Impact Factor ISSN: 1449-1907

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2018; 15(13):1508-1516. doi:10.7150/ijms.28106 This issue Cite
Research Paper
Simvastatin Protects Heart from Pressure Overload Injury by Inhibiting Excessive Autophagy
Feifei Su1 ![]() *, Miaoqian Shi2*, Jian Zhang3*, Qiangsun Zheng4, Dongwei Zhang1, Wei Zhang1, Haichang Wang1, Xue Li1
*, Miaoqian Shi2*, Jian Zhang3*, Qiangsun Zheng4, Dongwei Zhang1, Wei Zhang1, Haichang Wang1, Xue Li1
1. Department of Cardiology, Tangdu Hospital, Fourth Military Medical University, Xi'an, 710038, China.
2. Department of Cardiology, PLA Army General Hospital, No.5 Nanmen Cang, Dongcheng District, Beijing, 100700, China.
3. Department of Cardiology, Beijing Chest Hospital Heart Center, Capital Medical University, No.9. Beiguan Grand Street, Tongzhou District, Beijing, 101149, China.
4. Division of Cardiology, Second Affiliated Hospital of JiaoTong University, Xi'an, 710004, China.
* These authors contributed equally to this work.
Received 2018-6-25; Accepted 2018-8-29; Published 2018-10-20
Abstract
Cardiac hypertrophy is an independent predictor of cardiovascular morbidity and mortality. To identify the mechanisms by which simvastatin inhibits cardiac hypertrophy induced by pressure overload, we determined effects of simvastatin on 14-3-3 protein expression and autophagic activity. Simvastatin was administered intragastrically to Sprague-Dawley (SD) rats before abdominal aortic banding (AAB). Neonatal rat cardiomyocytes (NRCs) were treated with simvastatin before angiotensin II (AngII) stimulation. 14-3-3, LC3, and p62 protein levels were determined by western blot. Autophagy was also measured by the double-labeled red fluorescent protein-green fluorescent protein autophagy reporter system. Simvastatin alleviated excessive autophagy, characterized by a high LC3II/LC3I ratio and low level of p62, and blunted cardiac hypertrophy while increasing 14-3-3 protein expression in rats that had undergone AAB. In addition, it increased 14-3-3 expression and inhibited excessive autophagy in NRCs exposed to AngII. Our study demonstrated that simvastatin may inhibit excessive autophagy, increase 14-3-3 expression, and finally exert beneficial effects on cardioprotection against pressure overload.
Keywords: 14-3-3, autophagy, hydroxymethylglutaryl-CoA reductase inhibitors, simvastatin, hypertrophy
Introduction
Autophagy is a catabolic process that degrades damaged cellular proteins and organelles. It is essential for maintaining cellular homeostasis[1]. However, prolonged and excessive autophagy is maladaptive and can result in cell death[2]. Hypertrophic cardiomyocyte growth and dysfunction are associated with various forms of heart diseases, such as hypertension and coronary heart disease[3, 4]. Recently, accumulating evidence has revealed a tight link between cardiomyocyte autophagy and cardiac hypertrophy[5]. Excessive autophagy can occur in hypertrophied cardiac myocytes[6]. Thus, therapeutic strategies targeting autophagy for cardiac hypertrophy are attractive.
Hydroxymethylglutaryl coenzyme A reductase inhibitors (statins) have been shown to reduce cardiovascular-related mobility and mortality independently of their cholesterol-lowering function [7]. Simvastatin, a statin, has been shown to have pleiotropic effects, such as reducing cardiac hypertrophy [8, 9]. However, the effects of simvastatin on autophagy in the hypertrophied heart have not been clearly documented thus far.
Members of the 14-3-3 protein family regulate signal transduction, apoptosis, and checkpoint control pathways[10]. These proteins have been shown to modulate autophagy in cancers[11-13]. Recently, 14-3-3 proteins were indicated to play important roles in cardiomyocytes. Dominant-negative 14-3-3 promotes cardiac apoptosis [14]. 14-3-3 protects against diabetic cardiomyopathy [15]. Additionally, the 14-3-3γ protein attenuates lipopolysaccharide- induced cardiomyocyte injury through the Bcl-2 family pathway[16] and has a protective role against mitochondrion-mediated cardiomyocyte apoptosis [17]. Most previous studies were focused on the role of 14-3-3 in the diabetic heart, and less attention has been paid to the effects of the protein on cardiac hypertrophy.
Therefore, we hypothesized that simvastatin might prevent cardiomyocyte hypertrophy by regulating 14-3-3 and autophagy. To test the hypothesis, we used an in vitro model of angiotensin II (AngII)-induced neonatal rat ventricular cardiomyocytes (NRCs) and an in vivo model of abdominal aortic banding (AAB)-induced heart hypertrophy. We found that simvastatin partially reversed cardiomyocyte hypertrophy by regulating autophagy and 14-3-3 protein expression.
Materials and Methods
Ethical approval of the present study protocol
The experimental protocol was implemented according to the tenets of the Declaration of Helsinki and the procedures involving animals were approved by the Animal Care and Welfare Ethics Committee of the Fourth Military Medical University (FMMU, Xi'an, China).
AAB surgical procedures
Male Sprague-Dawley (SD) rats were purchased from the Animal Center of the FMMU. Simvastatin was purchased from Merck, Sharp & Dohme Inc. Other chemicals were of laboratory grade. Forty rats (weight, 180-220 g; age, 9-12 weeks) were divided into four groups (n = 10): SHAM group, comprising rats in which surgery was performed without banding of the aorta; SHAM+SIM group, comprising SHAM rats treated with simvastatin (40 mg/kg·day); AAB group, comprising rats in which AAB was performed; and AAB+SIM group, comprising AAB rats treated with simvastatin 1 day before the AAB procedure. AAB was performed as previously described with minor modifications[18]. All rats were anesthetized with an intraperitoneal injection of 45 mg/kg pentobarbital sodium. In brief, after anesthetization, the rats were restrained and a midline abdominal incision was made under sterile conditions. The abdominal aorta above the renal arteries was dissected. A ligature (4-0 silk sutures) was positioned around the aorta along with a blunt 22-gauge needle, which was then withdrawn, leading to constriction (approximately 50%) of the aorta. After surgery, each rat received 30 kU penicillin intramuscularly as antibiotic therapy. Simvastatin was dissolved in 0.9% saline solution. The rats received placebo (0.9% saline solution) or simvastatin intragastrically every day for 7 weeks before surgery.
M-mode echocardiography and left ventricular-weight index measurements
Echocardiograms were performed by Dr. Yunyan Duan (Department of Ultrasonography, Xijing Hospital, Xi'an, China) as previously reported [9, 19]. In brief, the rats were anesthetized as previously described with an intraperitoneal injection of 45 mg/kg pentobarbital sodium. A 2-D axis view of the left ventricle was obtained with a 7.5-MHz transducer and the M-mode tracings were then recorded to measure the indexes of the left ventricle: left ventricular posterior wall thickness of diastasis (LVPWTd) and interventricular septal thickness of diastasis (IVSTd). The hearts of the rats were removed and the left ventricles were dissected. The left ventricle was weighed and divided by body weight in order to calculate the left ventricle weight index (LVWI).
Isolation of adult rat ventricular cardiomyocytes
Adult rat ventricular cardiomyocytes were isolated as reported previously[20]. In brief, the rats were anesthetized with pentobarbital sodium 45mg/kg and the hearts were quickly removed from the chest; thereafter, retrograde aortic perfusion with a Ca2+-free bicarbonate-based buffer was performed at 37°C for 3 min. Enzymatic digestion was then initiated until the heart became swollen. The left ventricle was rapidly removed 5-10 min later, cut into several chunks, and further digested in a shaker for 10 min. The supernatant containing the dispersed myocytes was filtered, centrifuged at 500 rpm for 1 min , and re-suspended. Finally, the cardiomyocytes were plated on laminin-coated glass coverslips before measurements.
Confocal microscopy
Confocal microscopy was used to detect 14-3-3 expression in adult rat cardiomyocytes as described previously [21]. Briefly, adult rat cardiomyocytes were isolated and plated on laminin-coated 4-well chamber slides (Lab-Tek). Expression of the 14-3-3 protein was detected using a primary mouse antibody (Santa Cruz, 1:100), and α-sarcomeric actinin was detected using a rabbit antibody (Sigma-Aldrich, 1:100). The secondary antibodies used were Alexa Fluor 594-labeled goat anti-mouse antibody and Alexa Fluor 488-labeled goat anti-rabbit antibody (Invitrogen, 1:500), and the mounting medium contained DAPI (Vector Laboratories). A Carl Zeiss 710 confocal microscope with ZEN software was used for visualizing 14-3-3, α-actinin, and DAPI. Total laser intensity and photomultiplier gain were constant for all the groups and settings, and data were verified by two independent observers who were blinded to the experimental group. A minimum of three coverslips was used for each experimental group, and at least three cell images were acquired from each coverslip.
Neonatal rat cardiomyocyte culture and treatment
Monolayer cultures of NRCs were prepared as described previously [22]. Ventricular myocardium from neonatal SD rats (aged 1-3d, FMMU) was homogenized and dissociated with collagenase II. The cell suspension was plated onto a 10-cm dish for 1 h, allowing enrichment for cardiomyocytes by differential adhesion. The supernatant was then plated on to new dishes with Dulbecco's modified Eagle's medium (DMEM, Gibco-BRL) containing 1% penicillin-streptomycin and 10% fetal bovine serum (FBS) under 5% CO2 at 37°C. The remaining fibroblasts were minimized by the addition of 10 μM 5-bromodeoxyuridine for 24 h. NRCs were divided into five groups: CTRL group, comprising cells treated with dimethylsulfoxide (DMSO); CTRL+SIM group, comprising cells treated with simvastatin; AngII group, comprising cells treated with AngII; AngII+SIM group, comprising cells pretreated with simvastatin before exposure to AngII; and rapamycin group, comprising cells treated with rapamycin alone. The cells were treated with simvastatin and AngII as previously described [23]. Briefly, after 24 h of serum starvation, the cells were treated with 1 μM simvastatin (sodium salt, MERCK Biosciences) in a treatment medium (DMEM containing 0.5% serum). AngII (ALEXIS BIOCHEMICALS) was added to achieve a final concentration of 100 nM 12 h after the addition of simvastatin. The cells were then incubated in the presence of both drugs for a further 24 h. Rapamycin was added to achieve a final concentration of 0.5 mM for 2 h before the cells were harvested.
Western blots
Western blots were performed as described previously[9]. Briefly, the rats' left ventricle tissue and NRC samples were lysed in a lysis buffer containing 50 mM Tris, 150 mM NaCl, 1% Nonidet P-40, 0.25% superoxide dismutase, 1 mM EDTA, 1 mM NaF, 1 mM Na3VO3, 1 mM phenylmethylsulphonyl fluoride, and a proteinase inhibitor cocktail (Roche). The samples were evaluated by SDS-PAGE. Briefly, total protein from the samples was determined before being subjected to polyacrylamide gel electrophoresis and being transferred to a nitrocellulose (NC) membrane. The NC membrane was immunoblotted with an anti-14-3-3 antibody (Santa Cruz; 1:1,000), LC3 antibody (Santa Cruz; 1:1,000), p62 antibody (Santa Cruz; 1:1,000), and anti-β-actin antibody (Santa Cruz; 1:10,000). β-Actin protein served as a loading control. Protein bands were evaluated by densitometry using the Odyssey infrared imaging system (LI-COR).
Autophagy mRFP-GFP-LC3 report system
The autophagy flux was measured as described previously[2, 21]. In brief, the NRCs were plated on coverslips and infected with an mRFP-GFP-LC3 lentivirus (Hanbio. Co. LTD, Shanghai, China) at a multiplicity of infection of 8. The NRCs were exposed to the virus overnight, after which media were aspirated and fresh media were applied. Then the NRCs were subjected to different treatments and fixed with paraformaldehyde in phosphate-buffered saline (PBS). After rinsing with PBS, the cells were permeabilized with 0.3% Triton X-100 in 10% normal goat serum blocking solution (Invitrogen, Life Technologies) for 60 min. Coverslips were mounted on slides with Hardset anti-fade mounting medium (Vector Laboratories), and confocal imaging was performed as described above (mRFP 594 nm ex., 667 nm em.; GFP 488 nm ex., 543 nm em.). The puncta of six cells in each experimental group were counted after obtaining digital images. The number of yellow puncta in the merged channel represented the number of autophagosomes. The number of autolysosomes (as a result of autophagosome-lysosome fusion) was represented by the number of red puncta.
Statistical analysis
Data are presented as the mean ± SEM. One-way ANOVA was used for statistical analysis and P<0.05 was considered statistically significant.
Results
Simvastatin partially blunted hypertrophy in AAB rats
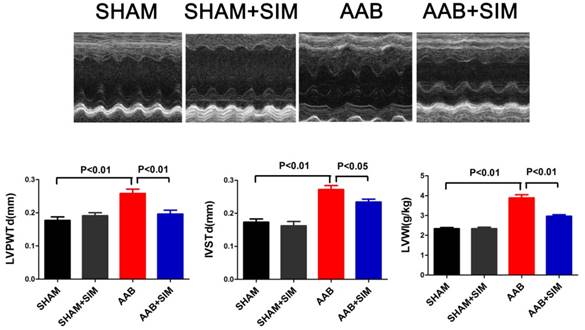
In the present study, the LVWI in the AAB group (3.89±0.16 g/kg; P<0.01) significantly increased, compared to that in the SHAM group (2.34±0.06 g/kg) 7 weeks after surgery. The LVPWTd (0.26±0.01 mm; P<0.01) and IVSTd (0.27±0.01 mm; P<0.01) in the AAB group markedly increased compared to that in the SHAM group (LVPWTd, 0.18±0.01 mm; IVSTd, 0.17±0.01) 7 weeks after surgery. Thus, AAB induced cardiac left ventricular hypertrophy in vivo 7 weeks after surgery, compared to that in the SHAM group. Simvastatin administration decreased the LVWI (2.97±0.08 g/kg; P<0.01), LVPWTd (0.20±0.01 mm; P<0.01), and IVSTd (0.23±0.01 mm; P<0.05) compared to that in the AAB group (Figure 1).
Simvastatin alleviated cardiac hypertrophy in AAB rats. The upper panel shows M-mode echocardiograms of rats from different groups. The bottom panel showed LVPWTd, IVSTd, and LVWI, respectively, from the SHAM (n=10), SHAM+SIM (n=10), AAB (n=10), and AAB+SIM (n=10) groups.
Simvastatin inhibited excessive autophagic activity in the myocardium of AAB rats
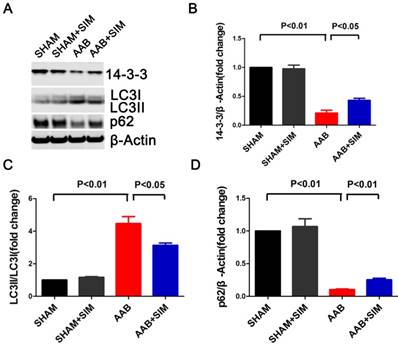
To assess the effects of simvastatin on autophagy in cardiac hypertrophy, we measured LC3 and p62 protein levels. The LC3II/LC3I ratio, which reflects autophagy, excessively increased in the AAB group (4.47±0.43-fold change; P<0.01) compared to that in the SHAM group. Additionally, the protein level of p62, indicating the amount of autophagy cargo delivered to lysosomes for degradation, significantly decreased in the AAB group (0.10±0.01-fold change; P<0.01) compared to that in the SHAM group. Simvastatin partially restored p62 protein level (0.25±0.02-fold change; P<0.01) while decreasing the LC3 II/LC3I ratio (3.13±0.15-fold change; P<0.05) compared to that in the AAB group (Figure 2). Thus, there was an excessive autophagy in the pressure-overloaded myocardium, which is consistent with the finding in a previous report [2]. Further, simvastatin inhibited this excessive autophagy in the pressure-overloaded myocardium.
Simvastatin inhibited autophagy and preserved 14-3-3 expression in the myocardium of AAB rats. A, Representative western blots of proteins in the myocardium of the SHAM (n=3), SHAM+SIM (n=3), AAB (n=3), and AAB+SIM (n=3) groups. B-D, Quantitative analysis of 14-3-3, LC3, and p62 proteins levels in different groups respectively. β-Actin served as the loading control and data were normalized to the protein level measured under SHAM conditions.
Simvastatin partially restored 14-3-3 protein expression in the myocardium of AAB rats
The 14-3-3 family proteins can regulate autophagy and play major roles in cardiomyopathy [10, 24, 25]. Therefore, we attempted to determine if simvastatin could regulate 14-3-3 protein expression in the rat hypertrophied heart. In the present study, AAB decreased 14-3-3 protein (0.21±0.05-fold change; P<0.01) in the myocardium compared to that in the SHAM group. Simvastatin significantly restored the 14-3-3 protein level (0.43±0.04-fold change; P<0.05) compared to that in the AAB group (Figure 2).
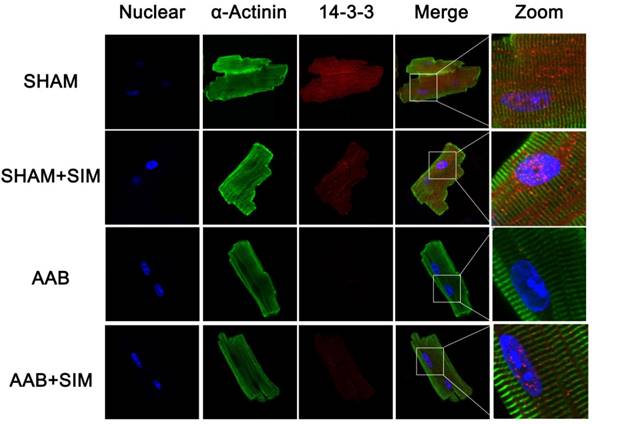
Furthermore, to confirm the protein expression of 14-3-3 morphologically in adult cardiomyocytes, we used confocal microscopy, the results of which were consistent with those of western blot, as seen in Figure 3. The 14-3-3 protein appeared more prominent in both the cytoplasm and nucleus of adult rat cardiomyocytes from the SHAM and SHAM+SIM groups. In contrast, adult cardiomyocytes from the AAB group showed decreased 14-3-3 protein expression. Simvastatin appeared to partially restore 14-3-3 protein in adult cardiomyocytes from the AAB+SIM group (Figure 3).
Confocal microscopy images showing preservation of 14-3-3 expression in isolated adult rat cardiomyocytes by simvastatin. Representative confocal microscopy images of 14-3-3 protein localization (red) in isolated adult rat cardiomyocytes from the SHAM (n=3), SHAM+SIM (n=3), AAB (n=3), and AAB+SIM (n=3) groups. The 14-3-3 expression level in different groups was confirmed by western blot in Figure 2. Blue staining represents DAPI-stained nuclei. Green staining represents α-sarcomeric actinin.
Simvastatin inhibited excessive autophagic activity in NRCs exposed to AngII
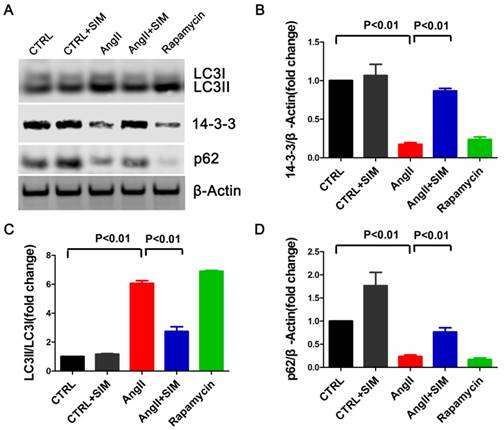
To assess whether the findings of studies on simvastatin in rats were relevant to NRCs in vitro, we measured LC3 and p62 protein levels in NRCs exposed to AngII. As seen in Figure 4, the LC3II/LC3I ratio significantly increased in the AngII group (6.07±0.18-fold change; P<0.01) compared to that in the CTRL group. In addition, the p62 protein level significantly decreased in the AngII group (0.23±0.03-fold change; P<0.01) compared to that in the CTRL group. Simvastatin partially restored the p-62 protein level (0.77±0.09-fold change; P<0.01) while decreasing the LC3 II/LC3I ratio (2.73±0.33-fold change; P<0.01) compared to those in the AngII group. The p62 protein represents a change in the actual amount of autophagy occurring in the cells, promotes aggresome formation, and protects cardiomyocytes against stress[26]. Thus, the findings indicated that simvastatin alleviated excessive autophagy in NRCs induced by AngII.
Simvastatin inhibited autophagy and preserved 14-3-3 expression in NRCs exposed to AngII. A, Representative western blots of proteins in NRCs from the CTRL (n=3), CTRL+SIM (n=3), AngII (n=3), AngII+SIM (n=3), and rapamycin (n=3) groups. B-D, Quantitative analysis of 14-3-3, LC3, and p62 protein levels from different groups respectively. β-Actin served as the loading control and data were normalized to the protein level measured under CTRL conditions.
To further confirm the alterations in protein levels of autophagic markers, such as LC3II/LC3I, we measured autophagic flux using a red fluorescent protein (RFP)-green fluorescent protein (GFP) double-labeled reporter system. The NRCs were transfected with RFP-GFP-LC3 adenovirus. GFP is sensitive to the acidic pH inside the lysosome whereas RFP is resistant to acidic pH. Thus, in the milieu of the autolysosome, the GFP fluorescence is quenched, leaving predominantly red puncta. Finally, the yellow puncta represent the merged fluorescence of GFP and RFP and reflect the presence of autophagosomes, whereas red puncta represent RFP alone. During excessive autophagy, there are more autophagosomes and autophagosome-lysosome fusions, resulting in an increase in the presence of both red and yellow puncta. Conversely, when autophagy is impeded, there are fewer autophagosomes and autophagosome-lysosome fusions, resulting in the presence of fewer red and yellow puncta[21]. Consistent with the results of western blots, the statistical analyses in this study indicated that AngII induced an increase in the numbers of both autophagosomes (yellow puncta, 30.20±1.88 puncta/cell; P<0.01) and autolysosomes (red puncta, 29.00±4.35 puncta/cell; P<0.01) compared to those in the CTRL group (yellow puncta, 2.40±0.51 puncta/cell; red puncta, 1.80±0.37 puncta/cell. On the other hand, simvastatin inhibited an increase in the numbers of both yellow (5.20±2.99 puncta/cell; P<0.01) and red puncta (5.60±2.23 puncta/cell; P<0.01), as seen in Figure 5.
Simvastatin inhibited excessive autophagy in NRCs exposed to AngII by mRFP-GFP-LC3 report system. The left panel represents fluorescent images of NRCs obtained by confocal microscopy. The right panel shows the quantification of the number of autophagosome (yellow) and autolysosome (red) puncta in NRCs from the CTRL (n=6), CTRL+SIM (n=6), AngII (n=6), AngII+SIM (n=6), and rapamycin (n=6) groups.
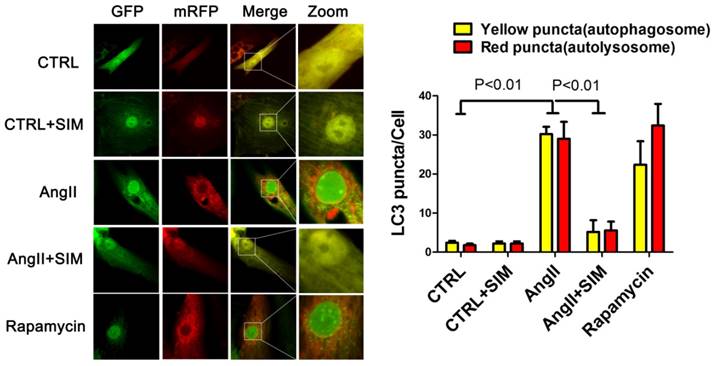
Simvastatin partially restored 14-3-3 protein level in NRCs exposed to AngII
To confirm if simvastatin could manipulate 14-3-3 in NRCs when the cells were exposed to AngII, we measured the 14-3-3 protein level. As seen in Figure 4, AngII effected a significant decrease in 14-3-3 protein expression (0.17±0.02-fold change; P<0.01), whereas simvastatin substantially increased the protein level of 14-3-3 (0.87±0.03 fold change; P<0.01). The result was consistent with the results obtained in vivo, supporting the hypothesis that simvastatin could increase the level of 14-3-3 during cell stress induced by AngII. Interestingly, rapamycin, the activator of autophagy, directly decreased the protein level of 14-3-3 (0.24±0.03-fold change) compared to that in the CTRL group (Figure 4).
Discussion
The main findings of the present study were as follows: 1) Simvastatin partially inhibited left ventricular hypertrophy induced by AAB in rats. 2) Autophagy excessively increased in the myocardium of AAB rats and was partially inhibited by simvastatin administration. The 14-3-3 protein level significantly decreased in the myocardium of AAB rats and was partially restored by simvastatin. 3) Simvastatin blunted the excessive autophagy and increased 14-3-3 protein level in NRCs exposed to AngII. 4) Rapamycin directly decreased 14-3-3 protein expression while activating autophagy in NRCs.
The beneficial effects of statins are far greater than those associated with cholesterol lowering alone, and they appear to afford cardiovascular protection at multiple levels, primarily as a result of their pleiotropic activity[27]. Simvastatin, a statin, has been shown to reduce cardiovascular-related morbidity and mortality independently of its cholesterol-lowering function in clinical trials. In our previous study, we reported that simvastatin inhibited AAB-induced left ventricular hypertrophy, which is an independent predictor of cardiovascular morbidity and mortality[9, 28]. Although it has been reported that statins inhibited autophagy by regulating the mTOR signaling pathway in diabetes [29], few studies have investigated whether simvastatin can regulate autophagy in cardiac hypertrophy. In the present study, simvastatin blunted left ventricular hypertrophy and reduced excessive autophagy, characterized by a high LC3 II/LC3I ratio and low p62 level in the myocardium of AAB rats. In addition, we found that 14-3-3 protein level significantly decreased in the myocardium of AAB rats. We also showed that simvastatin partially preserved 14-3-3 protein level in AAB rats. The present study indicates that 14-3-3 may be involved in the pleiotropic effects of simvastatin on hypertrophy, which is consistent with a previous study showing that 14-3-3 inhibits cardiomyocyte hypertrophy[24, 30]. In addition, as 14-3-3 inhibits autophagy in cancer cells, it may play a role in inhibiting autophagy in cardiomyocytes. Thus, simvastatin may regulate 14-3-3 protein expression, which could inhibit excessive autophagy and protect the heart against hypertrophy.
Autophagy is important in the heart for maintaining homeostasis, as modest autophagy can recycle proteins and nutrients in cells whereas excessive autophagy may lead to extreme degradation of critical cellular proteins and organelles, leading to cell death [2]. In the present study, in order to confirm that the alterations in the markers of autophagy in the myocardium are consistent with the changes in NRCs, we measured LC3 and p62 protein levels in NRCs exposed to AngII, which can induce cardiomyocyte hypertrophy[31]. AngII increased the LC3II/LC3I ratio and decreased the p62 protein level significantly in NRCs. Simvastatin blunted the effects of AngII on autophagy in NRCs. Furthermore, we also used an LC3 reporter system to measure autophagy. The system takes advantage of the fact that LC3I is post-translationally modified by a ubiquitin-like system that converts it to its lapidated LC3II form. LC3II is sequestered into autolysosomes in which it is degraded[32]. It showed that both autophagosomes and autolysosomes were induced significantly by AngII in NRCs, indicating excessive autophagy occurred, which is consistent with previous findings [2]. Simvastatin decreased autophagosomes and autolysosomes in NRCs exposed to AngII. In addition, AngII reduced 14-3-3 protein and simvastatin partially preserved 14-3-3 protein in NRCs. These results are consistent with the results in vivo.
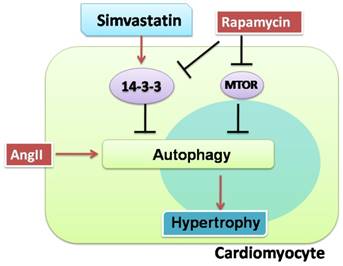
Another interesting observation was that rapamycin, an autophagy activator, not only activated excessive autophagy but also inhibited 14-3-3 protein expression in NRCs in the present study. While rapamycin inhibiting 14-3-3 in myocytes has not been reported, the finding is consistent with those of previous studies demonstrating that 14-3-3 inhibits autophagy in cancer cells[10] and rapamycin induces autophagy by removing the inhibitory effect of mechanistic target of rapamycin (mTOR) on autophagy induction[33]. Thus, it appears that rapamycin can also activate autophagy by regulating the 14-3-3 protein (Figure 6).
Proposed protective effects of simvastatin against cardiomyocyte hypertrophy. Cardiomyocytes are susceptible to hypertrophy when they were exposed to AngII. Simvastatin protected cardiomyocytes from hypertrophy, possibly by regulating 14-3-3 and autophagy. There may be a relationship between 14-3-3 and autophagy in cardiomyocytes. The red solid arrows show the positive effects, while the black solid arrows show the negative effects.
Despite successful efforts to prevent cardiac hypertrophy in different diseases, cardiac hypertrophy remains a major clinical problem that has failed to be influenced by multiple pharmacological approaches. The findings that simvastatin increased 14-3-3 expression and decreased excessive autophagy suggest that 14-3-3 and autophagy could serve as therapeutic targets for cardiac protection. Nonetheless, although increasing 14-3-3, inhibiting autophagy, or regulating both 14-3-3 and autophagy could protect cardiomyocytes against hypertrophy, these strategies have not resulted in medicines with safe therapeutic effects in vivo. Thus, mechanisms underlying the effects of statins on 14-3-3 and autophagy need to be evaluated. Hopefully, medicines that potentially modulate autophagy and 14-3-3 will be investigated for therapeutic purposes in the near future.
Abbreviations
SD: Sprague-Dawley; AAB: abdominal aortic banding; AngII: angiotensin II; NRCs: neonatal rat cardiomyocytes; LVPWTd: left ventricular posterior wall thickness of diastasis; IVSTd: interventricular septal thickness of diastasis; LVWI: left ventricle weight index; FBS: fetal bovine serum; DMSO: dimethylsulfoxide; NC: nitrocellulose; PBS: phosphate-buffered saline; mTOR: mechanistic target of rapamycin.
Sources of Funding
The present study was supported by the National Natural Science Foundation of China (Grant No. 81300077).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Ouyang C, You J, Xie Z. The interplay between autophagy and apoptosis in the diabetic heart[J]. J Mol Cell Cardiol. 2014;71:71-80
2. Cao T T, Chen H H, Dong Z, Xu Y W, Zhao P, Guo W, Wei H C, Zhang C, Lu R. Stachydrine Protects Against Pressure Overload-Induced Cardiac Hypertrophy by Suppressing Autophagy[J]. Cell Physiol Biochem. 2017;42(1):103-114
3. Trembley M A, Quijada P, Agullo-Pascual E, Tylock K M, Colpan M, Dirkx R J, Myers J R, Mickelsen D M, de Mesy B K, Rothenberg E, Moravec C S, Alexis J D, Gregorio C C, Dirksen R T, Delmar M, Small E M. Mechanosensitive Gene Regulation by Myocardin-Related Transcription Factors is Required for Cardiomyocyte Integrity in Load-Induced Ventricular Hypertrophy[J]. Circulation. 2018
4. Mohan M, McSwiggan S, Baig F, Rutherford L, Lang C C. Metformin and its effects on myocardial dimension and left ventricular hypertrophy in normotensive patients with coronary heart disease (the MET-REMODEL study): rationale and design of the MET-REMODEL study[J]. Cardiovasc Ther. 2015;33(1):1-8
5. Li Z, Wang J, Yang X. Functions of autophagy in pathological cardiac hypertrophy[J]. Int J Biol Sci. 2015;11(6):672-678
6. Baba S P, Zhang D, Singh M, Dassanayaka S, Xie Z, Jagatheesan G, Zhao J, Schmidtke V K, Brittian K R, Merchant M L, Conklin D J, Jones S P, Bhatnagar A. Deficiency of aldose reductase exacerbates early pressure overload-induced cardiac dysfunction and autophagy in mice[J]. J Mol Cell Cardiol. 2018;118:183-192
7. Randomised trial of cholesterol lowering in 4444 patients with coronary heart disease. the Scandinavian Simvastatin Survival Study (4S)[J]. Lancet. 1994;344(8934):1383-1389
8. Patel R, Nagueh S F, Tsybouleva N, Abdellatif M, Lutucuta S, Kopelen H A, Quinones M A, Zoghbi W A, Entman M L, Roberts R, Marian A J. Simvastatin induces regression of cardiac hypertrophy and fibrosis and improves cardiac function in a transgenic rabbit model of human hypertrophic cardiomyopathy[J]. Circulation. 2001;104(3):317-324
9. Su F, Shi M, Yan Z, Ou D, Li J, Lu Z, Zheng Q. Simvastatin modulates remodeling of Kv4.3 expression in rat hypertrophied cardiomyocytes[J]. Int J Biol Sci. 2012;8(2):236-248
10. Jia H, Liang Z, Zhang X, Wang J, Xu W, Qian H. 14-3-3 proteins: an important regulator of autophagy in diseases[J]. Am J Transl Res. 2017;9(11):4738-4746
11. Kidd M E, Shumaker D K, Ridge K M. The role of vimentin intermediate filaments in the progression of lung cancer[J]. Am J Respir Cell Mol Biol. 2014;50(1):1-6
12. Zhong J, Kong X, Zhang H, Yu C, Xu Y, Kang J, Yu H, Yi H, Yang X, Sun L. Inhibition of CLIC4 enhances autophagy and triggers mitochondrial and ER stress-induced apoptosis in human glioma U251 cells under starvation[J]. PLoS One. 2012;7(6):e39378
13. He C L, Bian Y Y, Xue Y, Liu Z X, Zhou K Q, Yao C F, Lin Y, Zou H F, Luo F X, Qu Y Y, Zhao J Y, Ye M L, Zhao S M, Xu W. Pyruvate Kinase M2 Activates mTORC1 by Phosphorylating AKT1S1[J]. Sci Rep. 2016;6:21524
14. Zhang S, Ren J, Zhang C E, Treskov I, Wang Y, Muslin A J. Role of 14-3-3-mediated p38 mitogen-activated protein kinase inhibition in cardiac myocyte survival[J]. Circ Res. 2003;93(11):1026-1028
15. Thandavarayan R A, Watanabe K, Ma M, Veeraveedu P T, Gurusamy N, Palaniyandi S S, Zhang S, Muslin A J, Kodama M, Aizawa Y. 14-3-3 protein regulates Ask1 signaling and protects against diabetic cardiomyopathy[J]. Biochem Pharmacol. 2008;75(9):1797-1806
16. Liu D, Yi B, Liao Z, Tang L, Yin D, Zeng S, Yao J, He M. 14-3-3gamma protein attenuates lipopolysaccharide-induced cardiomyocytes injury through the Bcl-2 family/mitochondria pathway[J]. Int Immunopharmacol. 2014;21(2):509-515
17. Sreedhar R, Arumugam S, Thandavarayan R A, Giridharan V V, Karuppagounder V, Pitchaimani V, Afrin R, Miyashita S, Nomoto M, Harima M, Gurusamy N, Suzuki K, Watanabe K. Myocardial 14-3-3eta protein protects against mitochondria mediated apoptosis[J]. Cell Signal. 2015;27(4):770-776
18. Berni R, Savi M, Bocchi L, Delucchi F, Musso E, Chaponnier C, Gabbiani G, Clement S, Stilli D. Modulation of actin isoform expression before the transition from experimental compensated pressure-overload cardiac hypertrophy to decompensation[J]. Am J Physiol Heart Circ Physiol. 2009;296(5):H1625-H1632
19. Weinberg E O, Schoen F J, George D, Kagaya Y, Douglas P S, Litwin S E, Schunkert H, Benedict C R, Lorell B H. Angiotensin-converting enzyme inhibition prolongs survival and modifies the transition to heart failure in rats with pressure overload hypertrophy due to ascending aortic stenosis[J]. Circulation. 1994;90(3):1410-1422
20. Zhou Y Y, Wang S Q, Zhu W Z, Chruscinski A, Kobilka B K, Ziman B, Wang S, Lakatta E G, Cheng H, Xiao R P. Culture and adenoviral infection of adult mouse cardiac myocytes: methods for cellular genetic physiology[J]. Am J Physiol Heart Circ Physiol. 2000;279(1):H429-H436
21. Su F, Myers V D, Knezevic T, Wang J, Gao E, Madesh M, Tahrir F G, Gupta M K, Gordon J, Rabinowitz J, Ramsey F V, Tilley D G, Khalili K, Cheung J Y, Feldman A M. Bcl-2-associated athanogene 3 protects the heart from ischemia/reperfusion injury[J]. JCI Insight. 2016;1(19):e90931
22. Guo W G, Yu Z B, Xie M J. [Protein kinase Cdelta is possibly involved in the transition from hypertrophy to apoptosis of myocardiocytes][J]. Sheng Li Xue Bao. 2006;58(3):269-274
23. Guo W G, Su F F, Yuan L J, Yang G D, Shi X Q, Li R Y, Shu Q, Liu X T, Lu Z F, Zheng Q S. Simvastatin inhibits angiotensin II-induced cardiac cell hypertrophy: role of Homer 1a[J]. Clin Exp Pharmacol Physiol. 2010;37(1):40-45
24. Liao W, Wang S, Han C, Zhang Y. 14-3-3 proteins regulate glycogen synthase 3beta phosphorylation and inhibit cardiomyocyte hypertrophy[J]. FEBS J. 2005;272(8):1845-1854
25. Thandavarayan R A, Watanabe K, Ma M, Veeraveedu P T, Gurusamy N, Palaniyandi S S, Zhang S, Muslin A J, Kodama M, Aizawa Y. 14-3-3 protein regulates Ask1 signaling and protects against diabetic cardiomyopathy[J]. Biochem Pharmacol. 2008;75(9):1797-1806
26. Zheng Q, Su H, Ranek M J, Wang X. Autophagy and p62 in cardiac proteinopathy[J]. Circ Res. 2011;109(3):296-308
27. Porter K E, Turner N A. Statins and myocardial remodelling: cell and molecular pathways[J]. Expert Rev Mol Med. 2011;13:e22
28. Su F F, Shi M Q, Guo W G, Liu X T, Wang H T, Lu Z F, Zheng Q S. High-mobility group box 1 induces calcineurin-mediated cell hypertrophy in neonatal rat ventricular myocytes[J]. Mediators Inflamm. 2012;2012:805149
29. Kubli D A, Gustafsson A B. Cardiomyocyte health: adapting to metabolic changes through autophagy[J]. Trends Endocrinol Metab. 2014;25(3):156-164
30. Thandavarayan R A, Watanabe K, Ma M, Veeraveedu P T, Gurusamy N, Palaniyandi S S, Zhang S, Muslin A J, Kodama M, Aizawa Y. 14-3-3 protein regulates Ask1 signaling and protects against diabetic cardiomyopathy[J]. Biochem Pharmacol. 2008;75(9):1797-1806
31. Frank D, Kuhn C, van Eickels M, Gehring D, Hanselmann C, Lippl S, Will R, Katus H A, Frey N. Calsarcin-1 protects against angiotensin-II induced cardiac hypertrophy[J]. Circulation. 2007;116(22):2587-2596
32. Barth S, Glick D, Macleod K F. Autophagy: assays and artifacts[J]. J Pathol. 2010;221(2):117-124
33. Dutta D, Xu J, Kim J S, Dunn W J, Leeuwenburgh C. Upregulated autophagy protects cardiomyocytes from oxidative stress-induced toxicity[J]. Autophagy. 2013;9(3):328-344
Author contact
![]() Corresponding author: Feifei Su, Department of Cardiology, Tangdu Hospital, Fourth Military Medical University, Xi'an, 710038, China. Telephone: +86 29 84777722; Email: shijianedu.cn
Corresponding author: Feifei Su, Department of Cardiology, Tangdu Hospital, Fourth Military Medical University, Xi'an, 710038, China. Telephone: +86 29 84777722; Email: shijianedu.cn