1, Anna Stainer1, Luca Sardella1, Giulia Pellegrino2, Giuseppe Francesco Sferrazza Papa2, Francesco Bini3, Bruno Dino Bodini4, Mauro Carone5, Sara Annoni6, Grazia Messinesi1, Alberto Pesci1 ![]()
1. School of Medicine and Surgery, University of Milano-Bicocca, Monza, Italy; Respiratory Unit, San Gerardo Hospital, ASST di Monza, Monza, Italy
2. Casa di Cura del Policlinico, Dipartimento di Scienze Neuroriabilitative, Milan, Italy
3. UOC Pulmonology, Department of Internal Medicine, Ospedale ASST-Rhodense, Garbagnate Milanese, Italy
4. Pulmonology Unit, Ospedale Maggiore della Carità, University of Piemonte Orientale, Novara, Italy
5. UOC Pulmonology and Pulmonary Rehabilitation, Istituti Clinici Scientifici Maugeri, IRCCS di Cassano Murge (BA), Italy
6. Physical therapy and Rehabilitation Unit, San Gerardo Hospital, ASST di Monza, Monza, Italy.
Received 2019-1-2; Accepted 2019-5-5; Published 2019-6-10
Abstract
Interstitial lung diseases (ILDs) may be complicated by chronic respiratory failure (CRF), especially in the advanced stages. Aim of this narrative review is to evaluate the current evidence in management of CRF in ILDs.
Many physiological mechanisms underlie CRF in ILDs, including lung restriction, ventilation/perfusion mismatch, impaired diffusion capacity and pulmonary vascular damage. Intermittent exertional hypoxemia is often the initial sign of CRF, evolving, as ILD progresses, into continuous hypoxemia. In the majority of the cases, the development of CRF is secondary to the worsening of the underlying disease; however, associated comorbidities may also play a role. When managing CRF in ILDs, the need for pulmonary rehabilitation, the referral to lung transplant centers and palliative care should be assessed and, if necessary, promptly offered. Long-term oxygen therapy is commonly prescribed in case of resting or exertional hypoxemia with the purpose to decrease dyspnea and improve exercise tolerance. High-Flow Nasal Cannula oxygen therapy may be used as an alternative to conventional oxygen therapy for ILD patients with severe hypoxemia requiring both high flows and high oxygen concentrations. Non-Invasive Ventilation may be used in the chronic setting for palliation of end-stage ILD patients, although the evidence to support this application is very limited.
Keywords: Interstitial lung diseases, idiopathic pulmonary fibrosis, chronic respiratory failure, non-invasive ventilation, oxygen therapy
1. Introduction
Interstitial lung diseases (ILDs) are a heterogeneous group including more than 200 diseases characterized by widespread fibrotic and/or inflammatory abnormalities of the lung parenchyma.[1] Respiratory failure is a common complication both in the advanced stages or following episodes of acute worsening of ILDs and can be classified on the basis of different parameters, including time of onset (acute or chronic), severity (mild to severe), and causes (reversible or irreversible).
This review is aimed to evaluate the current evidence in determining the best management of chronic respiratory failure (CRF) in ILDs. A search of relevant medical literature in the English language was conducted in Medline/PubMed and EMBASE databases including observational and interventional studies from 1990 through August 2018. Keywords used to perform the research are reported in Table 1. Studies targeting children and editorials, narrative, and conference abstracts have been excluded. For the purpose of this review, any kind of ILDs was included in the search.
Keywords used to perform the research
| Chronic respiratory failure (OR respiratory failure OR chronic respiratory worsening) AND interstitial lung diseases (OR IPF OR NSIP OR CTD-ILD OR chronic HP); |
| Pathophysiology AND chronic respiratory failure (OR respiratory failure) AND interstitial lung diseases (OR IPF OR NSIP OR CTD-ILD OR chronic HP); |
| Comorbidities (OR COPD, emphysema, CPFE, pulmonary hypertension, pulmonary embolism, venous thromboembolic disease, congestive heart failure, lung cancer, obstructive sleep apnea syndrome) AND interstitial lung diseases (OR IPF OR NSIP OR CTD-ILD OR chronic HP); |
| Rehabilitation (OR pulmonary rehabilitation) AND interstitial lung diseases (OR IPF OR NSIP OR CTD-ILD OR chronic HP); |
| Palliative care (OR palliation) AND interstitial lung diseases (OR IPF OR NSIP OR CTD-ILD OR chronic HP); |
| Lung transplantation (OR lung transplant) AND interstitial lung diseases (OR IPF OR NSIP OR CTD-ILD OR chronic HP); |
| Long term oxygen therapy (OR oxygen therapy, oxygen supplementation) AND interstitial lung diseases (OR IPF OR NSIP OR CTD-ILD OR chronic HP); |
| High flow oxygen (OR high-flow nasal cannula) AND interstitial lung diseases (OR IPF OR NSIP OR CTD-ILD OR chronic HP): |
| Non-invasive ventilation AND interstitial lung diseases (OR IPF OR NSIP OR CTD-ILD OR chronic HP) |
2. Pathophysiology of chronic respiratory failure in interstitial lung diseases
In ILD patients an impairment in gas exchange is a common finding, reflecting an increased alveolar-arterial oxygen gradient, which depends on the alteration of the ventilation-perfusion ratio and the diffusion capacity.[2] Pulmonary function tests (PFTs) are characterized by a restrictive pattern with decreased forced vital capacity (FVC) and total lung capacity (TLC), associated with decreased diffusing lung capacity for carbon monoxide (DLCO). The abovementioned lung alterations cause an increase in respiratory rate (RR) as a compensatory mechanism, with higher than normal minute ventilation, with hypercapnia developing only in the late disease stages. The reduction in lung compliance, as a consequence of the increased lung elastic recoil related to the extracellular matrix deposition, also contributes to the increase in RR due to the overloading of respiratory muscles that stimulates peripheral mechanoreceptors.[3] This breathing pattern aims to minimize the work of breathing; however, in the late stages of the disease and when exercise intensity increases, tidal volume accounts for a greater proportion of the diminished vital capacity and physiological dead space increases leading to increased respiratory request and possible development of hypercapnia.
Pulmonary vascular damage is another contributing factor to CRF. A sharp increase in pulmonary pressures often occurs during exercise, regardless of the presence of pulmonary hypertension (PH) at rest. Hypoxic pulmonary vasoconstriction is the initial factor responsible for the increased pulmonary pressure.[4] However, as the disease progresses, the vascular damage increases with an overall reduction in vascular bed, increase of right ventricular afterload and, ultimately, onset of heart failure. The combination of all these factors often leads to an early arterial oxyhemoglobin desaturation during exercise.[5]
Detailed evaluation of exercise capacity at rest and under stress with cardiopulmonary exercise test (CPET), 6-minute walking test (6MWT) and PFTs helps to provide insights into the physiological impairments. These measurements may suggest the best intervention, including supplemental oxygen or exercise training, and assess prognosis more accurately.
3. Chronic respiratory failure aetiologies and diagnostic work-up: complications and worsening of associated comorbidities
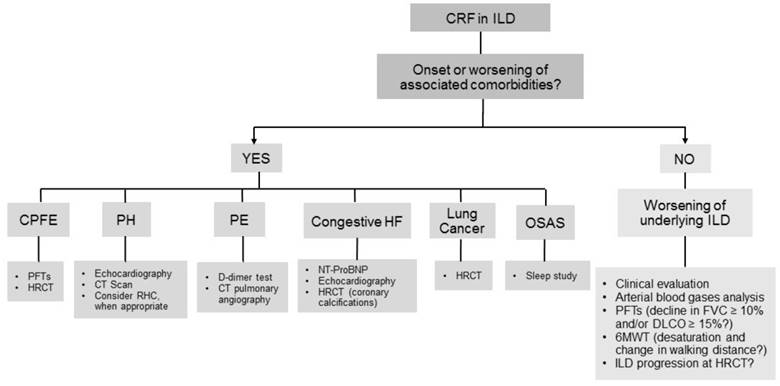
CRF often complicates the clinical course of ILDs, and usually is secondary to the worsening of the underlying disease; however, associated complications and comorbidities may also play a role (Figure 1 and Figure 2). We do not discuss here acute respiratory failure onset secondary to acute exacerbations of ILD (both idiopathic pulmonary fibrosis -IPF- and other than IPF), because it is subject of a previous review from our group.[6] Correct identification of the underlying cause of CRF is crucial in clinical practice both for prognostic implications and for different management. Thus, in the assessment of CRF, the onset or worsening of comorbidities should be always investigated.
Diagnostic work-up of chronic respiratory failure in interstitial lung diseases. Footnotes: 6MWT: six minute walking test; CPFE: combined pulmonary fibrosis and emphysema; CRF: chronic respiratory failure; CT: computed tomography; DLCO: diffusing lung capacity for carbon monoxide; FVC: forced vital capacity; HF: heart failure; HRCT: high resolution computed tomography; ILD: interstitial lung disease; NT-proBNP: N-terminal pro B-type natriuretic peptide; OSAS: obstructive sleep apnea syndrome; PE: pulmonary embolism; PFTs : pulmonary function tests; PH: pulmonary hypertension; RHC: right heart catheterization.
Causes of chronic respiratory failure in interstitial lung diseases and when to suspect them. Footnotes: CAD: coronary artery disease; COPD: chronic obstructive pulmonary disease; CTD: connective tissue disease; CRF: chronic respiratory failure; DLCO: diffusing lung capacity for carbon monoxide; FVC: forced vital capacity; HRCT: high resolution computed tomography; ILD: interstitial lung disease; LES: Systemic lupus erythematosus; PE: pulmonary embolism; PFTs: pulmonary function tests; PH: pulmonary hypertension; TLC: total lung capacity.
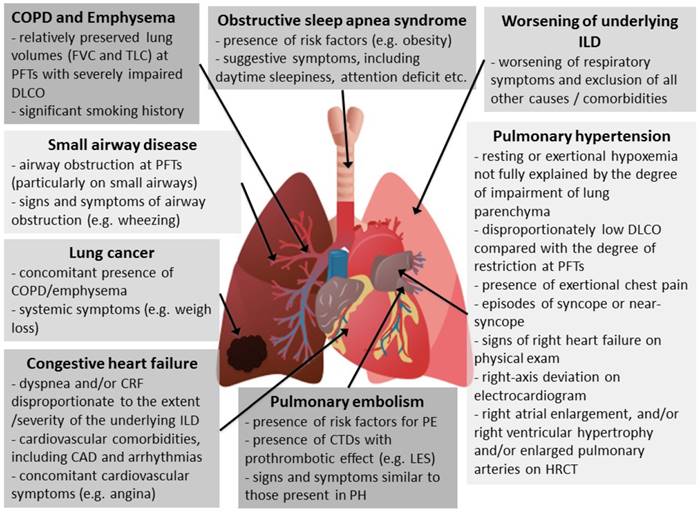
The most frequent complications and comorbidities associated with CRF are pulmonary hypertension, chronic obstructive pulmonary disease (COPD) and emphysema, pulmonary embolism (PE), congestive heart failure, lung cancer, obstructive sleep apnea syndrome (OSAS), and small airway disease.
- Worsening of underlying ILD
ILD natural history is affected by the development of CRF, which is often insidious and slowly progressive, while, more rarely, may occur as the consequence of an acute worsening of the underlying ILD. A prospective study conducted on IPF patients demonstrated that the development of CRF and the need for high oxygen flows were associated with higher mortality rates, regardless of PFTs.[7]
The interval from ILD diagnosis to development of respiratory failure is variable, with CRF occurring potentially at any stage of the disease. ILDs with a poorer prognosis and with a natural history characterized by acute exacerbations, e.g. IPF, show a higher rate and an earlier occurrence of CRF.[8,9] A possible worsening of the disease and development of progressive respiratory failure should be investigated and ruled out at every follow-up visit. Important instruments that may assist clinical evaluation include arterial blood gases analysis, PFTs and 6MWT. An absolute decline in FVC ≥ 10% or in DLCO ≥ 15% over 6 months is a reliable measure of disease progression in IPF and other ILD patients.[10] Desaturation at 6MWT and reduction of the walking distance at 6 months, despite a low reproducibility, have been correlated with mortality in IPF patients.[10] CPET at baseline may also have a prognostic role in ILD patients.[11,12]
Furthermore, HRCT could provide rapid, objective measurement of disease extent and change over time, both through qualitative visual assessment, limited by inter-observer and intra-observer variability, or by using the new computer-based methods for disease quantification.[13]
- Pulmonary Hypertension
PH, defined as mean pulmonary artery pressure ≥25 mm Hg confirmed by right heart catheterization (RHC), is a common complication in IPF, particularly as the disease progresses. The prevalence of PH in IPF ranges between 3% and 86%,[14] in sarcoidosis between 5% and 74%,[15] and in systemic sclerosis between 5% and 12%.[16] These wide prevalence ranges are due to differences in disease severity, variable definitions of PH and diagnostic methods used (echocardiography or RHC).
According to the American Thoracic Society (ATS) and the European Respiratory Society (ERS) guidelines,[17] PH associated with ILDs is categorized as group 3, which includes PH in chronic lung diseases and/or hypoxemia, and is associated with poor outcomes and high mortality. Furthermore, the most recent guidelines on lung transplant candidate selection cite the development of PH in IPF patients as a criterion to list for transplantation.[18]
Sarcoidosis, as well as Langherans cell histiocytosis-related PH, due to their multifactorial mechanism, are classified as group V PH.[17] Sarcoidosis-associated PH is not only related to hypoxic vasoconstriction/vascular rarefaction due to pulmonary fibrosis but also to compressive mediastinal infiltration or granulomatous involvement of pulmonary vessels.[19]
Although RHC remains the gold standard for PH detection, in clinical practice echocardiography is commonly used as screening tool. Nevertheless, there are no consensus recommendations regarding the timing for PH echocardiographic screening in ILD patients. The decision to refer a patient for RHC when echocardiography is suggestive for PH should be made on a case-by-case basis, particularly as treatment options are limited.
The optimization of supplementary long-term oxygen therapy (LTOT) to correct resting, nocturnal, and exertional hypoxia, diuretics and identification and treatment of contributing factors, such as OSAS, is crucial.
There are currently no approved therapies for the treatment of PH in IPF patients. The 2015 ATS/ERS Treatment Guidelines provided a strong recommendation against the use of selective endothelin receptor antagonist (Ambrisentan) in IPF, and a conditional recommendation against phosphodiesterase-5 inhibitors (Sildenafil) and dual endothelin receptor antagonists (Macitentan, Bosentan), regardless of the presence of PH.[20]
In ILD-PH, Sildenafil improved 6MWT distance and brain natriuretic peptide levels but showed no efficacy in reducing right ventricular systolic pressure after 6 months of treatment in small cohorts,[21] whereas in sarcoidosis Sildenafil improved mean pulmonary arterial pressure and cardiac output in repeated RHC 4 months after treatment.[22] Bosentan resulted to be ineffective in fibrotic idiopathic ILD-PH,[23] but was found to have beneficial effects in some sarcoidosis-PH patients, especially in those with limited ILD.[24-27] By contrast ambrisentan appeared to be poorly tolerated in sarcoidosis-PH.[28] More recently, Riociguat, a stimulator of the soluble guanylate cyclase, was found to increase cardiac output, decrease pulmonary vascular resistance and improve exercise capacity in an open-label, uncontrolled ILD-PH trial.[29] However, the RISE-IIP trial, a randomized controlled trial (RCT) on Riociguat in idiopathic ILDs, was stopped early due to increased severe adverse events and mortality.[30] PH targeted therapies proved to be effective in larger PH studies, including systemic sclerosis associated-ILD. Combination therapy with endothelin receptor antagonists, phosphodiesterase type-5 inhibitors and prostacyclin analogues did not affect survival but improved multiple outcome measures such as 6MWD, functional class and quality of life.[16]
In conclusion, PH treatment remains mainly supportive in ILDs with the exception of systemic sclerosis associated-ILD patients who have access to PH targeted therapies.
- COPD and emphysema
Combined pulmonary fibrosis and emphysema (CPFE) is defined as the coexistence of emphysema and pulmonary fibrosis.[31] This syndrome is frequently complicated by PH [32] and lung cancer. Resting and exertional hypoxemia are common and CRF is more frequent in CPFE compared with patients with pure emphysema or pure fibrosis, resulting in a poorer prognosis.
There is no specific therapy for CPFE, as no clinical trial directly addressing CPFE has been conducted, thus treatment recommendations are based on expert opinion. A subgroup analysis of the INPULSIS trials on Nintedanib, an antifibrotic agent approved for IPF treatment, found that the drug was effective in slowing disease progression also in IPF patients presenting emphysema.[33] In general, smoking cessation, vaccinations, supplemental oxygen and pulmonary rehabilitation should be prescribed when appropriate.
- Pulmonary embolism
ILD patients are at increased risk of venous thromboembolic disease,[34-36] mainly due to immobility secondary to dyspnea or to joint or muscle pain and stiffness in connective tissue disease associated ILDs (CTD-ILDs). The presence of a pro-coagulant microenvironment has been suggested in IPF and in non-IPF ILDs,[37] as well as a prothrombotic state has been shown to be more common in IPF patients than in healthy controls.[38]
Computed tomography pulmonary angiography is the gold standard for diagnosis, because ventilation/perfusion scanning is nonspecific for PE in ILDs, as perfusion defects are often present and correspond to honeycombing and emphysema.[39] Regarding PE treatment, warfarin is contraindicated in IPF patients,[20] because in a RCT it correlated to increased mortality.[40] However, there are no data suggesting that vitamin K antagonists are contraindicated for PE treatment in ILDs other than IPF.[20]
An increased risk of venous thromboembolism, compared to the general population, has been reported in CTDs that may present a pulmonary involvement,[41] in particular in systemic lupus erythematosus (SLE),[41,42] dermatopolymyositis,[43-45] granulomatosis with polyangiitis,[46] rheumatoid arthritis,[47] systemic sclerosis,[48-51] Sjögren syndrome.[52]
The increased thromboembolic risk in these diseases is thought to be secondary to the underlying inflammatory state, with proinflammatory cytokines causing endothelial dysfunction and playing a role in the activation of the coagulation cascade. Recurrent episodes of PE in SLE should prompt the exclusion of secondary antiphospholipid syndrome. Sarcoidosis has also been associated to increased risk of venous thromboembolism in population-based studies.[53,54]
- Congestive heart failure
Cardiovascular diseases, in particular coronary artery disease (CAD) and arrhythmias, represent a common comorbidity in ILD patients. CAD prevalence in IPF patients is as high as 60% and is directly proportional to the high prevalence of left ventricular diastolic dysfunction.[55,56] Although some Authors suggested to perform an extensive CPET in patients with IPF for both prognostic purposes and to detect potentially treatable cardiovascular alterations,[57] the cost-benefit ratio of these diagnostic exams still needs to be determined.
Recently, HRCT was found to be helpful in identifying CAD in IPF patients, since the presence of moderate-to-severe coronary calcifications has a high sensitivity and specificity for the presence of significant CAD, whereas the absence of calcifications has an extremely high negative predictive value.[58] Nathan et al. suggested that in case of angina or moderate-to-severe coronary calcifications on HRCT further cardiologic evaluation may be prudent.[58]
Finally, cardiac disease may result also from direct involvement of the heart as in sarcoidosis or in idiopathic inflammatory myopathies and systemic sclerosis.
- Lung cancer
The incidence of lung cancer is markedly increased among patients with smoking-related ILDs, particularly IPF. Pulmonary fibrosis appears to be a risk factor for lung cancer regardless of smoking history, which is a shared risk factor for the development of both diseases.[59] Lung cancer in IPF typically manifests as lower lung nodular lesions along the periphery of fibrotic areas. Squamous cell carcinoma is the most common histotype, followed by adenocarcinoma.
At present, there is neither evidence nor consensus regarding specific therapeutic approaches for patients with ILDs diagnosed with lung cancer. Therefore, management is based on risk/benefit considerations. Median survival after diagnosis is worse in patients with lung cancer and ILDs than in either ILDs or lung cancer alone.[60-62] All the available treatment options, including surgical resection, radiotherapy and chemotherapy, may provoke acute exacerbation of the underlying ILD.[63-65] Furthermore, since only a few studies have investigated the use of chemotherapy in ILD patients with lung cancer, the optimal therapeutic agents have yet to be determined.
- Obstructive sleep apnea syndrome
OSAS is common in ILD patients: reported prevalence ranges between 5.9% and 91% in IPF patients,[14,66-68] between 52 and 67% in sarcoidosis,[67,69-71] and between 56 and 66% in patients with systemic sclerosis.[67] This condition is more common during rapid eye movement sleep, when the only operative muscle is the diaphragm, while the intercostal muscles are inactive, leading to further reduction of functional residual capacity. This facilitates upper airway collapse during sleep in patients affected by ILDs.[72] However, development of OSAS cannot be totally explained with these changes and, most likely, multiple factors are involved. In sarcoidosis, the risk of OSAS may possibly be increased by the involvement of the upper airways themselves.[70] Oral corticosteroids, frequently used to treat some kind of ILDs, may lead to liquid accumulation and fat deposition in the pharyngeal wall and to myopathy of the pharyngeal muscles, possibly increasing the risk of OSAS. However, a retrospective study failed to find any differences in polysomnography data in patients treated or not with corticosteroids.[67] Obesity is also a predisposing condition to OSAS in IPF patients.[14,73]
In a prospective study on 31 IPF subjects evaluating the association between OSAS and mortality, intermittent sleep oxygen desaturation was directly correlated with survival, while apnea-hypopnea index was not. The Authors explained these findings mainly as the result of hypoxic vasoconstriction, leading to development or worsening of PH.[74]
A recent study conducted on IPF patients found a worse prognosis, both in terms of mortality and clinical deterioration, in patients with OSAS and sleep-related hypoxemia compared to patients without sleep breathing disorders and those with OSAS without nocturnal hypoxemia.[75]
Although international ILD guidelines do not recommend the execution of polysomnography in all patients, in case of development of CRF and/or other suggestive symptoms, such as daytime sleepiness and attention deficits, a sleep study should be considered.
- Small airway disease
Small airway disease is present in different ILDs typically associated with obstructive ventilatory defect, such as sarcoidosis, CPFE, lymphangioleiomyomatosis (LAM), hypersensitivity pneumonitis and pulmonary Langherans' cell histiocytosis. Radiological characteristics that may be observed at HRCT include mosaic pattern indicating air trapping and bronchial wall thickening. Other non-invasive methods to evaluate these alterations with less radiographic exposure are forced oscillation technique, ultrasonic pneumography and impulse oscillometry.[76,77]
The optimization of pharmacological treatment, for example with a trial of bronchodilator therapy, is crucial in these patients.[78-80]
4. Management and Treatment
The following therapeutic options may be considered for ILD patients with CRF together with different management approaches (Figure 3).
Management and therapeutic options in interstitial lung diseases patients with chronic respiratory failure. Footnotes: CRF: chronic respiratory failure.
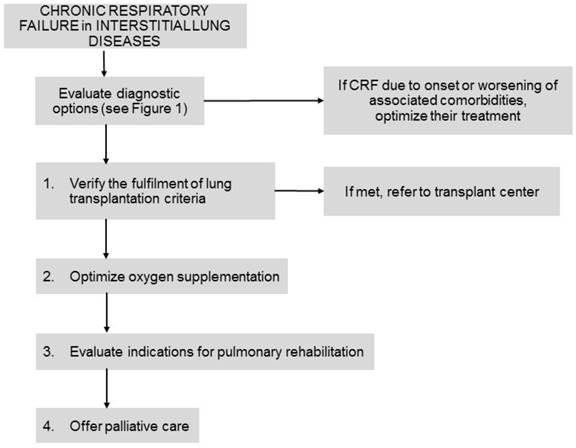
a) Therapies for the underlying disease
Currently there is no consensus on whether continuing pharmacologic treatment in end-stage ILD patients experiencing CRF. In fact, even in IPF, patients with severe disease (e.g., FVC<50% and/or DLCO<30-35%) were not enrolled in RCTs. Clinical observation experiences and preliminary results of long-term, open-label extensions of RCTs suggest that both Pirfenidone and Nintedanib may also slow or decrease progression in patients with severe IPF.[81-86] Furthermore, there is no consensus on management of severe-IPF patients experiencing functional progression, i.e. decline in FVC≥10% or DLCO≥15% over 6 months. Several options may be considered: switch to other antifibrotic drug, combination therapy, permanent discontinuation of the antifibrotic drug in order to avoid side-effects in the absence of a clear benefit, or continuing the antifibrotic treatment. Two studies demonstrated that in IPF patients who experienced a ≥10% absolute decline in FVC during the first 6 months of treatment, continued treatment with antifibrotics reduced the risk of further FVC decline or death compared with placebo.[87]
b) Therapies for dyspnea
- Oxygen therapy and high-flow nasal cannula
Clinical practice guidelines strongly recommend supplemental oxygen for hypoxemic patients with IPF,[20] despite the lack of evidence about a survival benefit.[88]
Oxygen administration improves exercise tolerance for patients with resting or exertional hypoxemia.[89,90] Although LTOT is likely to improve symptoms and overall quality of life in IPF patients,[20,91] a recent systematic review showed no effects of LTOT on exertional dyspnea in ILDs.[92]
The impact of LTOT on breathlessness is also influenced by patient's expectations.[93] Furthermore, despite a high compliance with LTOT prescription, most ILD patients experience significant anxiety because of social stigma and concerns that oxygen prescription is associated with end-stage disease.[94]
Recently, the ambulatory oxygen in fibrotic lung diseases (AmbOx) trial showed that ambulatory oxygen seemed to be associated with improved HRQoL in patients with isolated exertional hypoxia.[95]
High-flow nasal cannulae (HFNC) have also been recently introduced. HFNC work with an air oxygen blender allowing from 21% to 100% FIO2, heating and humidifying the inspiratory gas and generating up to 60 L/min in adults.[96] HFNC have also shown favorable physiological effects, such as pharyngeal dead space washout, reduction of nasopharyngeal resistance, positive end-expiratory pressure (PEEP) effect, and reduction of airway resistance.[96]
At present, the clinical applications of HFNC in ILD patients with CRF are subject of research. Bräunlich et al. demonstrated a PEEP effect in 13 IPF patients during HFNC oxygenation associated with reduced RR and reduced minute volume.[97]
Furthermore, based on the aforementioned advantages of HFNC compared with standard oxygen therapy, HFNC may have a role in the management of CRF, especially for end-stage ILD patients with severe hypoxemia, as palliative treatment.[98,99] In a retrospective cohort of 84 ILD patients with a do-not-intubate order, compared with non-invasive ventilation (NIV), treatment with HFNC was associated with an equivalent survival rate, better tolerance, less temporary interruption and discontinuation rates, allowing patients to eat and converse just before death.[100]
These findings seem to suggest that HFNC might be a reasonable add-on palliative treatment in ILD patients. However, to date, no cost-effectiveness analysis is available and further RCTs are needed to assess the clinical advantages of HFNC over standard oxygen therapy or NIV in ILD patients.
- Non-invasive ventilation
Neither oxygen application nor ventilation techniques modify the natural history of ILDs, however NIV may play a role in the chronic setting regarding the impact on the quality of life of patients with end-stage disease.
Some preliminary data seem to suggest that chronic use of nocturnal NIV in ILD patients with hypercapnic CRF (6 chronic hypersensitivity pneumonitis, 2 sarcoidosis and 2 undetermined ILDs) might decrease daytime arterial partial pressure of carbon dioxide and increase arterial partial pressure of oxygen.[101] However, given the retrospective nature of the study and the small cohort, the long-term benefits and the effect on patients' quality of life still need to be explored.
Another potential role for NIV regards the end-of-life care in ILDs, in which NIV is becoming increasingly common as a palliative treatment, although the evidence to support this application is limited.[102,103] In a recent paper Rajala et al. retrospectively analyzed healthcare documentation during the 6 months preceding death of 59 IPF patients from the Finnish prospective IPF cohort study (FinnishIPF).[103] The most common symptom manifested in the advanced stages of the disease was dyspnea and the most prescribed therapies to control it, during the last week before death, were opioids (71% of the patients), supplemental oxygen (81% of patients), and NIV, used in 29% of the cases.[104]
Despite the use of NIV together with opioids and benzodiazepines may seem incongruous, these two interventions may be associated with the aim to reduce the RR, optimize patient-NIV adaptation, and palliate severe refractory dyspnea with NIV application while adjusting sedative and dyspnea-reliever drugs.
Finally, NIV has also been applied to ILD patients during rehabilitation programs.[104] Moderno et al. compared the submaximal exercise tests (60% of maximum load) of 10 IPF patients in three different situations: without ventilatory support, with continuous positive airway pressure (CPAP) and with proportional assist ventilation (PAV).[104] Patients on PAV experienced increased submaximal test compared to those on CPAP or without ventilatory support, in addition to an improved oxygenation and lower subjective perception of effort. These preliminary results suggest that NIV may increase exercise tolerance and decrease dyspnea and cardiac effort in patients with IPF. Therefore, rehabilitation programs for ILD patients might benefit from NIV implementation to improve cardiopulmonary conditioning.
In conclusion, NIV in CRF in ILDs might present very different fields of application ranging from palliation to rehabilitation programs. However, given the limited number of studies and the low quality of the evidence available, future studies are needed to elucidate its best application.
- Palliative care
According to a World Health Organization definition dated 1990 “Palliative care is the active total care of patients whose disease is not responsive to curative treatment”.[105] The main goal of palliative care for patients with ILDs is to improve and maintain an acceptable quality of life during the advanced phases of the disease.
Despite the well-known ominous prognosis of some ILDs, in particular IPF, and the evidence on the applicability at an earlier stage in the disease course in association with antifibrotic treatment, palliative care remains largely understudied and underused in this population.[106-108] According to different European cohorts, many patients with advanced IPF still die in the hospital, although the hospital is hardly the favorite place of death, and end-of-life decisions are made very late in the course of the disease.[103,106,109,110]
Regardless of the cause or histological pattern, ILD clinical course is featured by a high burden of respiratory symptoms such as breathlessness and cough, accompanied in the advanced stages with fatigue, anxiety and depression.[111,112] These symptoms when uncontrolled lead to a reduced emotional wellbeing, resulting in severely impaired quality of life.[107,113,114]
Despite being the most debilitating symptom, there are cultural neglects, discomfort and unfamiliarity to report and quantify breathlessness.[111,113] Currently new multidimensional scales, such as the Dyspnoea-12 (D-12),[115] Table 2, the Multidimensional Dyspnea Profile,[116] and the Breathlessness and activities domain of the King's Brief Interstitial Lung Disease (K-BILD) questionnaire,[117] Table 3, are available to explore the breathing discomfort, sensory qualities, and the associated emotional responses.[118] Nevertheless, patients affected by ILDs receive poorer access to specialist end-of-life care services and seem to experience more breathlessness than patients with lung cancer.[112] A possible explanation for this lack of access to palliation is the high variability in clinical course among different ILDs, which makes challenging an appropriate counseling about disease's prognosis.
Dyspnoea-12 Questionnaire (115)
| Item | None | Mild | Moderate | Severe |
|---|---|---|---|---|
| 1. My breath does not go in all the way | ||||
| 2. My breathing requires more work | ||||
| 3. I feel short of breath | ||||
| 4. I have difficulty catching my breath | ||||
| 5. I cannot get enough air | ||||
| 6. My breathing is uncomfortable | ||||
| 7. My breathing is exhausting | ||||
| 8. My breathing makes me feel depressed | ||||
| 9. My breathing makes me feel miserable | ||||
| 10. My breathing is distressing | ||||
| 11. My breathing makes me agitated | ||||
| 12. My breathing is irritating |
This questionnaire is designed to help us learn more about how your breathing is troubling you. Please read each item and then tick in the box that best matches your breathing these days. If you do not experience an item tick the "none" box. Please respond to all items.
Scores: none (0), mild (1), moderate (2) and severe (3). Total scores range from 0 to 36, with higher scores corresponding to greater severity.
Breathlessness and activities domain of the King's Brief Interstitial Lung Disease (K-BILD) questionnaire (117)
| # 1. In the last 2 weeks, I have been breathless climbing stairs or walking up an incline or hill | ||||||
| 1. All of the time | 2. Most of the time | 3. A good bit of the time | 4. Some of the time | 5. A little of the time | 6. Hardly any of the time | 7. None of the time |
| # 4. In the last 2 weeks have you avoided doing things that make you breathless? | ||||||
| 1. All of the time | 2. Most of the time | 3. A good bit of the time | 4. Some of the time | 5. A little of the time | 6. Hardly any of the time | 7. None of the time |
| # 11. In the last 2 weeks has your lung condition interfered with your job or other daily tasks? | ||||||
| 1. All of the time | 2. Most of the time | 3. A good bit of the time | 4. Some of the time | 5. A little of the time | 6. Hardly any of the time | 7. None of the time |
| # 13. In the last 2 weeks, how much has your lung condition limited you carrying things, for example, groceries? | ||||||
| 1. All of the time | 2. Most of the time | 3. A good bit of the time | 4. Some of the time | 5. A little of the time | 6. Hardly any of the time | 7. None of the time |
This questionnaire is designed to assess the impact of your lung disease on various aspects of your life. Please circle the response that best applies to you for each question.
One of the main difficulties for the physician is to recognize the conditions still deserving of intensive therapeutic approaches vs 'end stage' manifestations, in which a parallel palliative care management should be planned. This is the concept of "integrated model" and "simultaneous care" in which palliative care accompanies specific treatments till the end-of-life phases, when palliative care substitutes the specific treatments. For chronic lung diseases, such as COPD and fibrosing ILDs, it is necessary to identify the moment when the severity of the disease makes remissions rarer and shorter and causes an increase in the number and duration of hospitalizations. Since timing for referral to palliative care specialists is not clearly scheduled, a practical approach could be to identify some referral criteria. LTOT prescription, age > 70 years, prior disease exacerbations, presence of “honeycomb” at HRCT and Usual Interstitial Pneumonia (UIP) histological pattern are associated with a poor prognosis and therefore may be considered as possible indicators for palliative care referral.[112] Furthermore, increased dependence in daily life activities and reduced functional autonomy, particularly when associated with higher degree of dyspnea, can also be considered useful indicators for palliative care initiation.[119-122]
Another important aspect to consider is that palliation is a dynamic process as patients' needs may change over time according to the phase of the disease.[123] The caregiver should also be involved and supported throughout the disease course. Patients should be fully informed about symptoms and treatment options early in the progression of the disease with special attention to patients' needs, preferences, culture, and beliefs.[123] Furthermore, both the patient and the caregiver need time to understand supportive treatment options and to choose setting of care. Van Manen et al. recently proposed a model (“the ABCDE of IPF care”) to tailor supportive care along the course of the disease in IPF patients.[124]
Most used therapies for palliation include morphine and opioids, such as fentanyl, oxycodone and hydromorphone, in different administration forms (oral, subcutaneous, intravenous, transdermal etc). Despite the wide choice of pharmacological agents, no RCTs have assessed the best drugs to control breathlessness in patients with end-stage ILDs and there are still many prejudices on the use of morphine and its derivatives in patients with end-stage chronic respiratory diseases.[125,126] Lack of guidelines and familiarity with these drugs and fear of downregulation of respiratory control centers are some of the factors that prevent implementation in clinical practice.[127]
In conclusion, similarly to oncologic diseases, in the late phases of ILDs there is a strong need to provide access to adequate palliative care and discuss treatment and supportive options in a dedicated multidisciplinary setting in order to improve quality of life. Further trials on the efficacy of symptomatic interventions for ILDs are advisable in order to elucidate the best models to take care of both patients and caregivers.[123]
c) Rehabilitation
Pulmonary rehabilitation (PR) is defined by the ATS and ERS as a “comprehensive intervention based on a thorough patient assessment followed by patient-tailored therapies that include, but are not limited to, exercise training, educational and behavioral changes, designed to improve the physical and psychological condition of people with chronic respiratory disease and to promote the long-term adherence of health-enhancing behaviors”.[128,129]
PR role and benefits have been well defined in patients with COPD and emerging evidence suggests that these benefits could be extended to other chronic respiratory conditions, such as ILDs.[130-132] Current guidelines for the management of IPF make a weak positive recommendation for PR because of inadequate reporting of methods and small number of participants in the available studies.[20] Nevertheless, a recent Cochrane review reported that PR seems to be safe for ILD patients and may reduce dyspnea, increase walking distance, increase maximum oxygen uptake and improve quality of life.[133] PR has also been shown to reduce exertional dyspnea in IPF patients as assessed by Borg scale,[134] MRC and Chronic Respiratory Disease Questionnaire (CRDQ),[135-138] but statistically significant results have not been unanimously achieved.[139,140]
PR also led to short-term improvements of health-related quality of life (HRQoL), as reported by different RCTs.[138,139,141,142] This was supported by other non-randomized studies,[135,137,140,143,144] while other authors did not report any positive effect on HRQoL.[145,146]
Functional exercise capacity, expressed as change in walking distance during 6MWT, significantly improved in IPF patients immediately after PR.[147] Similarly, gait speed over four metres (4MGS), a measure of usual walking speed widely used in gerontology as a measure of functional and lower limb performance,[148] improved significantly with PR.[143,149]
However, the role of PR as available therapeutic option has yet to be established, in particular it is not clear whether ILD etiology and severity may influence the response to PR.
The largest benefit following PR seems to occur in patients with asbestosis, followed by those with IPF, while patients with CTD-ILDs seem to receive less benefit from PR programs.[141,150,151]
There are opposite views regarding which patients may benefit most from PR programs: two studies suggest a greater treatment effect in those with less functional impairment,[152,153] while others found greater improvements in those with more severe impairment.[134,143] In IPF patients, the response to PR varied depending on the Medical Research Council (MRC) grade of dyspnea at baseline; specifically, a greater MRC grade of dyspnea is associated with greater functional improvement and lower hospitalization rate.[153]
The beneficial effects in ILDs, particularly in IPF, seem to be greater amongst patients with a shorter walking distance at baseline.[134,135,143,144] On the contrary, in the subgroups with severe ILDs and exercise desaturation, no definite improvements in walking distance and maximum oxygen uptake were demonstrated.[138,143,154,155] Moreover, oxygen users gained less from PR and had a higher mortality rate than non-oxygen users.[156] Furthermore, the presence of PH, as comorbidity, reduced the walking distance at 6MWT regardless of lung function.[157]
Shorter (6-8 weeks) exercise training programs have been shown to improve symptoms and physical activity levels both during and after rehabilitation.[158] Longer programs (3 months) seem able to maintain exercise oxygen uptake and lengthen constant load exercise time in patients with severe IPF.[141] Successful adherence is more likely in patients with milder disease resulting in greater treatment effects. When adherence to the PR protocol is limited by severe dyspnea or fear of adverse events, an interval training approach may be useful. Other strategies used to enhance the training effect on peripheral muscles in severe COPD patients, such as neuromuscular electrical stimulation and using step counters,[159] may also have a role in severe ILDs, although this has not yet been tested in RCTs. In CTD-ILDs, commonly associated with systemic manifestations such as joint pain and swelling, muscle weakness and pain, hydrotherapy or resistance training may be more suitable in achieving benefits.[141]
Uncertainties remain regarding how long the benefits after PR persist. Long-term benefits (up to 12-18 months) have not been consistently shown in patients with ILD.[133,138,141,145,150] A recent 12-week RCT on exercise training showed maintenance of improved quality of life and leg strength in IPF patients at 11 months, but not at 30 months of follow-up.[142]
Although the magnitude of improvement seems to be less evident than in COPD patients [130,145] and may not persist on long-term,[138] PR is safe and feasible in ILDs and appears to be a valuable adjunct therapy.
Further studies are needed to evaluate the best type (exercise, intensity, frequency, and duration) and timing of PR programs for ILD patients.
d) Transplantation
Regardless of the specific ILD, lung transplantation remains the only effective therapy in patients with advanced ILDs refractory to medical treatment.[160] The main limitation for lung transplantation is the number of available organs.[161,162]
A timely referral of the patient to the transplant center is fundamental to obtain an exhaustive formal evaluation and to timely place the patient in the active waiting list. It also warrants the patient time to collect the strong psychosocial support necessary to undergo this demanding procedure.[163]
According to the 2014 International Society for Heart and Lung Transplantation Consensus document, referral to transplant center must be considered in presence of any of the following conditions: “histopathological or radiographic evidence of UIP or fibrosing non-specific interstitial pneumonitis or lung function defect (i.e. “FVC <80% predicted or DLCO <40% predicted), dyspnea or functional limitation attributable to lung disease and oxygen requirement”, for inflammatory ILDs “failure to improve dyspnea, oxygen requirement, and/or lung function after a clinically indicated trial of medical therapy”.[18]
Although the median survival after lung transplantation in patients with ILDs is 4.7 years, this intervention still conveys a survival benefit and improvement in quality of life in suitable patients with advanced lung disease and hypoxemic CRF.[164]
Because of similar post transplantation survival rates between idiopathic and CTD-ILDs,[165] the current guidelines propose the application of the same criteria to those CTDs in which no extrapulmonary contraindication to transplantation coexist.[18]
Conclusions
Despite the known ominous prognosis of some ILDs, differential diagnosis of CRF should be properly assessed. Furthermore, in ILD patients management options, such as rehabilitation programs, and palliative care remain largely underused, thus, a personalized and multidisciplinary approach to CRF would be desirable and may improve the outcomes and, particularly, the quality of life of end-stage ILD patients.
Abbreviations
4MGS: gait speed over four metres; 6MWT: 6-minute walking test; ATS: American Thoracic Society; CAD: coronary artery disease; COPD: chronic obstructive pulmonary disease; CPAP: continuous positive airway pressure; CPET: cardiopulmonary exercise testing; CPFE: combined pulmonary fibrosis and emphysema; CRF: chronic respiratory failure; CRDQ: Chronic Respiratory Disease Questionnaire; CTD-ILD: connective tissue disease-associated interstitial lung disease; DLCO: diffusing lung capacity for carbon monoxide; ERS: European Respiratory Society; FIO2: inspiratory oxygen fraction; FVC: forced vital capacity; HFNC: high-flow nasal cannula; HRCT: high resolution computed tomography; HRQoL: health-related quality of life; ILDs: interstitial lung diseases; IPF: idiopathic pulmonary fibrosis; LAM: lymphangioleiomyomatosis; LTOT: long-term oxygen therapy; MRC: Medical Research Council; NIV: non-invasive ventilation; OSAS: obstructive sleep apnea syndrome; PAV: proportional assist ventilation; PEEP: positive end-expiratory pressure; PFT: pulmonary function test; PH: pulmonary hypertension; PR: pulmonary rehabilitation; RCT: randomized controlled trial; RHC: right heart catheterization; UIP: usual interstitial pneumonia.
Acknowledgements
We acknowledge that this research was partially supported by the Italian Ministry of University and Research (MIUR) - Department of Excellence project PREMIA (PREcision MedIcine Approach: bringing biomarker research to clinic).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Travis WD, Costabel U, Hansell DM, King TE, Lynch DA, Nicholson AG. et al. An official American Thoracic Society/European Respiratory Society statement: Update of the international multidisciplinary classification of the idiopathic interstitial pneumonias. Am J Respir Crit Care Med. 2013Sep15;188(6):733-48
2. Javaheri S, Sicilian L. Lung function, breathing pattern, and gas exchange in interstitial lung disease. Thorax. 1992Feb;47(2):93-7
3. Bag R, Suleman N, Guntupalli KK. Respiratory failure in interstitial lung disease. Curr Opin Pulm Med. 2004Sep;10(5):412-8
4. Agustí AG, Roca J, Gea J, Wagner PD, Xaubet A, Rodriguez-Roisin R. Mechanisms of gas-exchange impairment in idiopathic pulmonary fibrosis. Am Rev Respir Dis. 1991Feb;143(2):219-25
5. Gläser S, Noga O, Koch B, Opitz CF, Schmidt B, Temmesfeld B. et al. Impact of pulmonary hypertension on gas exchange and exercise capacity in patients with pulmonary fibrosis. Respir Med. 2009Feb;103(2):317-24
6. Faverio P, De Giacomi F, Sardella L, Fiorentino G, Carone M, Salerno F. et al. Management of acute respiratory failure in interstitial lung diseases: overview and clinical insights. BMC Pulm Med. 2018May15;18(1):70
7. Hook JL, Arcasoy SM, Zemmel D, Bartels MN, Kawut SM, Lederer DJ. Titrated oxygen requirement and prognostication in idiopathic pulmonary fibrosis. Eur Respir J. 2012Feb;39(2):359-65
8. Traila D, Oancea C, Tudorache E, Mladinescu OF, Timar B, Tudorache V. Clinical profile of unclassifiable interstitial lung disease: Comparison with chronic fibrosing idiopathic interstitial pneumonias. J Int Med Res. 2018Jan;46(1):448-56
9. Ryerson CJ, Vittinghoff E, Ley B, Lee JS, Mooney JJ, Jones KD. et al. Predicting survival across chronic interstitial lung disease: the ILD-GAP model. Chest. 2014Apr;145(4):723-8
10. Robbie H, Daccord C, Chua F, Devaraj A. Evaluating disease severity in idiopathic pulmonary fibrosis. Eur Respir Rev. 2017 Sep 30;26(145)
11. Layton AM, Armstrong HF, Kim HP, Meza KS, D'Ovidio F, Arcasoy SM. Cardiopulmonary exercise factors predict survival in patients with advanced interstitial lung disease referred for lung transplantation. Respir Med. 2017;126:59-67
12. Fell CD, Liu LX, Motika C, Kazerooni EA, Gross BH, Travis WD. et al. The prognostic value of cardiopulmonary exercise testing in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2009Mar1;179(5):402-7
13. Wu X, Kim GH, Salisbury ML, Barber D, Bartholmai BJ, Brown KK. et al. Computed Tomographic Biomarkers in Idiopathic Pulmonary Fibrosis: The Future of Quantitative Analysis. Am J Respir Crit Care Med. 2018 Jul 9
14. Raghu G, Amatto VC, Behr J, Stowasser S. Comorbidities in idiopathic pulmonary fibrosis patients: a systematic literature review. Eur Respir J. 2015Oct;46(4):1113-30
15. Corte TJ, Wells AU, Nicholson AG, Hansell DM, Wort SJ. Pulmonary hypertension in sarcoidosis: a review. Respirology. 2011Jan;16(1):69-77
16. Solomon JJ, Olson AL, Fischer A, Bull T, Brown KK, Raghu G. Scleroderma lung disease. Eur Respir Rev. 2013Mar1;22(127):6-19
17. Galiè N, Humbert M, Vachiery J-L, Gibbs S, Lang I, Torbicki A. et al. 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension: The Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT). Eur Respir J. 2015Oct;46(4):903-75
18. Weill D, Benden C, Corris PA, Dark JH, Davis RD, Keshavjee S. et al. A consensus document for the selection of lung transplant candidates: 2014-an update from the Pulmonary Transplantation Council of the International Society for Heart and Lung Transplantation. J Heart Lung Transplant. 2015Jan;34(1):1-15
19. Weatherald J, Savale L, Humbert M. Medical Management of Pulmonary Hypertension with Unclear and/or Multifactorial Mechanisms (Group 5): Is There a Role for Pulmonary Arterial Hypertension Medications? Curr Hypertens Rep. 2017Oct18;19(11):86
20. Raghu G, Rochwerg B, Zhang Y, Garcia CAC, Azuma A, Behr J. et al. An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline: Treatment of Idiopathic Pulmonary Fibrosis. An Update of the 2011 Clinical Practice Guideline. Am J Respir Crit Care Med. 2015Jul15;192(2):e3-19
21. Corte TJ, Gatzoulis MA, Parfitt L, Harries C, Wells AU, Wort SJ. The use of sildenafil to treat pulmonary hypertension associated with interstitial lung disease. Respirology. 2010Nov;15(8):1226-32
22. Milman N, Burton CM, Iversen M, Videbaek R, Jensen CV, Carlsen J. Pulmonary hypertension in end-stage pulmonary sarcoidosis: therapeutic effect of sildenafil? J Heart Lung Transplant. 2008Mar;27(3):329-34
23. Corte TJ, Keir GJ, Dimopoulos K, Howard L, Corris PA, Parfitt L. et al. Bosentan in pulmonary hypertension associated with fibrotic idiopathic interstitial pneumonia. Am J Respir Crit Care Med. 2014Jul15;190(2):208-17
24. Foley RJ, Metersky ML. Successful treatment of sarcoidosis-associated pulmonary hypertension with bosentan. Respiration. 2008;75(2):211-4
25. Pitsiou GG, Spyratos D, Kioumis I, Boutou AK, Nakou C, Stanopoulos I. Sarcoidosis-associated pulmonary hypertension: a role for endothelin receptor antagonists? Ther Adv Respir Dis. 2009Jun;3(3):99-101
26. Barnett CF, Bonura EJ, Nathan SD, Ahmad S, Shlobin OA, Osei K. et al. Treatment of sarcoidosis-associated pulmonary hypertension. A two-center experience. Chest. 2009Jun;135(6):1455-61
27. Sharma S, Kashour T, Philipp R. Secondary pulmonary arterial hypertension: treated with endothelin receptor blockade. Tex Heart Inst J. 2005;32(3):405-10
28. Judson MA, Highland KB, Kwon S, Donohue JF, Aris R, Craft N. et al. Ambrisentan for sarcoidosis associated pulmonary hypertension. Sarcoidosis Vasc Diffuse Lung Dis. 2011Oct;28(2):139-45
29. Hoeper MM, Halank M, Wilkens H, Günther A, Weimann G, Gebert I. et al. Riociguat for interstitial lung disease and pulmonary hypertension: a pilot trial. Eur Respir J. 2013Apr;41(4):853-60
30. Nathan SD, Behr J, Cottin V, Collard HR, Hoeper MM, Martinez FJ. et al. Idiopathic interstitial pneumonia-associated pulmonary hypertension: A target for therapy? Respir Med. 2017;122(Suppl 1):S10-3
31. Cottin V, Nunes H, Brillet P-Y, Delaval P, Devouassoux G, Tillie-Leblond I. et al. Combined pulmonary fibrosis and emphysema: a distinct underrecognised entity. Eur Respir J. 2005Oct;26(4):586-93
32. Mejía M, Carrillo G, Rojas-Serrano J, Estrada A, Suárez T, Alonso D. et al. Idiopathic pulmonary fibrosis and emphysema: decreased survival associated with severe pulmonary arterial hypertension. Chest. 2009Jul;136(1):10-5
33. Pfeifer M, Cottin V, Taniguchi H, Richeldi L, Collard HR, Kaye M. et al. Effect of baseline emphysema on reduction in FVC decline with nintedanib in the INPULSISTM trials. Pneumologie. 2015Mar;69(S 1):P254
34. Hubbard RB, Smith C, Le Jeune I, Gribbin J, Fogarty AW. The association between idiopathic pulmonary fibrosis and vascular disease: a population-based study. Am J Respir Crit Care Med. 2008Dec15;178(12):1257-61
35. Sprunger DB, Olson AL, Huie TJ, Fernandez-Perez ER, Fischer A, Solomon JJ. et al. Pulmonary fibrosis is associated with an elevated risk of thromboembolic disease. Eur Respir J. 2012Jan;39(1):125-32
36. Sode BF, Dahl M, Nielsen SF, Nordestgaard BG. Venous thromboembolism and risk of idiopathic interstitial pneumonia: a nationwide study. Am J Respir Crit Care Med. 2010May15;181(10):1085-92
37. Chambers RC. Procoagulant signalling mechanisms in lung inflammation and fibrosis: novel opportunities for pharmacological intervention? Br J Pharmacol. 2008Mar;153(Suppl 1):S367-378
38. Navaratnam V, Fogarty AW, McKeever T, Thompson N, Jenkins G, Johnson SR. et al. Presence of a prothrombotic state in people with idiopathic pulmonary fibrosis: a population-based case-control study. Thorax. 2014Mar;69(3):207-15
39. Strickland NH, Hughes JM, Hart DA, Myers MJ, Lavender JP. Cause of regional ventilation-perfusion mismatching in patients with idiopathic pulmonary fibrosis: a combined CT and scintigraphic study. AJR Am J Roentgenol. 1993Oct;161(4):719-25
40. Noth I, Anstrom KJ, Calvert SB, de Andrade J, Flaherty KR, Glazer C. et al. A placebo-controlled randomized trial of warfarin in idiopathic pulmonary fibrosis. Am J Respir Crit Care Med. 2012Jul1;186(1):88-95
41. Annangi S, Dammalapati TR, Nutalapati S, Henriques King MN. Prevalence of Pulmonary Embolism Among Systemic Lupus Erythematosus Discharges: A Decade of Analysis of the National Hospital Discharge Survey. J Clin Rheumatol. 2017Jun;23(4):200-6
42. Ahlehoff O, Wu JJ, Raunsø J, Kristensen SL, Khalid U, Kofoed K. et al. Cutaneous lupus erythematosus and the risk of deep venous thrombosis and pulmonary embolism: A Danish nationwide cohort study. Lupus. 2017Nov;26(13):1435-9
43. Li Y, Wang P, Li L, Wang F, Liu Y. Increased risk of venous thromboembolism associated with polymyositis and dermatomyositis: a meta-analysis. Ther Clin Risk Manag. 2018;14:157-65
44. Lee YH, Song GG. Idiopathic inflammatory myopathy and the risk of venous thromboembolism: a meta-analysis. Rheumatol Int. 2017Jul;37(7):1165-73
45. Carruthers EC, Choi HK, Sayre EC, Aviña-Zubieta JA. Risk of deep venous thrombosis and pulmonary embolism in individuals with polymyositis and dermatomyositis: a general population-based study. Ann Rheum Dis. 2016Jan;75(1):110-6
46. Merkel PA, Lo GH, Holbrook JT, Tibbs AK, Allen NB, Davis JC. et al. Brief communication: high incidence of venous thrombotic events among patients with Wegener granulomatosis: the Wegener's Clinical Occurrence of Thrombosis (WeCLOT) Study. Ann Intern Med. 2005Apr19;142(8):620-6
47. Choi HK, Rho Y-H, Zhu Y, Cea-Soriano L, Aviña-Zubieta JA, Zhang Y. The risk of pulmonary embolism and deep vein thrombosis in rheumatoid arthritis: a UK population-based outpatient cohort study. Ann Rheum Dis. 2013Jul;72(7):1182-7
48. Chung W-S, Lin C-L, Sung F-C, Hsu W-H, Yang W-T, Lu C-C. et al. Systemic sclerosis increases the risks of deep vein thrombosis and pulmonary thromboembolism: a nationwide cohort study. Rheumatology (Oxford). 2014Sep;53(9):1639-45
49. Ungprasert P, Srivali N, Kittanamongkolchai W. Systemic sclerosis and risk of venous thromboembolism: A systematic review and meta-analysis. Mod Rheumatol. 2015Nov;25(6):893-7
50. Schoenfeld SR, Choi HK, Sayre EC, Aviña-Zubieta JA. Risk of Pulmonary Embolism and Deep Venous Thrombosis in Systemic Sclerosis: A General Population-Based Study. Arthritis Care Res (Hoboken). 2016Feb;68(2):246-53
51. Johnson SR, Hakami N, Ahmad Z, Wijeysundera DN. Venous Thromboembolism in Systemic Sclerosis: Prevalence, Risk Factors, and Effect on Survival. J Rheumatol. 2018Jul;45(7):942-6
52. Aviña-Zubieta JA, Jansz M, Sayre EC, Choi HK. The Risk of Deep Venous Thrombosis and Pulmonary Embolism in Primary Sjögren Syndrome: A General Population-based Study. J Rheumatol. 2017;44(8):1184-9
53. Ungprasert P, Crowson CS, Matteson EL. Association of Sarcoidosis With Increased Risk of VTE: A Population-Based Study, 1976 to 2013. Chest. 2017;151(2):425-30
54. Ungprasert P, Srivali N, Wijarnpreecha K, Thongprayoon C. Sarcoidosis and risk of venous thromboembolism: A systematic review and meta-analysis. Sarcoidosis Vasc Diffuse Lung Dis. 2015Sep14;32(3):182-7
55. Kizer JR, Zisman DA, Blumenthal NP, Kotloff RM, Kimmel SE, Strieter RM. et al. Association between pulmonary fibrosis and coronary artery disease. Arch Intern Med. 2004Mar8;164(5):551-6
56. Nathan SD, Basavaraj A, Reichner C, Shlobin OA, Ahmad S, Kiernan J. et al. Prevalence and impact of coronary artery disease in idiopathic pulmonary fibrosis. Respir Med. 2010Jul;104(7):1035-41
57. Cicchitto G, Musella V, Acitorio M, Capuano N, Fiorenzano G, Owen CA. et al. Idiopathic pulmonary fibrosis and coronary artery disease. Multidiscip Respir Med. 2014;9(1):31
58. Nathan SD, Weir N, Shlobin OA, Urban BA, Curry CA, Basavaraj A. et al. The value of computed tomography scanning for the detection of coronary artery disease in patients with idiopathic pulmonary fibrosis. Respirology. 2011Apr;16(3):481-6
59. Hubbard R, Venn A, Lewis S, Britton J. Lung cancer and cryptogenic fibrosing alveolitis. A population-based cohort study. Am J Respir Crit Care Med. 2000Jan;161(1):5-8
60. Tomassetti S, Gurioli C, Ryu JH, Decker PA, Ravaglia C, Tantalocco P. et al. The impact of lung cancer on survival of idiopathic pulmonary fibrosis. Chest. 2015Jan;147(1):157-64
61. Girard N, Marchand-Adam S, Naccache J-M, Borie R, Urban T, Jouneau S. et al. Lung cancer in combined pulmonary fibrosis and emphysema: a series of 47 Western patients. J Thorac Oncol. 2014Aug;9(8):1162-70
62. Usui K, Tanai C, Tanaka Y, Noda H, Ishihara T. The prevalence of pulmonary fibrosis combined with emphysema in patients with lung cancer. Respirology. 2011Feb;16(2):326-31
63. Yamaguchi S, Ohguri T, Ide S, Aoki T, Imada H, Yahara K. et al. Stereotactic body radiotherapy for lung tumors in patients with subclinical interstitial lung disease: the potential risk of extensive radiation pneumonitis. Lung Cancer. 2013Nov;82(2):260-5
64. Minegishi Y, Kuribayashi H, Kitamura K, Mizutani H, Kosaihira S, Okano T. et al. The feasibility study of Carboplatin plus Etoposide for advanced small cell lung cancer with idiopathic interstitial pneumonias. J Thorac Oncol. 2011Apr;6(4):801-7
65. Minegishi Y, Sudoh J, Kuribayasi H, Mizutani H, Seike M, Azuma A. et al. The safety and efficacy of weekly paclitaxel in combination with carboplatin for advanced non-small cell lung cancer with idiopathic interstitial pneumonias. Lung Cancer. 2011Jan;71(1):70-4
66. Lancaster LH, Mason WR, Parnell JA, Rice TW, Loyd JE, Milstone AP. et al. Obstructive sleep apnea is common in idiopathic pulmonary fibrosis. Chest. 2009Sep;136(3):772-8
67. Pihtili A, Bingol Z, Kiyan E, Cuhadaroglu C, Issever H, Gulbaran Z. Obstructive sleep apnea is common in patients with interstitial lung disease. Sleep Breath. 2013Dec;17(4):1281-8
68. Mermigkis C, Stagaki E, Tryfon S, Schiza S, Amfilochiou A, Polychronopoulos V. et al. How common is sleep-disordered breathing in patients with idiopathic pulmonary fibrosis? Sleep Breath. 2010Dec;14(4):387-90
69. Mavroudi M, Papakosta D, Kontakiotis T, Domvri K, Kalamaras G, Zarogoulidou V. et al. Sleep disorders and health-related quality of life in patients with interstitial lung disease. Sleep Breath. 2018;22(2):393-400
70. Turner GA, Lower EE, Corser BC, Gunther KL, Baughman RP. Sleep apnea in sarcoidosis. Sarcoidosis Vasc Diffuse Lung Dis. 1997Mar;14(1):61-4
71. Bingol Z, Pihtili A, Gulbaran Z, Kiyan E. Relationship between parenchymal involvement and obstructive sleep apnea in subjects with sarcoidosis. Clin Respir J. 2015Jan;9(1):14-21
72. Schiza S, Mermigkis C, Margaritopoulos GA, Daniil Z, Harari S, Poletti V. et al. Idiopathic pulmonary fibrosis and sleep disorders: no longer strangers in the night. Eur Respir Rev. 2015Jun;24(136):327-39
73. Mermigkis C, Chapman J, Golish J, Mermigkis D, Budur K, Kopanakis A. et al. Sleep-related breathing disorders in patients with idiopathic pulmonary fibrosis. Lung. 2007Jun;185(3):173-8
74. Kolilekas L, Manali E, Vlami KA, Lyberopoulos P, Triantafillidou C, Kagouridis K. et al. Sleep oxygen desaturation predicts survival in idiopathic pulmonary fibrosis. J Clin Sleep Med. 2013Jun15;9(6):593-601
75. Bosi M, Milioli G, Fanfulla F, Tomassetti S, Ryu JH, Parrino L. et al. OSA and Prolonged Oxygen Desaturation During Sleep are Strong Predictors of Poor Outcome in IPF. Lung. 2017Oct;195(5):643-51
76. Mikamo M, Fujisawa T, Oyama Y, Kono M, Enomoto N, Nakamura Y. et al. Clinical Significance of Forced Oscillation Technique for Evaluation of Small Airway Disease in Interstitial Lung Diseases. Lung. 2016;194(6):975-83
77. Guerrero Zúñiga S, Sánchez Hernández J, Mateos Toledo H, Mejía Ávila M, Gochicoa-Rangel L, Miguel Reyes JL. et al. Small airway dysfunction in chronic hypersensitivity pneumonitis. Respirology. 2017;22(8):1637-42
78. Vassallo R, Harari S, Tazi A. Current understanding and management of pulmonary Langerhans cell histiocytosis. Thorax. 2017;72(10):937-45
79. Baldi BG, de Albuquerque ALP, Pimenta SP, Salge JM, Kairalla RA, Carvalho CRR. A pilot study assessing the effect of bronchodilator on dynamic hyperinflation in LAM. Respir Med. 2013Nov;107(11):1773-80
80. Le K, Steagall WK, Stylianou M, Pacheco-Rodriguez G, Darling TN, Vaughan M. et al. Effect of beta-agonists on LAM progression and treatment. Proc Natl Acad Sci USA. 2018:30 115(5):E944-53
81. Taguchi Y, Ebina M, Hashimoto S, Ogura T, Azuma A, Taniguchi H. et al. Efficacy of pirfenidone and disease severity of idiopathic pulmonary fibrosis: Extended analysis of phase III trial in Japan. Respir Investig. 2015Nov;53(6):279-87
82. Harari S, Caminati A, Albera C, Vancheri C, Poletti V, Pesci A. et al. Efficacy of pirfenidone for idiopathic pulmonary fibrosis: An Italian real life study. Respir Med. 2015Jul;109(7):904-13
83. Tzouvelekis A, Ntolios P, Karampitsakos T, Tzilas V, Anevlavis S, Bouros E. et al. Safety and efficacy of pirfenidone in severe Idiopathic Pulmonary Fibrosis: A real-world observational study. Pulm Pharmacol Ther. 2017Oct;46:48-53
84. Abe M, Tsushima K, Sakayori M, Suzuki K, Ikari J, Terada J. et al. Utility of nintedanib for severe idiopathic pulmonary fibrosis: a single-center retrospective study. Drug Des Devel Ther. 2018;12:3369-75
85. Harari S, Caminati A, Poletti V, Confalonieri M, Gasparini S, Lacedonia D. et al. A Real-Life Multicenter National Study on Nintedanib in Severe Idiopathic Pulmonary Fibrosis. Respiration. 2018;95(6):433-40
86. Wuyts WA, Kolb M, Stowasser S, Stansen W, Huggins JT, Raghu G. First Data on Efficacy and Safety of Nintedanib in Patients with Idiopathic Pulmonary Fibrosis and Forced Vital Capacity of ≤50 % of Predicted Value. Lung. 2016;194(5):739-43
87. Nathan SD, Albera C, Bradford WZ, Costabel U, du Bois RM, Fagan EA. et al. Effect of continued treatment with pirfenidone following clinically meaningful declines in forced vital capacity: analysis of data from three phase 3 trials in patients with idiopathic pulmonary fibrosis. Thorax. 2016;71(5):429-35
88. Crockett AJ, Cranston JM, Antic N. Domiciliary oxygen for interstitial lung disease. Cochrane Database Syst Rev. 2001(3):CD002883
89. Dowman LM, McDonald CF, Bozinovski S, Vlahos R, Gillies R, Pouniotis D. et al. Greater endurance capacity and improved dyspnoea with acute oxygen supplementation in idiopathic pulmonary fibrosis patients without resting hypoxaemia. Respirology. 2017;22(5):957-64
90. Ryerson CJ, Donesky D, Pantilat SZ, Collard HR. Dyspnea in idiopathic pulmonary fibrosis: a systematic review. J Pain Symptom Manage. 2012Apr;43(4):771-82
91. Egan JJ. Follow-up and nonpharmacological management of the idiopathic pulmonary fibrosis patient. Eur Respir Rev. 2011Jun;20(120):114-7
92. Bell EC, Cox NS, Goh N, Glaspole I, Westall GP, Watson A. et al. Oxygen therapy for interstitial lung disease: a systematic review. Eur Respir Rev. 2017 Jan;26(143)
93. Khor YH, Goh NSL, McDonald CF, Holland AE. Oxygen Therapy for Interstitial Lung Disease. A Mismatch between Patient Expectations and Experiences. Ann Am Thorac Soc. 2017Jun;14(6):888-95
94. Khor YH, Goh NSL, McDonald CF, Holland AE. Oxygen Therapy for Interstitial Lung Disease: Physicians' Perceptions and Experiences. Ann Am Thorac Soc. 2017Dec;14(12):1772-8
95. Visca D, Mori L, Tsipouri V, Fleming S, Firouzi A, Bonini M. et al. Effect of ambulatory oxygen on quality of life for patients with fibrotic lung disease (AmbOx): a prospective, open-label, mixed-method, crossover randomised controlled trial. Lancet Respir Med. 2018 Aug 28
96. Spoletini G, Alotaibi M, Blasi F, Hill NS. Heated Humidified High-Flow Nasal Oxygen in Adults: Mechanisms of Action and Clinical Implications. Chest. 2015Jul;148(1):253-61
97. Bräunlich J, Beyer D, Mai D, Hammerschmidt S, Seyfarth H-J, Wirtz H. Effects of nasal high flow on ventilation in volunteers, COPD and idiopathic pulmonary fibrosis patients. Respiration. 2013;85(4):319-25
98. Chatila W, Nugent T, Vance G, Gaughan J, Criner GJ. The effects of high-flow vs low-flow oxygen on exercise in advanced obstructive airways disease. Chest. 2004Oct;126(4):1108-15
99. Peters SG, Holets SR, Gay PC. High-flow nasal cannula therapy in do-not-intubate patients with hypoxemic respiratory distress. Respir Care. 2013Apr;58(4):597-600
100. Koyauchi T, Hasegawa H, Kanata K, Kakutani T, Amano Y, Ozawa Y. et al. Efficacy and Tolerability of High-Flow Nasal Cannula Oxygen Therapy for Hypoxemic Respiratory Failure in Patients with Interstitial Lung Disease with Do-Not-Intubate Orders: A Retrospective Single-Center Study. Respiration. 2018 Jun 28;1-7
101. Koschel D, Handzhiev S, Wiedemann B, Höffken G. Acute effects of NPPV in interstitial lung disease with chronic hypercapnic respiratory failure. Respir Med. 2010Feb;104(2):291-5
102. Quill CM, Quill TE. Palliative use of noninvasive ventilation: navigating murky waters. J Palliat Med. 2014Jun;17(6):657-61
103. Rajala K, Lehto JT, Saarinen M, Sutinen E, Saarto T, Myllärniemi M. End-of-life care of patients with idiopathic pulmonary fibrosis. BMC Palliat Care. 2016Oct12;15(1):85
104. Moderno EV, Yamaguti WPS, Schettino GPP, Kairalla RA, Martins MA, Carvalho CRR. et al. Effects of proportional assisted ventilation on exercise performance in idiopathic pulmonary fibrosis patients. Respir Med. 2010Jan;104(1):134-41
105. Cancer pain relief and palliative care. Report of a WHO Expert Committee. World Health Organ Tech Rep Ser. 1990;804:1-75
106. Lindell KO, Liang Z, Hoffman LA, Rosenzweig MQ, Saul MI, Pilewski JM. et al. Palliative care and location of death in decedents with idiopathic pulmonary fibrosis. Chest. 2015Feb;147(2):423-9
107. Bajwah S, Higginson IJ, Ross JR, Wells AU, Birring SS, Riley J. et al. The palliative care needs for fibrotic interstitial lung disease: a qualitative study of patients, informal caregivers and health professionals. Palliat Med. 2013Oct;27(9):869-76
108. Bajwah S, Koffman J, Higginson IJ, Ross JR, Wells AU, Birring SS. et al. “I wish I knew more..” the end-of-life planning and information needs for end-stage fibrotic interstitial lung disease: views of patients, carers and health professionals. BMJ Support Palliat Care. 2013Mar;3(1):84-90
109. Wijsenbeek M, Bendstrup E, Ross J, Wells A. Cultural Differences in Palliative Care in Patients With Idiopathic Pulmonary Fibrosis. Chest. 2015Aug;148(2):e56
110. Bajwah S, Ross JR, Wells AU, Mohammed K, Oyebode C, Birring SS. et al. Palliative care for patients with advanced fibrotic lung disease: a randomised controlled phase II and feasibility trial of a community case conference intervention. Thorax. 2015Sep;70(9):830-9
111. Carvajalino S, Reigada C, Johnson MJ, Dzingina M, Bajwah S. Symptom prevalence of patients with fibrotic interstitial lung disease: a systematic literature review. BMC Pulm Med. 2018May22;18(1):78
112. Ahmadi Z, Wysham NG, Lundström S, Janson C, Currow DC, Ekström M. End-of-life care in oxygen-dependent ILD compared with lung cancer: a national population-based study. Thorax. 2016;71(6):510-6
113. Kreuter M, Swigris J, Pittrow D, Geier S, Klotsche J, Prasse A. et al. Health related quality of life in patients with idiopathic pulmonary fibrosis in clinical practice: insights-IPF registry. Respir Res. 2017:14 18(1):139
114. Yount SE, Beaumont JL, Chen S-Y, Kaiser K, Wortman K, Van Brunt DL. et al. Health-Related Quality of Life in Patients with Idiopathic Pulmonary Fibrosis. Lung. 2016Apr;194(2):227-34
115. Yorke J, Moosavi SH, Shuldham C, Jones PW. Quantification of dyspnoea using descriptors: development and initial testing of the Dyspnoea-12. Thorax. 2010Jan;65(1):21-6
116. Meek PM, Banzett R, Parsall MB, Gracely RH, Schwartzstein RM, Lansing R. Reliability and validity of the multidimensional dyspnea profile. Chest. 2012Jun;141(6):1546-53
117. Patel AS, Siegert RJ, Brignall K, Gordon P, Steer S, Desai SR. et al. The development and validation of the King's Brief Interstitial Lung Disease (K-BILD) health status questionnaire. Thorax. 2012Sep;67(9):804-10
118. Yorke J, Swigris J, Russell A-M, Moosavi SH, Ng Man Kwong G, Longshaw M. et al. Dyspnea-12 is a valid and reliable measure of breathlessness in patients with interstitial lung disease. Chest. 2011Jan;139(1):159-64
119. O'Donnell DE, Aaron S, Bourbeau J, Hernandez P, Marciniuk DD, Balter M. et al. Canadian Thoracic Society recommendations for management of chronic obstructive pulmonary disease - 2007 update. Can Respir J. 2007 Sep;14 Suppl B:5B-32B
120. Mahler DA, Selecky PA, Harrod CG, Benditt JO, Carrieri-Kohlman V, Curtis JR. et al. American College of Chest Physicians consensus statement on the management of dyspnea in patients with advanced lung or heart disease. Chest. 2010Mar;137(3):674-91
121. Marciniuk DD, Goodridge D, Hernandez P, Rocker G, Balter M, Bailey P. et al. Managing dyspnea in patients with advanced chronic obstructive pulmonary disease: a Canadian Thoracic Society clinical practice guideline. Can Respir J. 2011Apr;18(2):69-78
122. Frankel SK, Schwarz MI. Update in idiopathic pulmonary fibrosis. Curr Opin Pulm Med. 2009Sep;15(5):463-9
123. Kreuter M, Bendstrup E, Russell A-M, Bajwah S, Lindell K, Adir Y. et al. Palliative care in interstitial lung disease: living well. Lancet Respir Med. 2017;5(12):968-80
124. van Manen MJG, Geelhoed JJM, Tak NC, Wijsenbeek MS. Optimizing quality of life in patients with idiopathic pulmonary fibrosis. Ther Adv Respir Dis. 2017;11(3):157-69
125. Kohberg C, Andersen CU, Bendstrup E. Opioids: an unexplored option for treatment of dyspnea in IPF. Eur Clin Respir J. 2016;3:30629
126. Allen S, Raut S, Woollard J, Vassallo M. Low dose diamorphine reduces breathlessness without causing a fall in oxygen saturation in elderly patients with end-stage idiopathic pulmonary fibrosis. Palliat Med. 2005Mar;19(2):128-30
127. Hadjiphilippou S, Odogwu S-E, Dand P. Doctors' attitudes towards prescribing opioids for refractory dyspnoea: a single-centred study. BMJ Support Palliat Care. 2014Jun;4(2):190-2
128. Spruit MA, Singh SJ, Garvey C, ZuWallack R, Nici L, Rochester C. et al. An official American Thoracic Society/European Respiratory Society statement: key concepts and advances in pulmonary rehabilitation. Am J Respir Crit Care Med. 2013Oct15;188(8):e13-64
129. Rochester CL, Vogiatzis I, Holland AE, Lareau SC, Marciniuk DD, Puhan MA. et al. An Official American Thoracic Society/European Respiratory Society Policy Statement: Enhancing Implementation, Use, and Delivery of Pulmonary Rehabilitation. Am J Respir Crit Care Med. 2015Dec1;192(11):1373-86
130. Holland AE, Wadell K, Spruit MA. How to adapt the pulmonary rehabilitation programme to patients with chronic respiratory disease other than COPD. Eur Respir Rev. 2013Dec;22(130):577-86
131. Galizia G, Balestrieri G, De Maria B, La Storia C, Monelli M, Salvaderi S. et al. Role of rehabilitation in the elderly after an acute event: insights from a real-life prospective study in the sub-acute care setting. Eur J Phys Rehabil Med. 2018 Jun 11
132. Arena R. Exercise testing and training in chronic lung disease and pulmonary arterial hypertension. Prog Cardiovasc Dis. 2011Jun;53(6):454-63
133. Dowman L, Hill CJ, Holland AE. Pulmonary rehabilitation for interstitial lung disease. Cochrane Database Syst Rev. 2014Oct6(10):CD006322
134. Ferreira A, Garvey C, Connors GL, Hilling L, Rigler J, Farrell S. et al. Pulmonary rehabilitation in interstitial lung disease: benefits and predictors of response. Chest. 2009Feb;135(2):442-7
135. Tonelli R, Cocconcelli E, Lanini B, Romagnoli I, Florini F, Castaniere I. et al. Effectiveness of pulmonary rehabilitation in patients with interstitial lung disease of different etiology: a multicenter prospective study. BMC Pulm Med. 2017Oct10;17(1):130
136. Vainshelboim B, Oliveira J, Yehoshua L, Weiss I, Fox BD, Fruchter O. et al. Exercise training-based pulmonary rehabilitation program is clinically beneficial for idiopathic pulmonary fibrosis. Respiration. 2014;88(5):378-88
137. Ozalevli S, Karaali HK, Ilgin D, Ucan ES. Effect of home-based pulmonary rehabilitation in patients with idiopathic pulmonary fibrosis. Multidiscip Respir Med. 2010Feb28;5(1):31-7
138. Holland AE, Hill CJ, Conron M, Munro P, McDonald CF. Short term improvement in exercise capacity and symptoms following exercise training in interstitial lung disease. Thorax. 2008Jun;63(6):549-54
139. Nishiyama O, Kondoh Y, Kimura T, Kato K, Kataoka K, Ogawa T. et al. Effects of pulmonary rehabilitation in patients with idiopathic pulmonary fibrosis. Respirology. 2008May;13(3):394-9
140. Rammaert B, Leroy S, Cavestri B, Wallaert B, Grosbois J-M. Home-based pulmonary rehabilitation in idiopathic pulmonary fibrosis. Rev Mal Respir. 2011Sep;28(7):e52-57
141. Dowman LM, McDonald CF, Hill CJ, Lee AL, Barker K, Boote C. et al. The evidence of benefits of exercise training in interstitial lung disease: a randomised controlled trial. Thorax. 2017;72(7):610-9
142. Vainshelboim B, Oliveira J, Fox BD, Soreck Y, Fruchter O, Kramer MR. Long-term effects of a 12-week exercise training program on clinical outcomes in idiopathic pulmonary fibrosis. Lung. 2015Jun;193(3):345-54
143. Ryerson CJ, Cayou C, Topp F, Hilling L, Camp PG, Wilcox PG. et al. Pulmonary rehabilitation improves long-term outcomes in interstitial lung disease: a prospective cohort study. Respir Med. 2014Jan;108(1):203-10
144. Huppmann P, Sczepanski B, Boensch M, Winterkamp S, Schönheit-Kenn U, Neurohr C. et al. Effects of inpatient pulmonary rehabilitation in patients with interstitial lung disease. Eur Respir J. 2013Aug;42(2):444-53
145. Kozu R, Senjyu H, Jenkins SC, Mukae H, Sakamoto N, Kohno S. Differences in response to pulmonary rehabilitation in idiopathic pulmonary fibrosis and chronic obstructive pulmonary disease. Respiration. 2011;81(3):196-205
146. Swigris JJ, Fairclough DL, Morrison M, Make B, Kozora E, Brown KK. et al. Benefits of pulmonary rehabilitation in idiopathic pulmonary fibrosis. Respir Care. 2011Jun;56(6):783-9
147. Swigris JJ, Wamboldt FS, Behr J, du Bois RM, King TE, Raghu G. et al. The 6 minute walk in idiopathic pulmonary fibrosis: longitudinal changes and minimum important difference. Thorax. 2010Feb;65(2):173-7
148. Steffen TM, Hacker TA, Mollinger L. Age- and gender-related test performance in community-dwelling elderly people: Six-Minute Walk Test, Berg Balance Scale, Timed Up & Go Test, and gait speeds. Phys Ther. 2002Feb;82(2):128-37
149. Nolan CM, Maddocks M, Maher TM, Canavan JL, Jones SE, Barker RE. et al. Phenotypic characteristics associated with slow gait speed in idiopathic pulmonary fibrosis. Respirology. 2018;23(5):498-506
150. Dale MT, McKeough ZJ, Munoz PA, Corte P, Bye PTP, Alison JA. Exercise training for asbestos-related and other dust-related respiratory diseases: a randomised controlled trial. BMC Pulm Med. 2014Nov18;14:180
151. Kenn K, Gloeckl R, Behr J. Pulmonary rehabilitation in patients with idiopathic pulmonary fibrosis-a review. Respiration. 2013;86(2):89-99
152. Holland AE, Hill CJ, Glaspole I, Goh N, McDonald CF. Predictors of benefit following pulmonary rehabilitation for interstitial lung disease. Respir Med. 2012Mar;106(3):429-35
153. Kozu R, Jenkins S, Senjyu H. Effect of disability level on response to pulmonary rehabilitation in patients with idiopathic pulmonary fibrosis. Respirology. 2011Nov;16(8):1196-202
154. Jackson RM, Gómez-Marín OW, Ramos CF, Sol CM, Cohen MI, Gaunaurd IA. et al. Exercise limitation in IPF patients: a randomized trial of pulmonary rehabilitation. Lung. 2014Jun;192(3):367-76
155. Vainshelboim B, Kramer MR, Izhakian S, Lima RM, Oliveira J. Physical Activity and Exertional Desaturation Are Associated with Mortality in Idiopathic Pulmonary Fibrosis. J Clin Med. 2016 Aug 18;5(8)
156. Johnson-Warrington V, Williams J, Bankart J, Steiner M, Morgan M, Singh S. Pulmonary rehabilitation and interstitial lung disease: aiding the referral decision. J Cardiopulm Rehabil Prev. 2013Jun;33(3):189-95
157. Andersen CU, Mellemkjær S, Hilberg O, Nielsen-Kudsk JE, Simonsen U, Bendstrup E. Pulmonary hypertension in interstitial lung disease: prevalence, prognosis and 6 min walk test. Respir Med. 2012Jun;106(6):875-82
158. Gaunaurd IA, Gómez-Marín OW, Ramos CF, Sol CM, Cohen MI, Cahalin LP. et al. Physical activity and quality of life improvements of patients with idiopathic pulmonary fibrosis completing a pulmonary rehabilitation program. Respir Care. 2014Dec;59(12):1872-9
159. Qiu S, Cai X, Wang X, He C, Zügel M, Steinacker JM. et al. Using step counters to promote physical activity and exercise capacity in patients with chronic obstructive pulmonary disease: a meta-analysis. Ther Adv Respir Dis. 2018Dec;12:1753466618787386
160. Ikezoe K, Handa T, Tanizawa K, Chen-Yoshikawa TF, Kubo T, Aoyama A. et al. Prognostic factors and outcomes in Japanese lung transplant candidates with interstitial lung disease. PLoS ONE. 2017;12(8):e0183171
161. Date H. Current status and problems of lung transplantation in Japan. J Thorac Dis. 2016Aug;8(Suppl 8):S631-636
162. Sato M, Okada Y, Oto T, Minami M, Shiraishi T, Nagayasu T. et al. Registry of the Japanese Society of Lung and Heart-Lung Transplantation: official Japanese lung transplantation report, 2014. Gen Thorac Cardiovasc Surg. 2014Oct;62(10):594-601
163. Smith PJ, Blumenthal JA, Trulock EP, Freedland KE, Carney RM, Davis RD. et al. Psychosocial Predictors of Mortality Following Lung Transplantation. Am J Transplant. 2016Jan;16(1):271-7
164. Brown AW, Kaya H, Nathan SD. Lung transplantation in IIP: A review. Respirology. 2016;21(7):1173-84
165. Park JE, Kim SY, Song JH, Kim YS, Chang J, Lee JG. et al. Comparison of short-term outcomes for connective tissue disease-related interstitial lung disease and idiopathic pulmonary fibrosis after lung transplantation. J Thorac Dis. 2018Mar;10(3):1538-47
Author contact
![]() Corresponding author: Paola Faverio, MD, Cardio-Thoracic-Vascular Department, University of Milan Bicocca, Respiratory Unit, San Gerardo Hospital, ASST di Monza, Via Pergolesi 33, 20900, Monza, Italy; E-mail: paola.faverioit; Tel: +393382185092; Fax: +390392336660
Corresponding author: Paola Faverio, MD, Cardio-Thoracic-Vascular Department, University of Milan Bicocca, Respiratory Unit, San Gerardo Hospital, ASST di Monza, Via Pergolesi 33, 20900, Monza, Italy; E-mail: paola.faverioit; Tel: +393382185092; Fax: +390392336660