Impact Factor ISSN: 1449-1907

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2019; 16(2):302-310. doi:10.7150/ijms.27829 This issue Cite
Research Paper
Imbalance of erythropoiesis and iron metabolism in patients with thalassemia
Yumei Huang1*, Yu Lei2*, Rongrong Liu1, Jiaodi Liu1, Gaohui Yang1, Zhifu Xiang3, Yuzhen Liang4, Yongrong Lai1 ![]()
1. Department of Hematology, the First Affiliated Hospital of Guangxi Medical University, Nanning, Guangxi, China
2. Department of Hematology, the First Affiliated Hospital of Guangxi University of Chinese Medicine, Guangxi, China
3. Division of Hematology/Oncology, University of Arkansas for Medical Sciences, Little Rock, AR, USA
4. Department of endocrinology, the Second Affiliated Hospital of Guangxi Medical University, Nanning , Guangxi, China
*Equal contributors
Received 2018-6-12; Accepted 2018-12-5; Published 2019-1-1
Abstract
Aim: This study aimed to evaluate the imbalance of erythropoiesis and iron metabolism in patients with thalassemia.
Methods: 192 patients with non-transfusion-dependent thalassemia (NTDT), 94 patients with transfusion-dependent thalassemia (TDT) and 101 healthy controls were recruited between June 2013 and December 2016 in the Hematology Department, the First Affiliated Hospital of Guangxi Medical University. The groups were compared in terms of levels of erythropoiesis biomarkers [growth differentiation factor 15 (GDF15), erythropoietin (EPO) and soluble transferrin receptor (sTfR)] and of iron overload biomarkers [serum ferritin (SF), liver iron concentration (LIC) and cardiac T2*] and hepcidin.
Results: The levels of GDF15, EPO, sTfR, LIC and SF were significantly higher in patients with thalassemia. The levels of GDF15 and EPO were significantly higher in patients with TDT compared to NTDT. Those with iron overload had higher EPO, GDF15, SF and sTfR levels compared with non-iron overload patients. Hepcidin levels and ratios of hepcidin to erythropoietic activity and to iron biomarker levels were lower in patients with β-thalassemia intermedia or hemoglobin (Hb) E/β-thalassemia than in patients with HbH disease. The hepcidin levels were correlated negatively with the levels of EPO, GDF15 and sTfR in patients with NTDT and TDT, but correlated positively with SF and Hb levels only in patients with TDT.
Conclusions: Patients with thalassemia showed iron overload, reduced hepcidin levels, and a greater extent of ineffective erythropoiesis. The hepcidin levels were more strongly related to ineffective erythropoiesis compared with iron overload. The imbalance between erythropoiesis and iron metabolism differed across different thalassemia types.
Keywords: Erythropoietic activity, hepcidin, iron overload, thalassemia
Introduction
Patients with non-transfusion-dependent thalassemia (NTDT) do not require lifelong, regular transfusions for survival. However, they may require occasional or frequent transfusions in certain clinical settings, usually for defined periods of time [1]. This type of thalassemia encompasses three clinically distinct forms: β-thalassemia intermedia, hemoglobin E/β-thalassemia (mild or moderate) and α-thalassemia intermedia, also known as hemoglobin H (HbH) disease [1, 2]. In contrast, patients with transfusion-dependent thalassemia (TDT) require regular, lifelong transfusions for survival.
Mortality and morbidity in patients with thalassemia result primarily from iron overload toxicity. In patients with NTDT, iron overload arises when ineffective erythropoiesis leads to inappropriately low hepcidin levels and increased absorption of intestinal iron [1, 3]. Hepcidin is a hormone, secreted by the liver, that regulates iron homeostasis [4]. In contrast, as humans have little ability to excrete iron, iron overload in patients with TDT arises when frequent blood transfusion leads to iron deposition in the liver and heart [1].
The combination of ineffective erythropoiesis and peripheral hemolysis leads to anemia, hypoxia and increased erythropoietin (EPO) production [5, 6]. In β-thalassemias, ineffective erythropoiesis induces the release of growth differentiating factor 15 (GDF15), twisted gastrulation protein homolog 1 (TWSG1), hypoxia-inducible factor and erythroferrone (ERFE), which inhibits hepcidin [5]. The expansion of the erythroid compartment leads to the overexpression of GDF15, which inhibits the expression of hepcidin, ultimately leading to iron overload [7]. A soluble form of the transferrin receptor (sTfR) is an erythropoiesis biomarker and is associated with overall morbidity in patients with NTDT [8].
Hence, thalassemias involve the perturbation of the balance between erythropoiesis and iron metabolism. The nature of the 'erythropoiesis-hepcidin- iron storage' axis may differ across different thalassemia types [9], and this axis is poorly understood in patients with NTDT, particularly in Southern China, where HbH disease is often associated with high morbidity.
The levels of erythropoietic biomarkers (GDF15, EPO and sTfR) and iron overload biomarkers [hepcidin, serum ferritin (SF), liver iron concentration (LIC) and cardiac T2*] were compared in patients with different types of thalassemia, as well as between patients and healthy controls, and several correlations were analyzed to shed light on the relationship between erythropoietic activity and iron metabolism in patients with thalassemia.
Methods
Study population
This cross-sectional study included 192 patients with NTDT and 94 patients with TDT who were diagnosed based on conventional clinical and hematologic criteria [1, 10]. The patients were enrolled between June 2013 and December 2016 in the Department of Hematology at the First Affiliated Hospital of Guangxi Medical University. In addition, 101 healthy volunteers were recruited from the medical examination center of the same hospital. All patients or their parents (if minors) provided written informed consent. This study was approved by the Medical Ethics Committee of the First Affiliated Hospital of Guangxi Medical University.
Clinical assessment
Interviews were conducted with patients to obtain the following clinical data: age, gender, age at the time of anemia diagnosis, blood transfusion history and previous chelation therapy. Transfusional iron intake mg Fe/kg/day was calculated according to the following formula:
[Whole blood (mL)*hematocrit% (65%)*1.08] / [365*kg of body weight]
Hematology and genotyping
Fasting venous blood samples were drawn by sterile venipuncture. Samples from patients receiving blood transfusion were drawn before transfusion of packed red blood cells. The hematologic analysis was performed using an automated blood cell analyzer (LH750 Beckman, USA). The SF levels were measured using an electro-chemiluminescence immunoassay (COBASE E601, Roche, USA), and the levels of hemoglobins, HbA, HbA2 and HbF were determined using the Bio-Rad Variant II high-performance liquid chromatography system. All patients were genotyped by the Shenzhen Huada Gene Medical Institute for 338 known mutations linked to α- or β-thalassemia.
Serum sampling and analysis
The serum was stored at -80°C for later batch analysis. Enzyme-linked immunosorbent assays were used to assay the serum levels of hepcidin-25, the main active peptide form of hepcidin (Bachem Group, CA, USA), sTfR and GDF15 (R&D Systems, MN, USA) and EPO (eBioscience, Vienna, Austria). Kits were used according to manufacturers' instructions. The range for GDF15 is 337-1060 pg/mL, the minimum detectable dose (MDD) is 2pg/mL and the coefficient of variability (CV) is 1.8-2.8%; the range for hepcidin is 0-25 ng/mL; the range for EPO is 1.6-100mIU/mL, the MDD is 0.17 mIU/mL and the CV is 6.2%; the range for sTfR is 95 - 111nmol/L, the MDD is 0.5nmol/L and the CV is 4.3-7.1%.
Analysis of LIC and myocardial iron deposition
LIC was measured using spin density projection-assisted R2 magnetic resonance imaging (1.5 T, Ferriscan-Resonance Health, Australia). Myocardial iron deposition was assessed using magnetic resonance imaging as previously described [11]. Cardiac T2* ≥20 ms was considered as a "conservative" normal value [12, 13]. LIC <3 mg/g (dry weight) was considered to be indicative of no significant iron load; 3-7 mg/g, mild load; LIC≥7 and <15 mg/g, moderate load; and ≥15 mg/g, severe load [11, 14].
Statistical analysis
Data were analyzed using SPSS 16.0 (IBM, IL, USA). The normal distribution was assessed using the Kolmogorov-Smirnov test. Data for continuous variables were reported as mean ± standard deviation or median (range); data for categorical variables were reported as frequency and percentage. The Student t test was used to compare the continuous data from two groups. Differences among three or more groups were assessed for significance using one-way analysis of variance (post hoc analysis was performed using least significant difference or Student-Newman-Keul test) or nonparametric tests (post hoc analysis was performed using Kruskal-Wallis test or Mann-Whitney U test). The chi-square test was used for categorical variables. A bivariate correlation analysis was performed using Pearson or Spearman correlation methods. A P value <0.05 was considered statistically significant.
Results
Patient characteristics
Most patients with NTDT had HbH disease (111, 57.8%) or β-thalassemia intermedia (59, 30.7%), while the remaining patients had Hb E/β-thalassemia (22, 11.5%). The patients with Hb E/β-thalassemia were younger those patients with NTDT, whereas age was similar between patients with HbH disease or β-thalassemia intermedia. Patients with TDT were significantly younger than patients with NTDT and controls.
The Hb levels were higher in healthy controls (132 g/L) than in patients with NTDT (85 g/L, P < 0.001) or those with transfusion-dependent disease (70.1 g/L, P < 0.001), and the difference between NTDT and TDT was statistically significant (P < 0.001) (Table 1). Among patients with non-transfusion-dependent disease, those with HbH disease had the highest Hb levels (Fig. 1). Age at first transfusion and transfusional iron intake differed significantly between patients with NTDT and those with TDT.
Characteristics and clinical parameters of patients with thalassemia and normal controls
| Variable | Controls | Patients | P | |
|---|---|---|---|---|
| NTDT | TDT | |||
| N | 101 | 192 | 94 | |
| Female n (%) | 43 (42.6%) | 82 (42.7%) | 39 (41.5%) | 0.243 |
| Median age (range), years | 20 (1-57) | 22 (1-63)★ | 7.0 (1-32)★ | <0.001 |
| Median Hb (range), g/L | 132(102-174) | 85 (33.8-132)★▲ | 70.1 (21.1-114.7)★● | <0.001 |
| Median Age at first transfusion (range), year | NA | 5 (1-45) | 1.4 (0.3-3) | 0.001 |
| Median total transfusion (range), mg/kg.year | NA | 2.26 (0-153.08) | 181.81 (41.03-419.05) | <0.001 |
| Previous chelation therapy [n(%)] | <0.001 | |||
| No | 101 | 157 (81.7%) | 26 (28%) | |
| Yes | None | 35 (18.3%) | 68 (72%) | |
| Serum ferritin, ng/mL [n(%)] | 101 | 192 | 94 | <0.001 |
| <1000 | 101(100%) | 115 (59.9) | 12 (12.8%) | |
| 1000-2500 | 0(0%) | 51 (26.6%) | 27 (28.7%) | |
| ≥2500 | 0(0%) | 26 (13.5%) | 55 (58.5%) | |
| Liver iron concentration, mg/g [n(%)] | None | 95 | 7 | 0.025 |
| <3.0 | 14 (14.7%) | 0 (0%) | ||
| 3.0-6.9 | 24 (25.3%) | 0 (0%) | ||
| 7-14.9 | 25 (26.3%) | 1 (14.3%) | ||
| ≥15 | 32 (33.7%) | 6 (85.7%) | ||
| Cardiac T2*, ms [n (%)] | None | 87 | 7 | <0.001 |
| <20 | 5 (5.7%) | 5 (71%) | ||
| ≥20 | 82 (94.3%) | 2 (29%) | ||
Abbreviations: NA, not available; NTDT, non-transfusion-dependent thalassemia; TDT, transfusion-dependent thalassemia. Significant difference between controls, patients with NTDT, and patients with TDT: ★significant difference compared with normal controls; ●significant difference compared with NTDT; ▲significant difference compared with TDT.
Comparison of indices of erythropoietic activity and iron overload in patients with NTDT. (A) Indices of erythropoietic activity; (B) indices of iron overload.
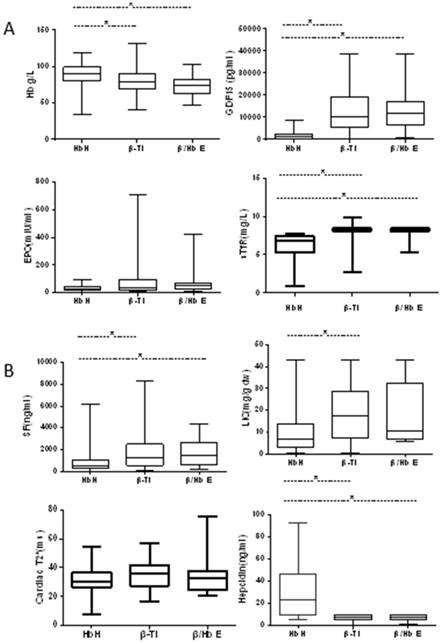
Greater ineffective erythropoiesis in patients with β-thalassemia
All patients showed higher erythropoietic activity compared with controls based on the levels of EPO, GDF15 and sTfR. The levels of EPO and GDF15 were significantly higher in patients with TDT than in patients with NTDT (P < 0.001 for EPO, P < 0.001 for GDF15) (Table 2). For patients with NTDT, the levels of GDF15 and sTfR were significantly higher in patients with β-thalassemia intermedia or Hb E/β-thalassemia than in patients with HbH disease, while the difference between patients with β-thalassemia intermedia and Hb E/β-thalassemia was not significant. Moreover, no difference in EPO levels was observed among patients with NTDT (Fig. 1).
Indices of erythropoietic activity and iron overload in patients with thalassemia and normal controls (median, range)
| Index | Controls | Patients | |
|---|---|---|---|
| NTDT | TDT | ||
| Hb, g/L | 132 (102-174) | 85 (33.8-132)★▲ | 70.1 (21.1-114.7)★● |
| Hepcidin, ng/mL | 115.7 (31.5-190.5) | 14.6 (0.0-136.7)★▲ | 31.6 (4.32-164.1)★● |
| EPO, mIU/mL | 4.4 (0.8-30.6) | 31.7 (1.73-1234.2)★▲ | 172.3 (9.0-1228.3)★● |
| GDF15, pg/mL | 141.6 (51.6-1448) | 2709.0 (77.91-38674.5)★▲ | 9647.6 (392.6-39043.8)★● |
| sTfR, mg/L | 1.6 (0.3-5.5) | 7.5 (0.9-9.87)★ | 7.9 (2.2-8.5)★ |
| Serum ferritin, ng/mL | 124.3 (30-726.6) | 718.5 (3.6-8300)★▲ | 2853 (136-26484)★● |
| Liver iron concentration, mg/g dw | NA | 9.9 (0.6-43)▲ | 43 (8.4-43)● |
| Cardiac T2*, ms | NA | 30.55 (7.46-75.08)▲ | 17 (11.6-40.6)● |
Abbreviations: EPO, Erythropoietin; GDF15, growth and differentiating factor 15; NA, not available; NTDT, nontransfusion-dependent thalassemia; sTFR, soluble form of the transferrin receptor; TDT, transfusion-dependent thalassemia;. Significant difference between controls, patients with NTDT, and patients with TDT: ★significant difference compared with controls; ●significant difference compared with NTDT; ▲significant difference compared with TDT.
Hepcidin suppression and iron overload in patients with thalassemia
The hepcidin levels in NTDT or TDT patient subgroups were significantly lower than those in healthy controls (14.6 vs 31.6 vs 115.7 ng/mL, P < 0.001); the levels were significantly higher in patients with TDT when compared to patients with NTDT (P = 0.03) (Table 2). For patients with NTDT, the levels of hepcidin were significantly lower in patients with β-thalassemia intermedia or Hb E/β-thalassemia than in patients with HbH disease. The hepcidin levels were similar between patients with β-thalassemia intermedia or Hb E/β-thalassemia (P = 0.836) (Fig. 1).
Iron overload was assessed based on SF level, LIC and cardiac T2* values. The latter two parameters were not measured in healthy controls due to economic reasons. Based on the magnetic resonance imaging (MRI) analysis of subsets of patients with HbH disease (55/111), β-thalassemia intermedia (28/59), Hb E/β-thalassemia (12/22), or TDT (7/94), the median SF levels were significantly higher in patients with thalassemia when compared to healthy controls, and in patients with TDT than in patients with NTDT (P < 0.001) (Table 2). For patients with NTDT, the median SF levels were significantly higher in patients with β-thalassemia intermedia (1453 ng/mL) and Hb E/β-thalassemia (2012 ng/mL) than in patients with HbH disease (553 ng/mL, P < 0.001). The levels were similar between patients with β-thalassemia intermedia or Hb E/β-thalassemia (P = 0.828) (Fig. 1).
The LIC levels were significantly higher in patients with TDT when compared to patients with NTDT (43 vs 9.9 mg/g dw, P = 0.001). For patients with NTDT, LIC levels were significantly higher in patients with β-thalassemia intermedia (14.8 mg/g dw) than in patients with HbH disease (7.2 mg/g dw, P = 0.004). The levels were similar between patients with β-thalassemia intermedia or Hb E/β-thalassemia (P = 0.661) and between patients with HbH disease and intermedia or Hb E/β-thalassemia (P = 0.06) (Fig. 1). Cardiac T2* values were significantly lower in patients with TDT (17 ms) than in patients with NTDT (30.55 ms, P = 0.002). The cardiac T2* values were not different among patients with NTDT (P = 0.124) (Fig. 1). These results indicated that nearly all patients with NTDT (85.3%) had adequate amounts of liver iron but few patients had cardiac iron (5.7%), whereas nearly all patients with transfusion-dependent disease had liver and cardiac iron overload (71%), although 72% were on chelation therapy.
The ratio of hepcidin levels to levels of erythropoietic markers was calculated as a marker of the appropriateness of hepcidin production driven by erythropoietic factors, and the ratio of hepcidin levels to levels of iron indices as a marker of the appropriateness of hepcidin production in relation to estimated body iron stores. Before calculation, data were log-transformed to fit a normal distribution. The ratios of hepcidin levels to levels of erythropoietic markers and to iron indices were lower in patients when compared to healthy controls (Figs. 1 and 2). Despite similar transfusion frequency in patients with NTDT, patients with β-thalassemia intermedia or Hb E/β-thalassemia showed significantly lower ratios of hepcidin to erythropoietic markers and of hepcidin to SF and LIC compared with patients with HbH disease. However, the ratio levels were similar between patients with β-thalassemia intermedia or Hb E/β-thalassemia.
Comparison of ratios of hepcidin levels to indices of erythropoietic activity and iron overload in patients with NTDT. (A) Ratios of hepcidin levels to indices of erythropoietic activity; (B) ratios of hepcidin levels to indices of iron overload.
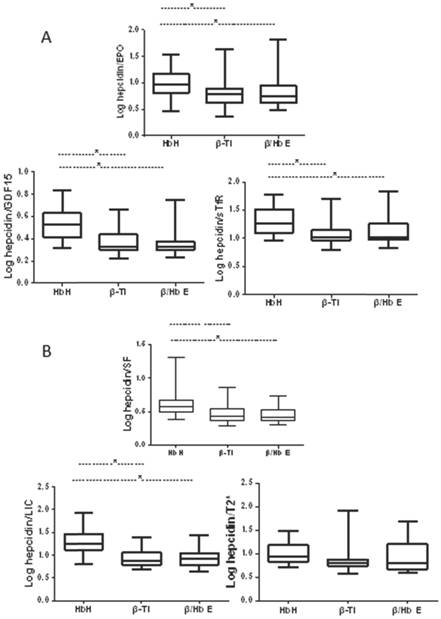
Patients with NTDT were grouped into with or without iron overload based on the LIC value: 81 (85.3%) patients with iron overload and 14 (14.7%) patients without iron overload. The erythropoietic activity and iron overload levels of the two groups were compared. The results showed that the patients with iron overload had higher erythropoietic activity based on the levels of EPO, GDF15, and sTfR and higher SF levels compared with patients with non-iron overload; however, the hepcidin and cardiac T2* levels were not significantly different (Table 3).
Indices of erythropoietic activity and LIC iron overload in NTDT patients (median, range)
| Index | Non-iron overload | Iron overload | P value |
|---|---|---|---|
| N | 14 (14.7%) | 81 (85.3%) | <0.001 |
| Hb, g/L | 101 (83-132) | 87 (48-117) | 0.001 |
| Hepcidin, ng/mL | 9.4 (5.7-104.0) | 13.2 (0.0-130.7) | 0.35 |
| EPO, mIU/mL | 16.1 (8.0-94.5) | 35.6 (8.1-1234.2) | 0.009 |
| GDF15, pg/mL | 716.9(275.4-2800) | 3179.5 (77.9-38660.5) | <0.001 |
| sTFR, mg/L | 6.8 (3.2-7.7) | 7.7(3.7-9.9) | 0.001 |
| Serum ferritinin, ng/mL | 321.5 (61-793) | 1453(3.6-8300) | <0.001 |
| Liver iron concentration, mg/g dw | 1.6 (0.6-2.9) | 12.1(3.0-43) | <0.001 |
| Cardiac T2*, ms | 32.1(23.4-48.2) | 30.4(7.5-75.1) | 0.503 |
Abbreviations: EPO, erythropoietin; GDF15, growth and differentiating factor 15; sTfR, soluble form of the transferrin receptor.
The requirement for blood transfusion of NTDT patients was defined according to the frequency of transfusion to maintain quality of life for the patient; regular (every 3 weeks to 3 months), occasional (every 4 months to once a year) and rare (none or once in several years) [15]. The transfusional iron intake was significantly different among the rare, occasional and regular groups (0.39 vs 11.33 vs 48.69 mg iron/kg/day, p<0.001).The levels of erythropoietic biomarkers and iron overload biomarkers were compared between the three groups. The results showed that the none/rare group had significantly higher Hb levels than the occasional or regular transfusion groups (90 vs 81 vs 75g/L. p=0.005), and the difference between the occasional and regular transfusion groups was not significant. However, the none/rare group had significantly lower SF levels than the occasional or regular transfusion groups (563vs 890 vs 1507ng/mL, p=0.004), the difference between the occasional and regular transfusion groups is also not significant (Table 4).
Comparison of Indices of erythropoietic activity and LIC iron overload of NTDT patients according to requirement for transfusion (median, range)
| Index | None/rare | Occasionally | Regularly | P value |
|---|---|---|---|---|
| N | 115(59.9%) | 54(28.1%) | 23(12.0%) | - |
| Median total transfusion (range), mg/kg.year | 0.39(0-30.77) | 11.33(4.02-49.17) ★▲ | 48.69(14.77-153.08) ★● | <0.001 |
| Hb, g/L | 90((41-132) | 81(33.8--105.9)★ | 75(56-96)★ | 0.005 |
| Hepcidin, ng/mL | 15.7(0.85-130.67) | 12.7(0.00-130.67 | 9.8(5.88-136.74) | 0.452 |
| EPO, mIU/mL | 26.6(1.73-1234.2) | 40.1(10.0-725.11) | 33.7(5.47-835.8) | 0.081 |
| GDF15, pg/mL | 2121.9(147.0-38660.6) | 3179.5(77.91-38660.5) | 8331.2(312.1-34153.5) | 0.098 |
| sTFR, mg/L | 7.3(1.41-9.87) | 7.7(0.9-9.8) | 7.7(1.81-8.48) | 0.072 |
| Serum ferritinin, ng/mL | 563.0(3.60-5011.0) | 890.0(20-6128.0)★ | 1507.0(293.0-8300)★ | 0.004 |
| Liver iron concentration, mg/g dw | 7.4(0.6-43.0) | 11.70(2.0-43) | 7.6(2.5-28.1) | 0.411 |
| Cardiac T2* ms | 30.1(7.46-56.98) | 30.1(17.94-75.1) | 32.8(17.37-45.69) | 0.92 |
Abbreviations: EPO, erythropoietin; GDF15, growth and differentiating factor 15; sTFR, soluble form of the transferrin receptor; ★significant difference compared with None/rare transfusion; ●significant difference compared with occasional transfusions; ▲significant difference compared with regular transfusions.
Association of hepcidin with iron status and erythropoietic markers in different types of thalassemia
Next the correlations of hepcidin levels with erythropoiesis markers or iron indices were examined (Table 5). The hepcidin levels were correlated positively with SF levels (r = 0.303, P = 0.002) and GDF15 (r = 0.276, P = 0.005) and sTfR (r = 0.264, P = 0.008) in controls. Among patients with thalassemia, the hepcidin levels were correlated positively with SF levels (r = 0.288, P = 0.005) and Hb levels (r = 0.565, P < 0.001) in patients with TDT, but not correlated with SF levels (r = 0.025, P = 0.732) in patients with NTDT; in both of them, the hepcidin levels were inversely correlated with the levels of EPO (r = -0.174, P = 0.016; r = -0.557, P < 0.001), GDF15 (r = -0.389, P < 0.001; r = -0.409, P < 0.001) and sTfR (r = -0.261, P < 0.001; r = -0.44, P < 0.001). The hepcidin levels in these patients were not correlated with LIC.
Correlation between hepcidin and biomarkers of erythropoietic activity and iron metabolism in patients with thalassemia
| Group | SF | LIC | Cardiac T2 | EPO | GDF15 | sTfR | Hb | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| r | P | r | P | r | P | r | P | r | P | r | P | r | P | |
| Controls | 0.303* | 0.002 | NA | NA | NA | NA | 0.001 | 0.990 | 0.276* | 0.005 | 0.264* | 0.008 | 0.109 | 0.276 |
| NTDT | 0.025 | 0.732 | 0.006 | 0.951 | -0.215* | 0.045 | -0.174* | 0.016 | -0.389* | 0.000 | -0.261* | 0.000 | 0.001 | 0.990 |
| TDT | 0.288* | 0.005 | 0.296 | 0.520 | -0.750 | 0.052 | -0.557* | 0.000 | -0.409* | 0.000 | -0.44* | 0.000 | 0.565* | 0.000 |
*Significant difference (P < 0.05).correlation coefficient(r).
Abbreviations: EPO, Erythropoietin; GDF15,growth and differentiating factor 15; Hb, hemoglobin; LIC, liver iron concentration; NA, not available; NTDT, non-transfusion-dependent thalassemia; SF, serum ferritin; sTfR, soluble form of the transferrin receptor; TDT, transfusion-dependent thalassemia.
Discussion
Erythropoiesis activity and iron metabolism vary among the different forms of thalassemia and even among different ethnic groups [16, 17]. This study provided detailed evidence that β-thalassemia was associated with a greater extent of ineffective erythropoiesis compared with HbH disease and various types of thalassemia were associated with hepcidin suppression and iron overload.
The hemoglobin levels of TDT patients in our study are lower than those pre-transfusion hemoglobin of the TDT guidelines. In China, TDT and NTDT patients were poorly-chelated and modestly transfused, our previous published paper has already proved that TDT patients in China are poorly-chelated and have a high burden of iron overload, 6 years old TDT patient was tested cardiac iron overload[11]. The above data indicated that due to insufficient blood and limited resources in the health insurance system, TDT and NTDT patients enrolled showed low hemoglobin levels and heavier iron load, which pose challenges to optimum care.
Patients with thalassemia in the present study showed significantly higher levels of EPO, GDF15 and sTfR compared with controls, indicating increased erythropoietic activity in the bone marrow. EPO is responsible for stimulating and regulating the erythropoiesis rate [18], and its production and secretion are stimulated by the combination of hypoxia and ineffective erythropoiesis. This can be resolved via blood transfusions [19]. The increased levels of GDF15 and sTfR indicate ineffective erythropoiesis [20]. Consistent with the results of the present study, a previous study reported significantly higher EPO levels in patients with thalassemia compared to normal subjects [18]. However, EPO levels did not differ significantly among different types of thalassemia in that study. The measurement of GDF15 expression in later stages of stress- or apoptosis-induced erythroid differentiation can predict ineffective or apoptotic erythropoiesis [21]. Previous studies reported higher levels of EPO, GDF15 and sTfR in patients with β-thalassemia intermedia in comparison to normal controls [16], and higher levels of GDF15 in patients with TDT than in patients with NTDT [22]. In this study, significantly higher levels of GDF15 were found in patients with TDT than in patients with NTDT, and also in patients with β-thalassemia intermedia compared to patients with HbH disease. GDF15 levels were correlated with anemia, iron overload and clinical severity score; they are significantly higher in splenectomized patients compared to nonsplenectomized patients [23]. GDF15 could identify patients with β-thalassemia intermedia who were at an increased risk of pulmonary and cardiovascular complications as well as subclinical atherosclerosis [22]. GDF-15 is a stress-induced cytokine and is not expressed in heart under normal physiological conditions but increases rapidly in response to cardiovascular injury, such as pressure overload, heart failure, ischemia/reperfusion and atherosclerosis [24]. GDF-15 activates Smad1 and reduces apoptotic cell death through upregulation of Bcl-xL and β-catenin. Xu et al. proposed a protective mechanism for GDF-15 against cardiac hypertrophy and cell death through Smad protein activation [24-26]. Therefore, further studies are needed to elucidate the potential mutual relationship between iron status, hepcidin and GDF15, as well as to their possible relevance in the pathogenesis of anemia in patients with cardiovascular pathology.
The levels of sTfR were found to be higher in patients with thalassemia patients compared to those in controls, but no significant differences were found between patients with TDT or NTDT. However, significantly higher levels of sTfR were observed in patients with β-thalassemia intermedia than in patients with HbH disease. In the presence of normal or elevated SF levels, sTfR levels reflect bone marrow erythropoietic activity [3]. Moreover, sTfR has shown impressive diagnostic accuracy in predicting risk of extramedullary hematopoiesis and clinical severity score [8, 27], age at first diagnosis and risk of therapeutic intervention and iron overload in patients with NTDT [28]. Consistent with the results of the present study, a previous study suggested greater erythropoietic activity in children with TDT than in children with β-thalassemia intermedia [28]. As in developing countries, our previous study reported that the TDT patients are more poorly-chelated, have a high burden of iron overload, and myocardial siderosis occurred in younger patients than in the other study. This was partly due to the lack of specific knowledge of the disease, shortages of blood, lower pre transfusion Hb levels and the limited availability of chelators [11].
Iron overload in patients with NTDT arises primarily because of increased iron absorption, whereas it occurs through iron accumulation in patients with TDT who undergo multiple transfusions. Lower hepcidin levels in patients than in controls supports a previous study on hereditary hemolytic anemia s[29], hemoglobin E/β-thalassemia and β-thalassemia trait disease [30]. In contrast, a previous study reported higher hepcidin levels in patients with β-thalassemia than those in controls [4]. Higher hepcidin levels were observed in patients with TDT compared to patients with NTDT, but no difference was found between patients with iron and non-iron overload. This might be explained by the hypothesis that regular transfusions in patients with transfusion-dependent disease inhibit the erythropoietic drive and induce high iron accumulation in tissues [4, 31, 32].
Reduced ratios of hepcidin with SF, LIC and cardiac T2 values were observed, indicating the inappropriate suppression of hepcidin synthesis. This hepcidin suppression, together with elevated sTfR and GDF15 levels, and reduced ratios of hepcidin with the erythropoiesis index suggested that, compared with controls, the hepcidin levels in thalassemia were also more sensitive to down-regulation by ineffective erythropoiesis, indicating a greater extent of ineffective erythropoiesis and signaling a need for a higher iron demand. Ultimately this leads to increased iron absorption and recycling, in agreement with studies on erythrocyte membrane defects and thalassemia traits [33].
Erythropoiesis and iron metabolism balance out with each other to match iron supply with globin synthesis, which is essential for normal production of red blood cells [33]. Hepcidin is the master regulator of systemic iron homeostasis: it facilitates iron balance by controlling intestinal iron absorption and recycling [32]. An increase in iron stores, transferrin saturation and inflammation trigger a homeostatic increase in liver hepcidin expression [34]. Conversely, hepcidin expression is suppressed in the event of iron deficiency or increased erythroid activity due to physiological erythropoiesis or pathological, ineffective erythropoiesis [6]. The specific metabolic steps of how erythropoiesis causes hepcidin suppression remains unclear.
The present study found that patients with thalassemia showed lower hepcidin levels but higher EPO levels compared with controls, raising the possibility that EPO regulates hepcidin levels. It is likely that other factors are also important in this regulation because human and animal studies have shown that anemia alone, even when EPO levels are elevated, is insufficient to suppress hepcidin levels [35-37]. GDF15 may help mediate the interaction between EPO and hepcidin. A negative correlation was observed between GFD15 and hepcidin in patients with thalassemia, which was consistent with the findings of a previous study [38]. Thus, it is possible that GDF15 over-expression inhibits hepcidin expression, contributing to iron overload in patients with thalassemia. Further studies are needed to examine this hypothesis, especially because a study in GDF15-knockout mice showed no reduction in hepatic hepcidin expression during increased erythropoietic activity [39].
The present study found that, like GDF15 levels, sTfR levels were correlated negatively with hepcidin levels in patients with hemoglobin E/β-thalassemia and TDT. This may shed light on the relationships between sTfR and hepcidin, which, to date, are poorly understood. The negative correlation in the present study was consistent with the idea that hepcidin suppression leads to higher iron demand and therefore to increased ineffective erythropoiesis. This finding was also consistent with the suggestion that the ratio of hepcidin or ferritin level to sTfR level may be useful for monitoring patients with thalassemia with respect to erythropoietic activity and the risk of iron overload[33]. Further studies are needed to clarify the contribution of sTfR to the regulation of hepcidin.
Another regulator of hepcidin expression that may help explain the present findings is the protein hormone, erythroferrone (ERFE), which participates in an EPO-induced pathway that suppresses the expression of hepcidin, twisted gastrulation protein 1 and GDF15[40]. Ablation of ERFE in an animal model of thalassemia restores hepatic hepcidin expression to wild-type levels [41]. ERFE may be responsible for the iron-loading phenotype observed in ineffective erythropoiesis and for regulation of iron absorption during periods of stress-induced erythropoiesis.
Limitations of the study
The limitations of this study were as follows: (1) it was a single-center study with a relatively small sample size, (2) MRI scans could not be performed in all cases and (3) TDT patients were transfused differently and not all patients received chelation therapy. Although the evaluation and biomarker correlations in these patients was a snapshot at a defined time point in their disease, we suggest that the data obtained justifies the implementation of a larger multicenter trial in the future.
Conclusions
The patients with thalassemia in the present study showed iron overload, as well as a greater extent of ineffective erythropoiesis and lower hepcidin levels, compared with healthy controls. The imbalance between erythropoiesis and iron metabolism differed across different thalassemia types. The hepcidin levels were correlated with ineffective erythropoiesis but not with iron overload. Prospective future studies are needed to explore the link between increased erythropoiesis and hepcidin suppression, including the role of the erythroid-derived hormone, ERFE.
Acknowledgements
This study was supported by research grants from the National Natural Science Foundation of China (grant no. 81260090 and 81460025), the Guangxi Natural Science Foundation (grant no. 1598011-1), and the Guangxi key laboratory of thalassemia research (grant no.16-380-34). The authors thank all participants involved in this study. The authors would like to thank Dr. Dev Sooranna, Imperial College London, for editing the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Musallam KM, Rivella S, Vichinsky E. et al. Non-transfusion-dependent thalassemias. Haematologica. 2013;98:833-44
2. Weatherall DJ. The definition and epidemiology of non-transfusion-dependent thalassemia. Blood Rev. 2012;26:S3-S6
3. Ginzburg Y, Rivella S. beta-thalassemia: a model for elucidating the dynamic regulation of ineffective erythropoiesis and iron metabolism. Blood. 2011;118:4321-30
4. Kaddah AM, Abdel-Salam A, Farhan MS. et al. Serum Hepcidin as a Diagnostic Marker of Severe Iron Overload in Beta-thalassemia Major. Indian journal of pediatrics. 2017
5. Makis A, Hatzimichael E, Papassotiriou I. et al. 2017 Clinical trials update in new treatments of beta-thalassemia. Am J Hematol. 2016;91:1135-45
6. Gardenghi S, Grady RW, Rivella S. Anemia, ineffective erythropoiesis, and hepcidin: interacting factors in abnormal iron metabolism leading to iron overload in beta-thalassemia. Hematology/oncology clinics of North America. 2010;24:1089-107
7. Tanno T, Bhanu NV, Oneal PA. et al. High levels of GDF15 in thalassemia suppress expression of the iron regulatory protein hepcidin. Nat Med. 2007;13:1096-101
8. Ricchi P, Ammirabile M, Costantini S. et al. Soluble form of transferrin receptor as a biomarker of overall morbidity in patients with non-transfusion-dependent thalassaemia: a cross-sectional study. Blood transfusion = Trasfusione del sangue. 2016;14:538-40
9. Origa R, Cazzola M, Mereu E. et al. Differences in the erythropoiesis-hepcidin-iron store axis between hemoglobin H disease and beta-thalassemia intermedia. Haematologica. 2015;100:e169-71
10. Galanello R, Origa R. Beta-thalassemia. Orphanet J Rare Dis. 2010;5:11
11. Yang G, Liu R, Peng P. et al. How early can myocardial iron overload occur in beta thalassemia major? Plos One. 2014;9:e85379
12. Ricchi P, Meloni A, Spasiano A. et al. Extramedullary hematopoiesis is associated with lower cardiac iron loading in chronically transfused thalassemia patients. Am J Hematol. 2015;90:1008-12
13. Wood JC, Enriquez C, Ghugre N. et al. MRI R2 and R2* mapping accurately estimates hepatic iron concentration in transfusion-dependent thalassemia and sickle cell disease patients. Blood. 2005;106:1460-5
14. Anderson LJ, Holden S, Davis B. et al. Cardiovascular T2-star (T2*) magnetic resonance for the early diagnosis of myocardial iron overload. Eur Heart J. 2001;22:2171-9
15. Sripichai O, Makarasara W, Munkongdee T. et al. A scoring system for the classification of beta-thalassemia/Hb E disease severity. Am J Hematol. 2008;83:482-4
16. Fertrin KY, Lanaro C, Franco-Penteado CF. et al. Erythropoiesis-driven regulation of hepcidin in human red cell disorders is better reflected through concentrations of soluble transferrin receptor rather than growth differentiation factor 15. Am J Hematol. 2014;89:385-90
17. Schotten N, Laarakkers CM, Roelofs RW. et al. EPO and hepcidin plasma concentrations in blood donors and beta-thalassemia intermedia are not related to commercially tested plasma ERFE concentrations. Am J Hematol. 2017;92:E29-E31
18. Butthep P, Wisedpanichkij R, Jindadamrongwech S. et al. Elevated erythropoietin and cytokines levels are related to impaired reticulocyte maturation in thalassemic patients. Blood cells, molecules & diseases. 2015;54:170-6
19. Camaschella C, Nai A. Ineffective erythropoiesis and regulation of iron status in iron loading anaemias. Br J Haematol. 2016;172:512-23
20. Ronzoni L, Sonzogni L, Duca L. et al. Growth Differentiation Factor 15 expression and regulation during erythroid differentiation in non-transfusion dependent thalassemia. Blood cells, molecules & diseases. 2015;54:26-8
21. Tanno T, Noel P, Miller JL. Growth differentiation factor 15 in erythroid health and disease. Curr Opin Hematol. 2010;17:184-90
22. Tantawy AA, Adly AA, Ismail EA. et al. Growth differentiation factor-15 in children and adolescents with thalassemia intermedia: Relation to subclinical atherosclerosis and pulmonary vasculopathy. Blood cells, molecules & diseases. 2015;55:144-50
23. Musallam KM, Taher AT, Duca L. et al. Levels of growth differentiation factor-15 are high and correlate with clinical severity in transfusion-independent patients with beta thalassemia intermedia. Blood cells, molecules & diseases. 2011;47:232-4
24. Adela R, Banerjee SK. GDF-15 as a Target and Biomarker for Diabetes and Cardiovascular Diseases: A Translational Prospective. Journal of diabetes research. 2015;2015:490842
25. Xu J, Kimball TR, Lorenz JN. et al. GDF15/MIC-1 functions as a protective and antihypertrophic factor released from the myocardium in association with SMAD protein activation. Circulation research. 2006;98:342-50
26. Schillaci G, Verdecchia P, Porcellati C. et al. Continuous relation between left ventricular mass and cardiovascular risk in essential hypertension. Hypertension. 2000;35:580-6
27. Ricchi P, Ammirabile M, Costantini S. et al. A useful relationship between the presence of extramedullary erythropoeisis and the level of the soluble form of the transferrin receptor in a large cohort of adult patients with thalassemia intermedia: a prospective study. Ann Hematol. 2012;91:905-9
28. Ragab SM, Safan MA, Badr EA. Study of serum haptoglobin level and its relation to erythropoietic activity in Beta thalassemia children. Mediterranean journal of hematology and infectious diseases. 2015;7:e2015019
29. El Beshlawy A, Alaraby I, Abdel Kader MS. et al. Study of serum hepcidin in hereditary hemolytic anemias. Hemoglobin. 2012;36:555-70
30. Jones E, Pasricha SR, Allen A. et al. Hepcidin is suppressed by erythropoiesis in hemoglobin E beta-thalassemia and beta-thalassemia trait. Blood. 2015;125:873-80
31. Pasricha SR, Frazer DM, Bowden DK. et al. Transfusion suppresses erythropoiesis and increases hepcidin in adult patients with beta-thalassemia major: a longitudinal study. Blood. 2013;122:124-33
32. Pasricha SR, McHugh K, Drakesmith H. Regulation of Hepcidin by Erythropoiesis: The Story So Far. Annual review of nutrition. 2016;36:417-34
33. Sulovska L, Holub D, Zidova Z. et al. Characterization of iron metabolism and erythropoiesis in erythrocyte membrane defects and thalassemia traits. Biomedical papers of the Medical Faculty of the University Palacky, Olomouc, Czechoslovakia. 2016;160:231-7
34. Ganz T. Systemic iron homeostasis. Physiological reviews. 2013;93:1721-41
35. Diaz V, Gammella E, Recalcati S. et al. Liver iron modulates hepcidin expression during chronically elevated erythropoiesis in mice. Hepatology. 2013;58:2122-32
36. Gammella E, Diaz V, Recalcati S. et al. Erythropoietin's inhibiting impact on hepcidin expression occurs indirectly. American journal of physiology Regulatory, integrative and comparative physiology. 2015;308:R330-5
37. Rybinska I, Cairo G. Mutual Cross Talk Between Iron Homeostasis and Erythropoiesis. Vitamins and hormones. 2017;105:143-60
38. Theurl I, Finkenstedt A, Schroll A. et al. Growth differentiation factor 15 in anaemia of chronic disease, iron deficiency anaemia and mixed type anaemia. Br J Haematol. 2010;148:449-55
39. Casanovas G, Vujic Spasic M, Casu C. et al. The murine growth differentiation factor 15 is not essential for systemic iron homeostasis in phlebotomized mice. Haematologica. 2013;98:444-7
40. Kautz L, Jung G, Valore EV. et al. Identification of erythroferrone as an erythroid regulator of iron metabolism. Nat Genet. 2014;46:678-84
41. Kautz L, Jung G, Du X. et al. Erythroferrone contributes to hepcidin suppression and iron overload in a mouse model of beta-thalassemia. Blood. 2015;126:2031-7
Author contact
![]() Corresponding author: Yongrong Lai, PhD MD. Department of Hematology, the First Affiliated Hospital of Guangxi Medical University, Nanning, Guangxi 530021, China. TEL: (86)0771 5352681; Fax (86)0771 5352681; Email: laiyongrongcom
Corresponding author: Yongrong Lai, PhD MD. Department of Hematology, the First Affiliated Hospital of Guangxi Medical University, Nanning, Guangxi 530021, China. TEL: (86)0771 5352681; Fax (86)0771 5352681; Email: laiyongrongcom