Impact Factor ISSN: 1449-1907

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2019; 16(2):276-284. doi:10.7150/ijms.28110 This issue Cite
Research Paper
Von Willebrand Factor Adhesive Activity and ADAMTS13 Protease Activity in HIV-1-Infected Men
Susan M. Graham1,2,3, Junmei Chen4, Jennie Le4, Minhua Ling4, Dominic W. Chung4, W. Conrad Liles1,2,5,6, José A. López1,4,7,8 ![]()
1. Department of Medicine, University of Washington, Seattle, WA USA
2. Department of Global Health, University of Washington, Seattle, WA USA
3. Department of Epidemiology, University of Washington, Seattle, WA USA
4. Bloodworks Research Institute, Seattle, WA, USA
5. Department of Pathology, University of Washington, Seattle, WA USA
6. Department of Pharmacology, University of Washington, Seattle, WA, USA
7. Department of Biochemistry, University of Washington, Seattle, WA USA
8. Department of Mechanical Engineering, University of Washington, Seattle, WA, USA
Received 2018-6-25; Accepted 2018-10-18; Published 2019-1-1
Abstract
Background: Endothelial activation caused by HIV-1 infection leads to release of von Willebrand factor (VWF), which enters the circulation or attaches to vessel walls and self-assembles into strings and fibers, enabling platelet adhesion; this adhesive activity is regulated by the VWF-cleaving protease ADAMTS13. Our objective was to assess VWF adhesive activity and ADAMTS13 protease activity in HIV-1 infection.
Methods: We measured levels of VWF antigen, VWF activation factor (a measure of adhesive activity), ADAMTS13 antigen, ADAMTS13 activity, and apolipoprotein A1 (which interferes with VWF self-association) in serum samples from HIV-1-infected men whose infections were acute (n=10), chronic untreated (n=10), or chronic treated (n=10), compared to uninfected controls (n=10). Means across groups were compared using analysis of variance with contrasts, and Pearson correlations were calculated.
Results: Plasma viral load was positively correlated with VWF adhesive activity, which was elevated in acute relative to chronic treated HIV-1 infection. ADAMTS13 antigen and activity were both positively correlated with plasma viral load, and ADAMTS13 activity was significantly higher in men with acute HIV infection than in uninfected controls, and in both acute and chronic untreated HIV infection relative to chronic treated infection.
Conclusion: These findings suggest that even in the setting of increased ADAMTS13 protease activity, VWF in HIV-1 infection is hyperadhesive, which may favor development of microvascular and arterial thromboses and thereby contribute to increased cardiovascular risk in HIV-1-infected individuals.
Keywords: HIV-1, von Willebrand factor, ADAMTS13 protein, apolipoprotein A1, thrombosis
Introduction
HIV-1 infection is associated with inflammation and activation of the coagulation system and the vascular endothelium, and these phenomena persist during antiretroviral therapy (ART), despite modest improvements [1-4]. These processes are known to play mechanistic roles in microangiopathy [5], especially in the context of infection [6,7]. Activation of endothelial cells is accompanied by the release and persistent attachment to the vessel wall of von Willebrand factor (VWF), a multimeric adhesive protein that mediates the first step of platelet adhesion [8]. Over time, VWF persistence on the intact endothelium and attendant platelet adhesion can promote atherosclerosis [9], possibly contributing to increased cardiovascular risk in HIV-1-infected persons.
Several investigators have reported elevated levels of circulating VWF in untreated HIV-1-infected individuals [10,11]. VWF antigen levels are negatively correlated with CD4 count and positively correlated with plasma viral load [12-14], and higher VWF antigen levels are associated with increased all-cause mortality [15]. Of note, VWF antigen levels decrease after effective ART [14,16], as do levels of other endothelial activation biomarkers [2]. Studies are lacking, however, of VWF adhesive activity and of circulating levels and activity of one of the primary regulators of VWF activity, the VWF-cleaving protease ADAMTS13 (a disintegrin and metalloproteinase with a thrombospondin type 1 motif, member 13). Such studies could provide a better understanding of the connection between HIV-1 infection, accelerated atherosclerosis [17], and mortality from cardiovascular disease [18,19].
Our objective was to assess VWF activation factor (a measure of adhesive activity) and ADAMTS13 activity in stored samples from men with acute, chronic untreated, and chronic treated HIV-1 infection, compared to uninfected controls, and to evaluate associations between these parameters and HIV-1 disease biomarkers. We also analyzed associations of HIV-1 biomarkers, VWF adhesive activity, and ADAMTS13 protease activity with levels of apolipoprotein A1 (ApoA1), which has been shown to interfere with VWF self-association [20], potentially decreasing platelet and monocyte recruitment to atherosclerosis-prone regions of the vasculature.
Materials and Methods
Study populations
Randomly selected, stored serum samples were requested from three sources affiliated with the University of Washington (UW). Serum samples collected in 1995-2002 from 10 ART-naïve men with acute HIV-1 infection (Feibig stages I, II, or III) were obtained from the Seattle Primary Infection Project (SeaPIP) cohort, which enrolls and follows individuals diagnosed within 30 days of the onset of symptoms of acute HIV-1 infection. Serum samples collected in 2010-2013 from 10 men with chronic untreated HIV-1 infection and 10 men with chronic treated HIV-1 infection were obtained from the UW Center for AIDS Research (CFAR) HIV Specimen Repository, which stores samples from HIV-1-infected patients receiving care at UW HIV clinics. Control samples were collected from 10 consecutively recruited HIV-uninfected men participating in the Bloodworks Northwest Research Institute Normal Control Registry and Repository.
Clinical data associated with the stored specimens were obtained from the primary investigative teams, including participant age, race/ethnicity, CD4 count and plasma viral load (if HIV-1-infected), and platelet count, if available. CD4 count, plasma viral load, and platelet testing was performed according to the methods in use at each clinical site laboratory at the time of sample collection. When laboratory results were not available on the same date as the stored specimen, the closest value within 6 months was used; these measures were not repeated for the purpose of this study. For participants with acute HIV-1 infection, date of infection was estimated; for treated participants, treatment regimen data were obtained. Participants were all outpatients at the time of sample collection; men with a diagnosis of cancer were excluded.
Laboratory testing
VWF antigen was measured by enzyme-linked immunosorbent assay (ELISA) using a polyclonal VWF antibody as the capture antibody, and the bound VWF was detected by a horseradish peroxidase (HRP)-conjugated polyclonal VWF antibody (DAKO North America, Inc., Carpinteria, CA, USA). VWF activation factor was determined by ELISA using the llama nanobody AU/VWFa-11 (which detects a gain-of-function conformation of the VWF A1 domain) as the capture antibody, as previously described [21]. Total active VWF (TA-VWF), a measure of total VWF reactivity, was calculated by multiplying VWF antigen by VWF activation factor, as described by Chen et al [22]. ADAMTS13 antigen was measured using an ELISA (American Diagnostica, Stamford, CT, USA) per the manufacturer's instructions. ADAMTS13 proteolytic activity was measured using an HRP-conjugated peptide substrate, as previously described [23]. The ApoA1 antigen concentration was measured by ELISA using a monoclonal ApoA1 antibody as the capture antibody and an HRP-conjugated polyclonal ApoA1 antibody as the detection antibody (LS-C11247 and LS-C11248 respectively, LifeSpan Biosciences, Inc., Seattle, WA, USA). All parameters were assigned values for participant serum relative to normal values generated from healthy volunteer samples (i.e., fold-normal values). All sample testing was carried out blinded to participant characteristics. Of note, all biomarkers tested are stable in cold storage and resistant to degradation during four freeze-thaw cycles (Dominic Chung, unpublished data).
Statistical analysis
Descriptive statistics were used for biomarker distributions, and scatter plots were used to present biomarker data by participant group. The Shapiro-Wilk test was used to test whether measured biomarkers were distributed normally. VWF antigen and VWF activation factor were not normally distributed, and were therefore log2-transformed for statistical testing, so that each unit increase represents a doubling in biomarker level. Plasma viral load was log10-transformed, consistent with the usual practice for this biomarker. Analysis of variance (ANOVA) with contrasts was used to compare mean values across the four patient populations. Pairwise Pearson coefficients were calculated to assess correlation between biomarkers, along with p values for significance. P values <0.05 were considered significant, with no adjustment for multiple comparisons. Stata version 14.2 was used for analysis.
Post-hoc power
With 10 uninfected controls and 30 HIV-1-infected cases, the minimal detectable difference between HIV-1-infected and uninfected participants at 80% power and α=0.05 was 0.906 for ln2-transformed VWF activation factor (standard deviation in controls = 0.863) and 0.160 for ADAMTS13 activity (standard deviation in controls = 0.152).
Ethics statement
All study participants had provided written, informed consent including permission to store and test samples for factors associated with HIV-1 disease progression and adverse outcomes. SEA-PIP and the CFAR HIV specimen repository were approved by the University of Washington Human Subjects Division. The Bloodworks Northwest Research Institute Normal Control Registry and Repository was approved by the Western Institutional Review Board.
Results
Participant characteristics
Table 1 presents characteristics of the 40 participants, along with descriptive statistics for the parameters measured in their serum samples. As expected, plasma viral load differed across the HIV-1-infected groups, being very high in acutely infected men, somewhat lower in men with chronic untreated infection, and below the level of detection in all treated men. CD4 counts were similar in all groups of HIV-1-infected men, with means ranging from 515 cells/μL in the chronic untreated group to 591 cells/μL in the men with acute infection. Mean platelet counts ranged from 207.0 x 109/L in the chronic untreated group to 238.1 x 109/L in the treated group.
Characteristics of Participants by Group
| Characteristic | Acute HIV (n = 10) Mean (SD) | Chronic Untreated HIV (n = 10) Mean (SD) | Treated HIV (n = 10) Mean (SD) | HIV-Negative Control (n = 10) Mean (SD) |
|---|---|---|---|---|
| Age, years | 33.0 (8.1) | 39.3 (8.6) | 41.8 (9.8) | 32.7 (8.3) |
| Race/ethnicity | ||||
| Non-hispanic white | 9 (90%) | 6 (60%) | 7 (70%) | 4 (40%) |
| Hispanic | 0 | 1 (10%) | 0 | 3 (30%) |
| Black | 0 | 3 (30%) | 2 (20%) | 1 (10%) |
| Asian | 1 (10%) | 0 | 0 | 2 (20%) |
| American Indian | 0 | 0 | 1 (10%) | 0 |
| Days since infection | 16.8 (13.7) | — | — | NA |
| ART regimen type | NA | NA | NA | |
| PI-based | 5 (50) | |||
| NNRTI-based | 2 (20) | |||
| PI- and INSTI-based | 3 (30) | |||
| Abacavir-containing | 2 (20) | |||
| Log10 plasma viral load, copies/mL | 5.92 (0.87) | 4.29 (1.11) | 1.30* | NA |
| CD4 count, cells/μL | 591.3 (269.7) | 515.0 (169.8) | 537.0 (249.3) | NA |
| Platelet count x 109/L | 231.5 (59.7) | 207.0 (58.1) | 238.1 (68.2) | — |
| VWF antigen, fold-normal† | 1.52 (0.77) | 2.14 (0.69) | 1.75 (0.71) | 1.01 (0.37) |
| VWF activation factor, fold-normal† | 1.11 (0.60) | 0.66 (0.45) | 0.59 (0.33) | 0.92 (0.60) |
| TA-VWF, fold-normal† | 1.90 (1.58) | 1.57 (1.62) | 1.19 (0.97) | 0.98 (0.73) |
| ADAMTS13 antigen, fold-normal† | 1.19 (0.21) | 1.15 (0.18) | 1.04 (0.30) | 1.18 (0.29) |
| ADAMTS13 activity, fold-normal† | 1.41 (0.22) | 1.23 (0.19) | 1.01 (0.36) | 1.13 (0.15) |
| ADAMTS13 specific activity† | 1.20 (0.15) | 1.09 (0.18) | 0.99 (0.20) | 0.98 (0.17) |
| ApoA1 antigen, fold-normal† | 1.10 (0.21) | 0.98 (0.25) | 1.01 (0.19) | 0.99 (0.21) |
* All treated men had plasma viral load <40 copies/mL.
† Parameters were assigned values for participant serum relative to normal serum values (i.e., fold-normal values). Total active VWF is VWF antigen multiplied by the VWF activation factor. ADAMTS13 specific activity is the ratio of ADAMTS13 activity to ADAMTS13 antigen.
Abbreviations used: ADAMTS13 = a disintegrin and metalloproteinase with a thrombospondin type 1 motif member 13, ApoA1 = apolipoprotein A1, ART = antiretroviral therapy, HIV = human immunodeficiency virus, INSTI = integrase strand transfer inhibitor, NA = not applicable, NNRTI = non-nucleoside reverse transcriptase inhibitor, PI = protease inhibitor, SD = standard deviation, TA-VWF = total active von Willebrand factor, VWF = von Willebrand factor.
VWF antigen
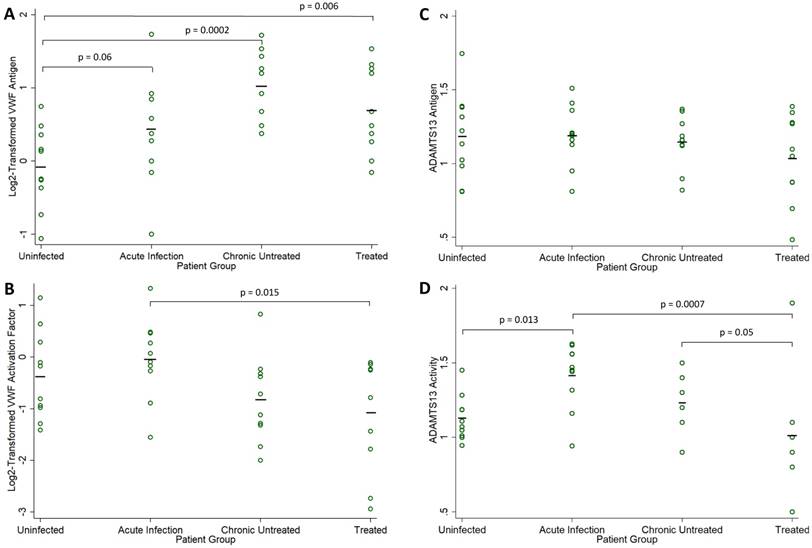
VWF antigen was elevated in the 30 HIV-1-infected men relative to the 10 uninfected controls (mean, 0.72 vs -0.08 log2 fold-normal, p=0.001). VWF antigen levels were elevated at all stages of infection (Figure 1A), with significant differences compared to the uninfected controls in both chronic infection groups (ANOVA p=0.06 for acute infection, p=0.0002 for chronic untreated infection, and p=0.006 for treated infection). Among the 30 HIV-1-infected men, VWF antigen levels did not correlate with CD4 count (r = -0.179, p=0.34) or plasma viral load (r = 0.016, p=0.93). Among the 20 men with untreated HIV infection, there was a borderline negative correlation between VWF antigen levels and CD4 count (r = -0.423, p=0.06), but no correlation with plasma viral load (r = -0.024, p=0.92).
Biomarker Levels in HIV-Infected Compared to Uninfected Men. Fold-normal values from sera of HIV-infected participants in each group (acute infection, chronic untreated infection, and treated infection) are compared to values from sera obtained from uninfected healthy volunteers: (A) log2-transformed VWF antigen, (B) log2-transformed VWF activation factor, (C) ADAMTS13 antigen, and (D) ADAMTS13 activity. Individual measures are indicated with open circles and group means are indicated by a solid black line. P values are derived from one-way analysis of variance and only shown for borderline significant and statistically significant comparisons to the uninfected group.
VWF activation factor
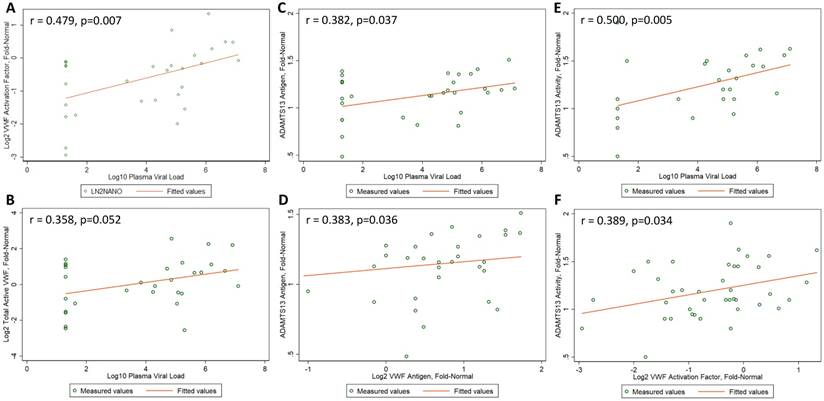
VWF activation factor levels were not greater in the 30 HIV-1-infected men relative to the 10 uninfected controls (mean, -0.362 vs -0.642 log2 fold-normal, p=0.43). There were no differences in VWF activation factor levels relative to the uninfected controls (ANOVA p=0.42 in acute infection, p=0.26 in chronic untreated infection, p=0.09 in treated infection). However, VWF activation levels were significantly higher in acute compared to chronic treated HIV infection (p=0.015, Figure 1B). Among the 30 HIV-1-infected men, there was a positive correlation between VWF activation factor levels and plasma viral load (r = 0.479, p=0.007, Figure 2A) but no correlation between VWF activation factor levels and CD4 count (r = 0.227, p=0.23). Among the 20 men with untreated HIV infection, VWF activation factor was also positively correlated with plasma viral load (r = 0.595, p=0.006), but had no correlation with CD4 count (r = -0.072, p=0.76). Total active VWF, the product of VWF antigen level and activation factor, had a borderline positive correlation with plasma viral load (r = 0.358, p = 0.052, Figure 2B), but was not correlated with CD4 count (r = 0.082, p = 0.67). Among the 20 men with untreated HIV infection, the correlation between total active VWF and plasma viral load remained of borderline significance (r = 0.415, p=0.07), and there was again no correlation with CD4 count (r = -0.281, p=0.23).
Correlations between Biomarker Levels. Pearson correlation coefficients (r values) and corresponding p values are presented for (A) log2-transformed VWF activation factor and log10-transformed plasma viral load, (B) log2-transformed total active VWF and log10-transformed plasma viral load, (C) ADAMTS13 antigen levels and log10-transformed plasma viral load, (D) ADAMTS13 antigen levels and log2-transformed VWF antigen levels, (E) ADAMTS13 activity and log10-transformed plasma viral load, and (F) ADAMTS13 activity and log2-transformed VWF activation factor. Measured values are present as open circles and dashed lines depict fitted values.
ADAMTS13 antigen
ADAMTS13 antigen levels were no different in the 30 HIV-1-infected men relative to the 10 uninfected controls (mean, 1.12 vs 1.18 fold-normal, p=0.53). ADAMTS13 antigen levels did not differ across the four groups (ANOVA p=0.92 for acute infection, p=0.75 for chronic untreated infection, and p=0.20 for treated infection, Figure 1C). Among the 30 HIV-1-infected men, ADAMTS13 antigen levels did not correlate with CD4 count (r = 0.135, p=0.48); however, there was a positive correlation between ADAMTS13 antigen levels and plasma viral load (r = 0.382, p=0.04, Figure 2C). Similarly, among the 20 men with untreated HIV infection, there was a positive correlation between ADAMTS13 antigen levels and plasma viral load (r = 0.451, p=0.046), but not CD4 count (r = -0.077, p=0.75). Overall in the 30 HIV-1-infected men, ADAMTS13 antigen had a positive correlation with VWF antigen (r=0.383, p=0.04, Figure 2D) and with VWF activation factor (r=0.549, p=0.002, not shown).
ADAMTS13 activity
ADAMTS13 activity was not greater in the 30 HIV-1-infected men relative to the 10 uninfected controls (mean, 1.22 vs 1.13 fold-normal, p=0.39). However, ADAMTS13 activity was higher among men with acute infection relative to the uninfected men (ANOVA p=0.013 in acute infection, p=0.36 in chronic untreated infection, p=0.29 in treated infection). In addition, ADAMTS13 activity was significantly higher in acute compared to chronic treated HIV infection (p=0.0007) and was borderline higher in chronic untreated compared to chronic treated HIV infection (p=0.05, Figure 1D). Among the 30 HIV-1-infected men, ADAMTS13 activity correlated positively with plasma viral load (r = 0.500, p=0.005, Figure 2E) but not with CD4 count (r = 0.295, p=0.11). Among the 20 men with untreated infection, ADAMTS13 activity was not significantly correlated with either viral load or CD4 count (data not shown). ADAMTS13 activity had a positive correlation with VWF activation factor (r = 0.389, p=0.03, Figure 2F).
ApoA1 levels
ApoA1 levels were not associated with HIV infection overall, nor with CD4 count or plasma viral load in the 30 HIV-1-infected men. Similarly, there were no associations between ApoA1 levels and CD4 count or plasma viral load among the 20 men with untreated infection. There were no correlations between ApoA1 levels and VWF antigen, VWF activation factor, ADAMTS13 antigen, or ADAMTS13 protease activity.
Discussion
Platelet adhesion to the vessel wall is involved in the initiation of cardiovascular disease (atherosclerosis) and in arterial thrombosis, the most common disease-defining cardiovascular event [24]. This process is influenced by a number of physiological factors, including the plasma concentration and adhesive activity of the primary molecule mediating platelet adhesion, VWF. VWF's adhesive activity is enhanced by self-association of the molecule into larger molecular structures with a high density of platelet binding sites. Self-association, in turn, is attenuated by proteolysis (ADAMTS13) and by high-density lipoproteins (HDL) and increases as VWF concentration increases. In this study comparing three groups of American men at various stages of HIV-1 infection to uninfected controls, we confirmed that VWF antigen levels were elevated in HIV infection, including during suppressive ART. VWF activation factor, a measure of VWF adhesive activity, was positively correlated with plasma viral load, and was elevated in acute infection relative to chronic treated infection. ADAMTS13 antigen and activity were both positively correlated with plasma viral load, and ADAMTS13 activity was significantly higher in men with acute HIV infection than in uninfected controls, and in both acute and chronic untreated HIV infection relative to chronic treated infection. ApoA1 levels were not different in HIV-1-infected men than in controls and not correlated with any other biomarker studied.
In a recent systematic review of the clinical utility of biomarkers of endothelial activation and coagulation [25], we identified six studies that investigated associations between VWF antigen and clinical outcomes or vascular dysfunction in HIV infection [14,15,26-29]. In one study, higher VWF antigen levels were associated with increased all-cause mortality in HIV-1-infected patients [15]. In addition, two studies suggested an indirect association between VWF antigen and clinical outcomes. In the first, a marked rise in VWF levels was associated with HIV disease progression, with a positive correlation between VWF levels and plasma viral load [14]. In the second study, VWF levels decreased while carotid intima media thickness increased and arterial stiffness decreased during ART [26]. A study published more recently reported an association between VWF antigen levels and both first and recurrent venous thromboembolic events in HIV-1-infected patients [30]. These studies suggest that the known elevation of VWF antigen levels in HIV infection may adversely affect clinical outcomes.
To our knowledge, this is the first study of VWF adhesive activity in HIV infection. Because VWF adhesive activity has been shown to be an independent risk factor for a first ST-elevation myocardial infarction [31], further research on the utility of VWF antigen and VWF adhesive activity as biomarkers of cardiovascular risk in HIV-1-infected patients appears warranted. In this relatively small study, we found that VWF adhesive activity was positively correlated with plasma viral load, with the highest activation factor level noted in acute infection, when viral load is highest. VWF adhesive activity was significantly lower among men with chronic treated HIV relative to those with acute infection, suggesting that successful virologic suppression with ART may lower VWF adhesive activity. Further study and validation of these findings and their implications for HIV-related cardiovascular disease are needed.
It has long been recognized that HIV infection can lead to thrombotic thrombocytopenic purpura (TTP) [32,33]. HIV-associated TTP occurs in the setting of profound CD4 deficiency and altered ADAMTS13 protease activity [34,35], and can be complicated by myocardial infarction and stroke [36,37]. Little research has been conducted on ADAMTS levels and activity in HIV-1-infected persons without frank TTP. In a small study of HIV-1-infected stroke patients, higher VWF antigen levels and lower ADAMTS13 antigen levels were present in the stroke patients than in HIV-1-infected, non-stroke controls, but both groups had lower ADAMTS13 antigen levels than uninfected controls [38]. While this finding suggests that lower ADAMTS13 antigen levels increase cardiovascular disease risk, the blood samples in that study were collected after the stroke event, and therefore may have been impacted by the event itself. It remains unclear whether ADAMTS13 antigen levels or activity could be a useful biomarker for the prediction of HIV-related cardiovascular disease.
In our study, we found that both ADAMTS13 antigen and activity correlated positively with plasma viral load. Men with acute HIV had higher ADAMTS13 activity than did uninfected controls. In addition, men with acute infection and those with chronic untreated infection had significantly higher ADAMTS13 activity than did treated men. Both ADAMTS13 antigen and activity correlated positively with VWF activation factor in our study, which is unexpected. This relationship is usually inversely correlated, given that ADAMTS13 proteolysis regulates VWF adhesive activity. However, the regulation of ADAMTS13 antigen and activity and its relationship with VWF adhesive activity appears not to be straight-forward. In sickle cell disease, for example, we have found many cases in which ADAMTS13 activity is elevated when measured with a small peptide substrate (as is standard in clinical laboratories), but severely defective in its ability to cleave multimeric plasma VWF [39]. Because of limited sample volume, we were unable to determine if a similar situation exists in HIV infection. However, our study provides circumstantial evidence that the ADAMTS13 response may be impaired in HIV infection; additional investigation of this finding seems warranted.
Although HDL cholesterol levels have been used to identify HIV-1-infected patients at increased risk for cardiovascular disease [40], ApoA1, the major protein in HDL particles, may be a better predictor of cardiovascular risk [41]. In this small study, we found that ApoA1 levels were not associated with HIV infection or any of the biomarkers studied. Several larger studies have reported lower ApoA1 levels in HIV-1-infected patients than in uninfected controls [42-44], but some have not [45,46]. One study that did not find lower ApoA1 levels in HIV-1-infected patients did report a positive correlation between ApoA1 levels and CD4 count, suggesting that reduced ApoA1 levels were associated with higher degrees of immunosuppression than were found in our study population [45]. Another study reported that ApoA1 levels increase in response to ART [47]. Both of these findings are consistent with the observed reduction and remodeling of HDL in inflammatory states [48,49]. Because ApoA1 is likely to be protective against cardiovascular risk, its potential use as a biomarker to assist in risk stratification of patients merits further study.
This study had several limitations that should be considered in interpreting our results. First, the sample size was small, with only 10 participants in each group, limiting power to detect small differences and weak correlations. Second, patients in the treated group were not all on the same antiretroviral regimen, which may have increased biomarker variability. Third, we used available HIV biomarker data because our small budget and limited sample availability did not permit for retesting of these measures. Fourth, we did not have detailed data on potential confounders of the relationship between HIV disease stage and the biomarkers studied, and the limited sample size would have precluded robust multivariable analysis had we had this data. Therefore, although many of our findings are consistent with previously published results, this study should be considered exploratory and our results should be confirmed in larger studies. The strength of this work is its novelty in being the first study of VWF adhesive activity and ADAMTS13 activity in HIV-1-infected patients and uninfected controls. Given the expense and complexity of measuring these indices of hemostasis and thrombosis, our results are novel and should be of interest to the scientific community.
Overall, our findings indicate that during HIV infection, VWF antigen quantity and adhesive activity are elevated, in spite of an apparent corresponding increase in the quantity and activity of its regulating protease, ADAMTS13. These elevations are associated with higher plasma viral load, and the net effect is a moderately prothrombotic state. Over time, this prothrombotic state may favor the accelerated development of atherosclerosis, microangiopathy, and arterial thrombosis, elevating cardiovascular risk in HIV-1-infected individuals, including those receiving effective ART. Prior reviews of HIV-1-related cardiovascular disease pathogenesis have focused on the role of traditional cardiovascular risk factors, dyslipidemia, inflammation, and immune activation in the development of atherogenic lesions [50-52]. Our findings suggest that VWF self-assembly and related platelet adhesion could also play key roles in this process.
Abbreviations
ADAMTS13: a disintegrin and metalloproteinase with a thrombospondin type 1 motif member 13; ANOVA: analysis of variance; ApoA1: apolipoprotein A1; ART: antiretroviral therapy; CI: confidence interval; ELISA: enzyme-linked immunosorbent assay; HIV: human immunodeficiency virus; HRP: horseradish peroxidase; INSTI: integrase strand transfer inhibitor; NA: not applicable; NNRTI: non-nucleoside reverse transcriptase inhibitor; PI: protease inhibitor; SD: standard deviation; SEAPIP: Seattle Primary Infection Project; TA VWF: total active von Willebrand factor; University of Washington: UW; VWF: von Willebrand factor.
Acknowledgements
The authors would like to acknowledge Michalina Montaño and the other members of the Clinical Epidemiology and Health Services Core and the Clinical and Retrovirology Research Core teams for providing data and specimens from the UW CFAR HIV specimen repository; Jim Mullins, Haiying Zhu, Scott Breen, Breana Hall, and Janine Maenza for providing data and specimens from the HIV Primary Infection cohort; and Angela Dove for providing data and specimens from the Bloodworks Northwest Research Institute Normal Control Registry and Repository.
Funding
Sample analysis was supported by NIH grants R21 HL129526, an HIV and Aging pilot grant through R24 AG044325, R01 HL112633, and R01 HL117639, and by institutional funds from Bloodworks Northwest. The SeaPIP cohort is supported by NIH grant P01 AI57005. The Center for AIDS Research HIV Specimen Repository is funded by NIH grant AI027757. The Bloodworks Northwest Research Institute Normal Control Registry and Repository is supported by institutional funds from Bloodworks Northwest.
Author contributions
SMG, JC, DWC, WCL, and JAL conceived and designed the study; JC, DWC, ML, and JL carried out biochemical analyses; SMG, JC, and DWC analyzed the data; SMG drafted the manuscript; JC, DWC, WCL, and JAL provided input for the final version.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Graham SM, Rajwans N, Jaoko W. et al. Endothelial activation biomarkers increase after HIV-1 acquisition: plasma vascular cell adhesion molecule-1 predicts disease progression. AIDS. 2013;27:1803-1813
2. Graham SM, Rajwans N, Tapia KA. et al. A prospective study of endothelial activation biomarkers, including plasma angiopoietin-1 and angiopoietin-2, in Kenyan women initiating antiretroviral therapy. BMC Infect Dis. 2013;3:263
3. Kuller LH, Tracy R, Belloso W. et al. Inflammatory and coagulation biomarkers and mortality in patients with HIV infection. PLoS Med. 2008;5:e203
4. Nixon DE, Landay AL. Biomarkers of immune dysfunction in HIV. Curr Opin HIV AIDS. 2010;5:498-503
5. Matevosyan K, Sarode R. Thrombosis, microangiopathies, and inflammation. Semin Thromb Hemost. 2015;41:556-562
6. Levi M, van der Poll T, Schultz M. New insights into pathways that determine the link between infection and thrombosis. Neth J Med. 2012;70:114-120
7. Levi M, van der Poll T, Schultz M. Infection and inflammation as risk factors for thrombosis and atherosclerosis. Semin Thromb Hemost. 2012;38:506-514
8. Sadler JE. Biochemistry and genetics of von Willebrand factor. Annu Rev Biochem. 1998;67:395-424
9. Davies PF, Civelek M, Fang Y, Fleming I. The atherosusceptible endothelium: endothelial phenotypes in complex haemodynamic shear stress regions in vivo. Cardiovasc Res. 2013;99:315-327
10. Drouet L, Scrobohaci ML, Janier M, Baudin B. Endothelial cells: target for the HIV1 virus? Nouv Rev Fr Hematol. 1990;32:103-106
11. Janier M, Flageul B, Drouet L. et al. Cutaneous and plasma values of von Willebrand factor in AIDS: a marker of endothelial stimulation? J Invest Dermatol. 1988;90:703-707
12. Lafeuillade A, Alessi MC, Poizot-Martin I. et al. Endothelial cell dysfunction in HIV infection. J Acquir Immune Defic Syndr. 1992;5:127-131
13. Seigneur M, Constans J, Blann A. et al. Soluble adhesion molecules and endothelial cell damage in HIV infected patients. Thromb Haemost. 1997;77:646-649
14. Aukrust P, Bjornsen S, Lunden B. et al. Persistently elevated levels of von Willebrand factor antigen in HIV infection. Downregulation during highly active antiretroviral therapy. Thromb Haemost. 2000;84:183-187
15. Schved JF, Gris JC, Arnaud A. et al. von Willebrand factor antigen, tissue-type plasminogen activator antigen, and risk of death in human immunodeficiency virus 1-related clinical disease: independent prognostic relevance of tissue-type plasminogen activator. J Lab Clin Med. 1992;120:411-419
16. Wolf K, Tsakiris DA, Weber R. et al. Antiretroviral therapy reduces markers of endothelial and coagulation activation in patients infected with human immunodeficiency virus type 1. J Infect Dis. 2002;185:456-462
17. Shrestha S, Irvin MR, Grunfeld C, Arnett DK. HIV, inflammation, and calcium in atherosclerosis. Arterioscler Thromb Vasc Biol. 2014;34:244-250
18. Miller CJ, Baker JV, Bormann AM. et al. Adjudicated morbidity and mortality outcomes by age among individuals with HIV infection on suppressive antiretroviral therapy. PLoS One. 2014;9:e95061
19. Feinstein MJ, Bahiru E, Achenbach C. et al. Patterns of cardiovascular mortality for HIV-infected adults in the United States: 1999 to 2013. Am J Cardiol. 2016;117:214-220
20. Chung DW, Zheng Y, Chen J. et al. High density lipoprotein and apolipoprotein A-I bind von Willebrand factor and prevent its self-association into thick fibers. Blood. 2013;122:194
21. Hulstein JJ, de Groot PG, Silence K. et al. A novel nanobody that detects the gain-of-function phenotype of von Willebrand factor in ADAMTS13 deficiency and von Willebrand disease type 2B. Blood. 2005;106:3035-3042
22. Chen J, Hobbs WE, Le J. et al. The rate of hemolysis in sickle cell disease correlates with the quantity of active von Willebrand factor in the plasma. Blood. 2011;117:3680-3683
23. Wu JJ, Fujikawa K, Lian EC. et al. A rapid enzyme-linked assay for ADAMTS-13. J Thromb Haemost. 2006;4:129-136
24. Mastenbroek TG, van Geffen JP, Heemskerk JW, Cosemans JM. Acute and persistent platelet and coagulant activities in atherothrombosis. J Thromb Haemost. 2015;13:S272-280
25. Graham SM, Mwilu R, Liles WC. Clinical utility of biomarkers of endothelial activation and coagulation for prognosis in HIV infection: A systematic review. Virulence. 2013;4:564-571
26. van Vonderen MG, Hassink EA, van Agtmael MA. et al. Increase in carotid artery intima-media thickness and arterial stiffness but improvement in several markers of endothelial function after initiation of antiretroviral therapy. J Infect Dis. 2009;199:1186-1194
27. Jong E, Louw S, van Gorp EC. et al. The effect of initiating combined antiretroviral therapy on endothelial cell activation and coagulation markers in South African HIV-infected individuals. Thromb Haemost. 2010;104:1228-1234
28. Musselwhite LW, Sheikh V, Norton TD. et al. Markers of endothelial dysfunction, coagulation and tissue fibrosis independently predict venous thromboembolism in HIV. AIDS. 2011;25:787-795
29. Tungsiripat M, El-Bejjani D, Rizk N. et al. Carotid intima media thickness, inflammatory markers, and endothelial activation markers in HIV Patients with lipoatrophy increased at 48 weeks regardless of use of rosiglitazone or placebo. AIDS Res Hum Retroviruses. 2011;27:295-302
30. van den Dries LW, Gruters RA, Hovels-van der Borden SB. et al. von Willebrand factor is elevated in HIV patients with a history of thrombosis. Front Microbiol. 2015;6:180
31. Rutten B, Maseri A, Cianflone D. et al. Plasma levels of active Von Willebrand factor are increased in patients with first ST-segment elevation myocardial infarction: a multicenter and multiethnic study. Eur Heart J Acute Cardiovasc Care. 2015;4:64-74
32. Benjamin M, Terrell DR, Vesely SK. et al. Frequency and significance of HIV infection among patients diagnosed with thrombotic thrombocytopenic purpura. Clin Infect Dis. 2009;48:1129-1137
33. Brecher ME, Hay SN, Park YA. Is it HIV TTP or HIV-associated thrombotic microangiopathy? J Clin Apher. 2008;23:186-190
34. Park YA, Hay SN, Brecher ME. ADAMTS13 activity levels in patients with human immunodeficiency virus-associated thrombotic microangiopathy and profound CD4 deficiency. J Clin Apher. 2009;24:32-36
35. Malak S, Wolf M, Millot GA. et al. Human immunodeficiency virus-associated thrombotic microangiopathies: clinical characteristics and outcome according to ADAMTS13 activity. Scand J Immunol. 2008;68:337-344
36. Visagie GJ, Louw VJ. Myocardial injury in HIV-associated thrombotic thrombocytopenic purpura (TTP). Transfus Med. 2010;20:258-264
37. Rakhmanina N, Wong EC, Davis JC, Ray PE. Hemorrhagic stroke in an adolescent female with HIV-associated thrombotic thrombocytopenic purpura. J AIDS Clin Res. 2014;5:311
38. Allie S, Stanley A, Bryer A. et al. High levels of von Willebrand factor and low levels of its cleaving protease, ADAMTS13, are associated with stroke in young HIV-infected patients. Int J Stroke. 2015;10:1294-1296
39. Chen J, Özpolat T, Wang Y. et al. Potential mechanisms for enhanced activity of Von Willebrand Factor in patients with sickle cell disease. Blood. 2016;128:3716. (Abstract)
40. Duprez DA, Kuller LH, Tracy R. et al. Lipoprotein particle subclasses, cardiovascular disease and HIV infection. Atherosclerosis. 2009;207:524-529
41. Walldius G, Jungner I. Apolipoprotein A-1 versus HDL cholesterol in the prediction of risk for myocardial infarction and stroke. Curr Opin Cardiol. 2007;22:359-367
42. Haarburger D, Bergstrom J, Pillay TS. Serum proteome changes following human immunodeficiency virus infection. Clin Lab. 2013;59:639-646
43. Constans J, Pellegrin JL, Peuchant E. et al. Plasma lipids in HIV-infected patients: a prospective study in 95 patients. Eur J Clin Invest. 1994;24:416-420
44. Syed SS, Balluz RS, Kabagambe EK. et al. Assessment of biomarkers of cardiovascular risk among HIV type 1-infected adolescents: role of soluble vascular cell adhesion molecule as an early indicator of endothelial inflammation. AIDS Res Hum Retroviruses. 2013;29:493-500
45. Rose H, Hoy J, Woolley I. et al. HIV infection and high density lipoprotein metabolism. Atherosclerosis. 2008;199:79-86
46. Rose H, Low H, Dewar E. et al. The effect of HIV infection on atherosclerosis and lipoprotein metabolism: a one year prospective study. Atherosclerosis. 2013;229:206-211
47. Baker JV, Neuhaus J, Duprez D. et al. Inflammation predicts changes in high-density lipoprotein particles and apolipoprotein A1 following initiation of antiretroviral therapy. AIDS. 2011;25:2133-2142
48. Feingold KR, Grunfeld C. The effect of inflammation and infection on lipids and lipoproteins. In: (ed.) De Groot LJ, Chrousos G, Dungan K. et al. Endotext [Internet]. South Dartmouth, MA: MDText.com, Inc. 2015:2000 -
49. van der Westhuyzen DR, de Beer FC, Webb NR. HDL cholesterol transport during inflammation. Curr Opin Lipidol. 2007;18:147-151
50. d'Ettorre G, Ceccarelli G, Pavone P. et al. What happens to cardiovascular system behind the undetectable level of HIV viremia? AIDS Res Ther. 2016;13:21
51. Haser GC, Sumpio B. Systemic and cell-specific mechanisms of vasculopathy induced by human immunodeficiency virus and highly active antiretroviral therapy. J Vasc Surg. 2017;65:849-859
52. Nou E, Lo J, Hadigan C, Grinspoon SK. Pathophysiology and management of cardiovascular disease in patients with HIV. Lancet Diabetes Endocrinol. 2016;4:598-610
Author contact
![]() Corresponding author: Susan M. Graham, MD, MPH, PhD, University of Washington, Box 359909, 325 Ninth Avenue, Seattle, WA 98104-2499. E-mail: grahamsmwashington.edu; Phone: (206) 543-4278; Fax: (206) 543-4818
Corresponding author: Susan M. Graham, MD, MPH, PhD, University of Washington, Box 359909, 325 Ninth Avenue, Seattle, WA 98104-2499. E-mail: grahamsmwashington.edu; Phone: (206) 543-4278; Fax: (206) 543-4818