Impact Factor ISSN: 1449-1907

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2018; 15(12):1384-1396. doi:10.7150/ijms.24702 This issue Cite
Research Paper
B355252, A Novel Small Molecule, Confers Neuroprotection Against Cobalt Chloride Toxicity In Mouse Hippocampal Cells Through Altering Mitochondrial Dynamics And Limiting Autophagy Induction
Uchechukwu Chimeh*, Mary Ann Zimmerman*, Nailya Gilyazova, P. Andy Li ![]()
Department of Pharmaceutical Sciences, Biomanufacturing Research Institute Biotechnology Enterprise (BRITE), North Carolina Central University, Durham, NC USA
*Equal Contributions
Received 2018-1-2; Accepted 2018-4-12; Published 2018-9-7
Abstract
Cerebral hypoxia as often occurs in cases of stroke, hemorrhage, or other traumatic brain injuries, is one of the leading causes of death worldwide and a main driver of disabilities in the elderly. Using a chemical mimetic of hypoxia, cobalt chloride (CoCl2), we tested the ability of a novel small molecule, 4-chloro-N-(naphthalen-1-ylmethyl)-5-(3-(piperazin-1-yl)phenoxy)thiophene-2-sulfonamide (B355252), to alleviate CoCl2-induced damage in mouse hippocampal HT22 cells. A dose-dependent decrease in cell viability was observed during CoCl2 treatment along with increases in mitochondrial membrane potential and generation of reactive oxygen species (ROS). B355252 conferred protection against these changes. We further found that mitochondrial dynamics, the balance between mitochondrial fusion and fission, were perturbed by CoCl2 treatment. Mitochondrial fusion, which was assessed by measuring the expression of proteins optic atrophy protein 1 (OPA1) and mitofusin 2 (Mfn2), declined due to CoCl2 exposure, but B355252 addition was able to elevate Mfn2 expression while OPA1 expression was unchanged. Mitochondrial fission, measured by phosphorylated dynamin-related protein 1 (p-DRP1) and fission protein 1 (FIS1) expression, also decreased following CoCl2 exposure, and was stabilized by B355252 addition. Finally, autophagy was assessed by measuring the conversion of cytosolic microtubule-associated protein 1A/1B-light chain three-I (LC3-I) to autophagosome-bound microtubule-associated protein 1A/1B-light chain three-II (LC3-II) and was found to be increased by CoCl2. B355252 addition significantly reduced autophagy induction. Taken together, our results indicate B355252 has therapeutic potential to reduce the damaging effects caused by CoCl2 and should be further evaluated for applications in cerebral ischemia therapy.
Keywords: Hypoxia, mitochondrial dynamics, cobalt chloride, B355252, fusion, fission
Introduction
Cerebral ischemia, or stroke, is the fifth leading cause of death in the United States, and the second highest cause of death globally. It is also the primary cause of disability in adults. Given that its prevalence is expected to increase 20.5% by the year 2030, stroke will continue to pose a significant burden on our healthcare system and economy, not to mention the personal toll it takes on families affected [1, 2].
Stroke most commonly results from a clot or rupture of blood vessels in the brain and subsequently causes an interruption in the supply of oxygen and nutrients that perfuse the brain [2, 3]. This shortage of blood and nutrients causes damage and death of the oxygen-deficient brain cells [2, 3]. The overall effect on the body varies, depending on which part of the brain is affected and the severity and duration of the injury [3, 4]. Furthermore, delays in reperfusion and treatment serve to expand the ischemic core area and can lead to irreversible damage [5, 6]. As of today, the only approved effective therapy for stroke is recombinant tissue plasminogen activator (rTPA), which breaks down the clot obstructing blood flow [4, 7]. Unfortunately, rTPA treatment is only effective when administered within 4.5 hours after a stroke incident. This narrow therapeutic window limits the application of rTPA to only about 5% of patients, therefore, expanding the therapeutic window of stroke therapies is a critical goal of stroke research [7].
B355252 is a phenoxy thiophene sulphonamide small molecule from an in-house library, which was synthesized by Williams et el. and shown to potentiate Nerve Growth Factor-induced neurite outgrowth [8]. Thus, B355252 was speculated to have neuroprotective functions. Previous studies by Gilyazova et al indicated anti-apoptotic effects of B355252 during glutamate-induced excitotoxicity, as well as in a Parkinson's disease (PD) model in the murine hippocampal cell line HT22. They further demonstrated that glutamate- and PD-induced oxidative stress were significantly reduced with B355252 treatment [9]. Given these results, we hypothesized that B355252 could confer protection against neuronal damage induced by hypoxia. We tested this hypothesis using a hypoxia model that employs the compound, CoCl2, to chemically mimic hypoxia induction in cells.
CoCl2 has been used in vitro to chemically induce hypoxia in various cell types, including rat cardiomyoblasts, human embryonic kidney cells, and mouse hippocampal neuronal cells [10-13]. Cobalt is a transition metal which, upon binding, stabilizes the hypoxia-induced transcription factor, HIF-1α. HIF-1α under normoxic conditions is continually degraded, but becomes stable during hypoxia where it plays a central role in activating many hypoxia-induced cell pathways. Thus this stabilization of HIF-1α by CoCl2 greatly mimics the cellular effects seen during hypoxia from lack of oxygen and is a cost effective and highly reproducible model [14].
Many of these cellular effects can be particularly devastating to neurons which need a lot of energy to function given their highly active, highly specialized nature. Most of the energy utilized by cerebral neurons is obtained from ATP generation during oxidative phosphorylation in mitochondria [15, 16]. Mitochondrial morphology and function are regulated by a balance between mitochondrial fusion and fission, referred to as mitochondrial dynamics [17].
Mitochondrial fusion leads to preservation of mitochondrial DNA and transmission of membrane potential across multiple mitochondria [17]. It enables survival of damaged mitochondria by transferring DNA and metabolites from neighboring mitochondria [18]. Fusion is activated primarily by dynamin family GTPases Mitofusin 1 & 2 (Mfn1/2) and OPA1 [18]. Fission is involved in the mitotic fragmentation of mitochondria, transportation of mitochondria to regions in the cell that require energy, and elimination of damaged mitochondria [17, 18]. Mitochondrial fission is controlled by the interaction of DRP1 with outer mitochondrial membrane proteins such as FIS1 [17]. An imbalance between fusion and fission can lead to a decrease in ATP production and mitochondrial mobility, generation of damaging ROS, deletion of mitochondrial DNA, and eventually neuronal death [15]. Disruption of the fusion/fission equilibrium leads to mitochondrial dysfunction and is linked to cancer, metabolic, cardiac and neurodegenerative diseases, including stroke [17, 19].
The purpose of this project is to elucidate the mechanism of disruption of mitochondrial dynamics by using CoCl2 to mimic ischemia in murine hippocampal cells. A previous study by Peng et al has already given a glimpse of this effect by showing a decrease in expression of the fusion-associated mitochondrial protein, Mfn2, following CoCl2 treatment [12]. In addition, mitochondrial fission seems to have a role in increasing autophagy following cerebral ischemia, but this mechanism isn't entirely clear [19]. Complicating matters, the role of autophagy itself has been controversial. Autophagy is the process of degradation and recycling of organelles and proteins in the cell and, while it is important for neuronal homeostasis, it can also over-activate to kill the cell [20]. The involvement of apoptotic and necrotic cell death in cases of cerebral hypoxia have been well documented, but whether the increase in autophagy seen during ischemia serves to promote or protect against cell death remains under debate [20, 21].
However, an increase in autophagy markers has been seen in neuroblastoma [22] and cardiomyoblasts following CoCl2-induced hypoxia [10] and we hypothesized that CoCl2 induces cytotoxicity in hippocampal cells by altering mitochondrial dynamics to activate autophagy. The main objectives of this work are to, first, investigate the effect of the hypoxia mimetic, CoCl2, on mitochondrial oxidative stress, mitochondrial dynamics and autophagy and, secondly, to test the effects of the neuroprotective compound, B355252, on cells exposed to CoCl2. Our aim is to provide proof-of-concept research as a starting point to further explore the therapeutic efficacy of this agent as a potential treatment for cerebral hypoxia.
Materials and Method
Materials
Mouse hippocampal HT22 cells were kindly provided by Dr. Jun Panee at the University of Hawaii [23]. Dulbecco's Modified Eagles Medium (DMEM) High Glucose medium, and Phosphate Buffered Saline solution (PBS) were purchased from GE Healthcare Life Sciences (Logan, UT). Fetal Bovine Serum (FBS), L-Glutamine 200 mM (100X) Solution, and Penicillin/Streptomycin Solution (10,000 units/mL penicillin, 10,000 μg/mL streptomycin) were purchased from Thermo Fisher Scientific (Logan, UT). Trypsin-Versene Mixture was obtained from Lonza Walkersville, Inc. (Walkersville, MD). Cobalt (II) chloride hexahydrate was purchased from Sigma-Aldrich (St. Louis, MO). B355252 was synthesized at North Carolina Central University's Biomanufacturing Research Institute and Technology Enterprise by A.L. Williams et al. [8]. Cell viability was measured with resazurin sodium salt purchased from Acros Organics (Fair Lawn, NJ). CellROX Deep Red Reagent obtained from Life Technologies Corporation (Carlsbad, CA) was used for oxidative stress detection. Mitochondrial membrane potential was determined with Tetramethylrhodamine methyl ester (TMRM) purchased from Life Technologies Corporation (Carlsbad, CA). M-PER Mammalian Protein Extraction Reagent, Pierce Protease Inhibitor Mini Tablets, Halt Phosphatase Inhibitor Single-Use Cocktail (100X), and the Pierce BCA Protein Assay Kit were purchased from Thermo Fisher Scientific (Rockford, IL). NuPAGE LDS Sample buffer, NuPAGE Sample Reducing Agent (10X), NuPAGE Antioxidant, NuPAGE Novex 4-12% Bis-Tris Protein Gels, NuPAGE MES SDS Running Buffer (20X), and NuPAGE Transfer Buffer (20X) were purchased from Life Technologies Corporation (Carlsbad, CA). Methanol (Certified ACS), and Tween 20 were obtained from Fisher Scientific (Fair Lawn, NJ). Sodium Dodecyl Sulfate (SDS), 20% Solution was purchased from AMRESCO, LLC (Solon, OH). Odyssey Blocking Buffer (PBS), IRDye 800CW Donkey anti-Rabbit antibody, IRDye 680LT Donkey anti-Mouse antibody and Odyssey Protein Molecular Weight Marker were purchased from LI-COR, Inc. (Lincoln, NE). Purified Mouse Anti-OPA1 monoclonal antibody was obtained from BD Transduction Laboratories (Franklin Lake, NJ). Rabbit Anti-Mfn2 polyclonal antibody was purchased from Santa Cruz Biotechnology, Inc. (Dallas, TX). Mouse Anti β-Actin monoclonal antibody, Rabbit Anti-Beclin-1 monoclonal antibody, Rabbit Anti-LC3A/B polyclonal antibody, and Rabbit Anti-Phospho-DRP1 polyclonal antibody were purchased from Cell Signaling Technology (Danvers, MA). Rabbit Anti-Fis1 polyclonal antibody was obtained from MBL International Corporation (Woburn, MA).
Cell Culture
HT22 neuronal cells, derived from mouse hippocampus and immortalized, were cultured in Dulbecco's Modified Eagles Medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 2 mM L-glutamine, 200 units/ml penicillin G and 200 µg/ml streptomycin. The cells were grown at 90 - 95% humidity in a 5% CO2 incubator at 37oC.
Cobalt Chloride Dose Response
For dose response assays, 1×104 HT22 cells per well were seeded in 96 well plates and allowed to settle for 24 hours. Cells were then treated with DMEM media containing multiple concentrations of CoCl2, 100-500 µM, to mimic hypoxia [11]. Cells were incubated in CoCl2 at 37oC for 24 hours prior to assessing cell viability using a resazurin assay as described below. A concentration of 300 μM CoCl2 produced 70% cell viability and was used for subsequent experiments unless otherwise stated.
B355252 Dose Response
1×104 HT22 cells were plated in 96 well plates and allowed to settle for 24 hours. After settling, HT22 cells were pretreated for 2 hours with various concentrations of B355252 (0.625-20 µM). This was followed by the addition of 300 μM CoCl2 and incubation at 37oC for 24 hours. At the end of this time, cell viability was determined by resazurin assay as described below.
Cell Viability
Cell viability was measured using a resazurin (7-Hydroxy-3H-phenoxazin-3-one 10-oxide) assay. A stock solution of resazurin was prepared in diH2O at a concentration of 1 mg/mL and added to assay plates to achieve a final concentration of 0.1 mg/mL. After treatment with CoCl2 and B355252, 10 μL of the dye was added to 100 μL of DMEM in each well. After 3 hours of incubation in 5% CO2 at 37oC, the cells were equilibrated to room temperature for 15 minutes. Fluorescence was measured with a PHERAstar Microplate Reader (BMG Labtech, Durham, NC) using the 540-20/590-20 filter. The relative fluorescence of the untreated, control cells was arbitrarily converted to 100% cell viability and experimental groups were converted to their corresponding percentages relative to the control.
Reactive Oxygen Species (ROS) Assay
HT22 cells were plated at a density of 1×104 cells per well in 96 well plates and allowed to incubate for 24 hours. After this settling period, cells were either untreated, treated with 300 μM CoCl2, or treated with 300 µM CoCl2 plus 2.5 μM B355252 for 24 hours. During the last 30 min, 5 μM CellROX Deep Red Reagent in DMEM was added to each treatment well and then placed back in incubation at 37oC for the duration of the 24 hour CoCl2/B355252 treatment period. CellROX Deep Red Reagent is a fluorogenic dye, which is non-fluorescent in its reduced state, but becomes fluorescent at excitation and emission maxima of 640/665 when oxidized by ROS. Additional treatment sets without CellROX Deep Red Reagent were also used for subtraction of fluorescent background. At the end of the incubation period, media was removed and cells were washed twice with PBS. A final volume of 100 µl PBS was added to each well prior to scanning plates. Fluorescence was read using a PHERAstar Microplate Reader with a 590-50/675-50 filter. To compensate for fluorescence changes caused by cell death, resazurin cell viability assays, as described above, were performed in parallel using the same CoCl2 and B355252 treatments used here to measure ROS production. The CellROX fluorescent measurements were normalized against the cell viability to calculate the relative fluorescence values in which an increase in fluorescence is indicative of an increase in ROS production.
Mitochondrial Membrane Potential Assay
HT22 cells were plated at a density of 1×104 cells per well in 96 well plates and allowed to incubate for 24 hours. Cells were then either untreated, treated with 300 μM CoCl2, or treated with 300 µM CoCl2 plus 2.5 μM B355252 for 24 hours. Following 24 hour treatment, 500 nM tetramethylrhodamine, methyl ester (TMRM) in DMEM medium was added to each well. TMRM is a fluorogenic dye which penetrates the cell and gathers in active mitochondria that maintain their membrane potential. Because of this, the TMRM fluorescent signal is weak when mitochondria lose their membrane potential through depolarization, or the signal can become stronger indicating hyperpolarization of the membrane. In either case, an alteration of the mitochondrial membrane can contribute to the cell's demise. HT22 cells were incubated with TMRM for 30 minutes at 37oC. Afterwards, the media was removed and cells were washed twice with PBS with a final volume of 100 µL PBS being added to each well. Fluorescence was read in a PHERAstar Microplate Reader (BMG Labtech, Durham, NC) using a 590-50 675-50 filter. To compensate for fluorescence changes caused by cell death, resazurin cell viability assays, as described above, were performed in parallel using the same CoCl2 and B355252 treatments used here to measure the mitochondrial membrane potential. Relative TMRM fluorescence values were calculated by normalizing TMRM fluorescent measurements against cell viability measurements.
Western Blotting
For western blot analysis, 2×106 HT22 cells were seeded in 100 mm plates and allowed to settle for 24 hours prior to treatments. At the end of various CoCl2/B355252 treatment times, cells were lysed in Mammalian Protein Extraction Reagent (M-PER) (Thermo Fisher Scientific) supplemented with protease and phosphatase inhibitors for 5 min on ice, scraped, and centrifuted at 20,000 ×g for 10 min at 4oC to remove cellular debris. Resultant protein concentrations were measured using a BCA assay (Thermo Fisher Scientific). Protein lysates (15 µg per well) were separated using 4-12% Bis-Tris NuPAGE gels (Invitrogen) according to the manufacturer's instructions. The Bio-Rad Mini Trans-Blot system was used to transfer the separated protein to nitrocellulose membranes. After the transfer, membranes were blocked in a 1:1 solution of LI-COR Odyssey Blocking Buffer and 1X PBS. Membranes were then probed using the following primary antibodies at 1:500 dilutions in blocking buffer: anti-beclin-1, anti-LC3A/B, anti-OPA1, anti-Mfn2, anti-Fis1, and anti-phospho-DRP1. A 1:2000 dilution of anti-β-actin antibody was used as an internal loading control for all blots. After overnight incubation at 4ºC, blots were washed three times with PBS-0.01%Tween before adding anti-mouse or anti-rabbit secondary antibodies, as listed in the materials section above, diluted 1:15,000 in blocking buffer. After a 1 hour incubation at room temperature, blots were again washed three times with PBS-0.01%Tween and a final wash with PBS before imaging. Fluorescence of secondary antibodies was detected using the LI-COR Odyssey Classic Imaging System scanner. A molecular weight marker, listed in the materials above, was utilized to confirm bands were selected for analysis at the expected protein weights. Images obtained using this scanner were analyzed with the LI-COR Image Studio Software version 5.2.5 (Lincoln, NB) with signals for the proteins of interest being normalized to signals for β-actin.
Statistical Analysis
Each experiment described above was repeated a minimum of three times. Data is presented as mean values ± standard deviation (SD), or as a percentage of the control. Each parameter in all data sets involving more than three groups was compared by one-way Analysis of Variance (ANOVA) or two-way ANOVA, followed by Bonferroni's multiple comparison test. GraphPad Prism 5 software (GraphPad Software, Inc., La Jolla, CA) was used for all data analysis. A p < 0.05 value was considered statistically significant.
Results
CoCl2-induced hypoxia decreases cell viability in murine hippocampal cells
To obtain a working concentration of CoCl2 capable of inducing hypoxia, we performed a dose response experiment in HT22 cells. After 24 hours exposure to CoCl2, cell viability was measured using the cell permeable, fluorogenic dye, resazurin, as described in the Materials and Method. Figure 1a shows that cell viability decreased in a dose-dependent manner from 0 to 500 µM CoCl2 with the highest dose killing 54.2% of cells (p<0.001). HT22 cell populations treated with 300 μM CoCl2 had an average of 70% viability (p<0.001) after 24 hours, when compared to the untreated control group. This concentration was chosen for subsequent CoCl2 experiments unless otherwise indicated. The effect of 300 μM CoCl2 treatment on HT22 cell growth and morphology was observed under a light microscope and representative images are presented in Figure 1b. Twenty-four hour CoCl2 treatment resulted in decreased cell growth and elongated cell shapes consistent with a starved or a deficient mitochondrial metabolic state.
CoCl2-induced hypoxia reduces HT22 cell viability and impedes recovery in a dose dependent manner A) Cell viability assay showing CoCl2 treatment decreases cell viability in a dose dependent manner. HT22 cells were treated for 24 hours with 100 - 500 μM CoCl2 as indicated. B) Photo images showing decreased cell growth and distribution in HT22 cells after 24h treatment with 300 μM CoCl2. Images were obtained using a routine inverted light microscope at 10X magnification. C) Cell viability assay showing continued dose-dependent decreases in cell viability after removal of CoCl2. HT22 cells were treated 24h with the indicated CoCl2 concentrations, then washed and incubated in media sans CoCl2 for an additional 24h recovery period. One-way ANOVA and Bonferroni's post-tests were used for analysis in A and C. ### p<0.001 vs. control.
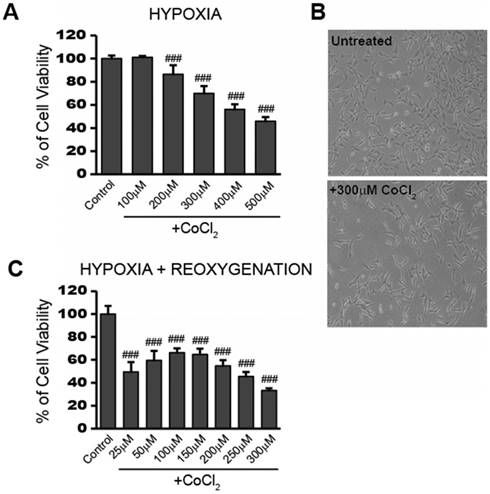
In addition to the reduced viability seen during CoCl2 exposure, cell viability continued to further decrease after removal of CoCl2 for the 24 hour reoxygenation period. This is in line with the continued stress and cellular damage commonly observed during the reperfusion stage after an ischemic attack. Instead of recovering lost viability, cells previously treated with 300 µM CoCl2, showed a further loss in viability going from 70% viable cells to around 30% viable cells at the end of the 24 hour reoxygenation period (Figure 1c). Even a lower initial dose of 100 μM CoCl2 resulted in 66% cell viability (p<0.001) after 24 hours reoxygenation, indicating the damage triggered by CoCl2-induced hypoxia continued to affect cell viability even after the initial hypoxic stimulus was removed.
B355252 improves cell viability during CoCl2-induced hypoxia
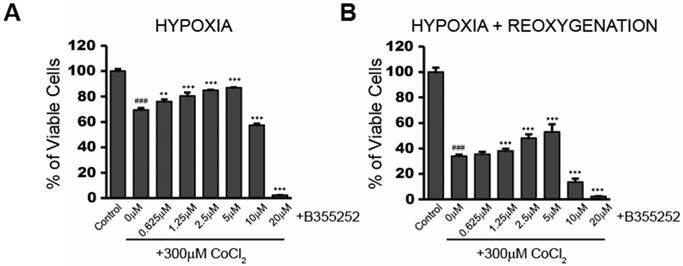
B355252 (4-chloro-N-(naphthalen-1-ylmethyl)-5-(3-(piperazin-1-yl) phenoxy) thiophene-2-sulfonamide) is an aryl thiophene sulfonamide, which has shown neuroprotection during glutamate-induced oxidative injury in HT22 cells [9]. We tested the protective effect of B355252 against CoCl2-induced hypoxia. Figure 2a shows the effect on cell viability when cells were treated with increasing concentrations of B355252 for two hours prior to addition of CoCl2. From 0.625 to 5 µM, B355252 increased cell viability in a dose-dependent manner, protecting against CoCl2-induced hypoxia. A similar dose-dependent, protective effect was observed during the 24 hour reoxygenation period in which CoCl2 and B355252 were both removed (Figure 2b). Maximum protection was observed with 5 µM B35525 which increased cell viability by 19% (p<0.001). Above 5 μM, B355252 became toxic to the cells and, synergistically with CoCl2, decreased cell viability. To avoid B355252 toxicity, we chose the sublethal dose of 2.5 μM B355252 together with 300 μM CoCl2 for subsequent experiments.
B355252 confers protection against CoCl2-induced decreases in cell viability during hypoxia and reoxygenation A) Cell viability assay showing addition of B355252 protects against a CoCl2-induced decrease in cell viability. HT22 cells were pretreated with the indicated concentrations of B355252 for 2h prior to adding 300 μM CoCl2 for 24h. B) Cell viability assay showing addition of B355252 also mitigates damage that further occurs after CoCl2 is removed. For this reoxygenation phase, cells were treated as in A, then washed and incubated with fresh, drug-free media for an additional 24h before assessing cell viability. One-way ANOVA followed by Bonferroni's post-test were used for analysis. ### p<0.001 vs. control. *** p<0.001. ** p<0.01 vs. CoCl2 alone.
B355252 suppresses the increased ROS production seen during CoCl2-induced hypoxia and restores mitochondrial membrane potential to its pre-hypoxic state
While some generation of ROS occurs under normal cellular functions, these ROS are typically detoxified by antioxidant enzymes before they can cause any significant cellular damage. However, ROS production becomes excessive during times of mitochondrial dysfunction and hypoxia. This excess of ROS can lead to DNA damage and destabilization of the mitochondrial membrane potential, resulting in cell death.
The presence of reactive oxygen species (ROS) in CoCl2-treated cells was determined using CellROX Deep Red Reagent. As shown in Figure 3a, CoCl2 caused a significant two-fold increase in ROS production when compared to the untreated control (p<0.001). Simultaneous treatment with B355252 and CoCl2 resulted in a roughly 50% decrease in CoCl2-induced ROS production (p<0.001). This suggests that the neuroprotection of B355252 involves reducing ROS generated as a result of CoCl2-induced hypoxia. Since increased generation of ROS often goes hand-in-hand with disruption of the mitochondrial membrane potential, we next used tetramethylrhodamine, methyl ester (TMRM) to measure changes in the relative mitochondrial membrane potential of treated cells.
B355252 reduces both production of ROS and restores mitochondrial membrane potential after CoCl2-induced hypoxia A) B355252 decreases the elevated ROS production observed during CoCl2-induced hypoxia. The presence of ROS was determined using CellROX Deep Red Reagent. HT22 cells treated for 24h with 300 μM CoCl2 and 2.5 μM B355252 (2h prior) before adding 5 µM CellRox Deep Red Reagent and incubating at 37°C for 30 min. B) Mitochondrial membrane potential is restored after B355252 treatment. Quantification of the mitochondrial membrane potential was done using Tetramethylrhodamine, methyl ester (TMRM) in HT22 cells. Cells were treated with CoCl2 and B355252 as in A before adding 500 nM TMRM and incubating cells at 37oC for 30 min. In both A and B, fluorescence was measured using a PHERAstar Microplate Reader with a 590-50/675-50 filter. The results of both the CellRox and TMRM fluorescence measurements were normalized against the cell viability to obtain the relative fluorescence values with control cells set at 100% fluorescence. One-way ANOVA followed by Bonferroni's post-test was used for analysis. ### p<0.001 vs. control. ** p<0.01 vs CoCl2 alone. *** p<0.001 vs. CoCl2 alone.
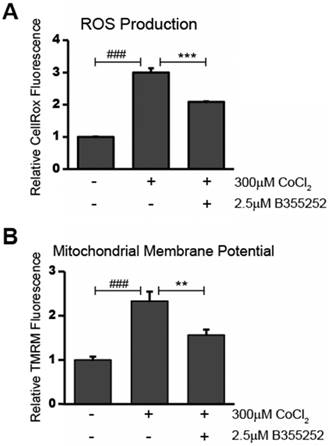
Alterations in the mitochondrial membrane, such as depolarization or hyperpolarization, were assessed using the fluorogenic dye, TMRM, as described in the material and methods. Figure 3b shows the results of measuring TMRM fluorescence in HT22 cells. Treatment with CoCl2 alone resulted in a 133% increase in TMRM fluorescence (p<0.001). This suggests that the mitochondria of the CoCl2-treated cells were in a hyperpolarized state. Administering B355252 with CoCl2 reduced the elevated fluorescence by 57% (p<0.01). The observed decrease in hyperpolarization as a result of B355252 protection corresponds with the lower ROS generation shown in Figure 3a. Taken together, this suggests that CoCl2-induced hypoxia works to damage hippocampal cells by targeting mitochondria to increase ROS production and dissipate the mitochondrial membrane potential and that B355252 may be a potent defense against these causes of cellular distress.
CoCl2-induced hypoxia and B355252 treatment modulate the expression of mitochondrial fusion proteins, OPA1 and Mfn2
In addition to increasing oxidative stress and mitochondrial membrane disruption, CoCl2-induced hypoxia may further contribute to cell death by altering the careful balance between mitochondrial fission and fusion referred to as mitochondrial dynamics. Western blotting experiments were performed to assess changes in the expression of proteins involved in mitochondrial dynamics as a result of CoCl2-induced hypoxia. Expression of OPA1 and Mfn2 were measured to study altered mitochondrial fusion.
A representative blot for OPA1 expression in the various experimental groups is shown in Figure 4a. Figure 4b shows the relative fluorescence of OPA1 obtained from normalizing the OPA1 signal to β-actin. Overall, there were significant decreases in OPA1 expression after 10h (20% decrease, p<0.001) and 24h (43% decrease, p<0.001) CoCl2 exposure, however, OPA1 expression was not restored by B355252 treatment. This suggests that CoCl2-induced hypoxia may reduce mitochondrial fusion and that B355252 confers protection by other means, for example Mfn2.
B355252 protects against CoCl2-induced alterations in mitochondrial fusion proteins OPA1 and Mfn2 A) Western blot of mitochondrial fusion protein, OPA1, and loading control, β-actin, in HT22 cells. Total protein lysates were obtained from untreated control and cells treated for 10 and 24hr with 300 μM CoCl2 with or without 2.5 μM B355252. Picture is representative of three separate experiments. B) Quantification of average OPA1 expression when normalized to β-actin. C) Western blot of mitochondrial fusion protein, Mfn2, and loading control, β-actin, in HT22 cells. Protein samples were obtained as indicated in A and picture is representative of three separate experiments. D) Quantification of average Mfn2 expression when normalized to β-actin. One-way ANOVA followed by Bonferroni's post-test was used for analysis. ### p<0.001 vs. control. *** p<0.001 vs. CoCl2 alone.
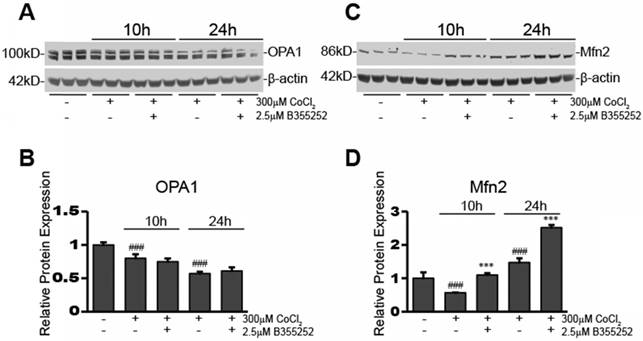
Expression of this second mitochondrial fusion protein, Mfn2, was also assessed and a representative blot is presented in Figure 4c. The average relative protein expression of Mfn2, normalized to β-actin, is presented in Figure 4d. There was an initial 43% decrease in Mfn2 expression after 10 hours of CoCl2-induced hypoxia (p<0.001). B355252 treatment restored Mfn2 expression to its pre-hypoxia level. However, Mfn2 expression significantly increased by 47% (p<0.001) from the pre-hypoxia control level after 24 hours of CoCl2 exposure. Curiously, B355252 co-treatment further increased Mfn2 expression by an additional 71% (p<0.001) after 24 hours. The initial decrease in Mfn2 at 10h, followed by a significant increase after 24h, may indicate that hypoxic stimuli cause an initial drop in Mfn2, but is followed by cellular countermeasures that serve to increase Mfn2 to protect against CoCl2-induced hypoxia. Most notably, B355252 increasing Mfn2 at both time points correlated with the increases in cell viability observed with B355252 treatment.
CoCl2-induced hypoxia reduces the expression of mitochondrial fission proteins, p-DRP1 and FIS1, while B355252 restores them
Perturbing mitochondrial dynamics is often through disturbing the ratio of fusion to fission mitochondrial proteins. Altering the balance of these proteins can have detrimental consequences to the cell by causing over- or under-promotion of either of these events. After seeing changes in mitochondrial fusion proteins upon exposure to CoCl2 and B355252, we next ascertained the effects on mitochondrial fission proteins, p-DRP1 and FIS1. Figure 5a shows a representative Western blot image for three individual experiments and Figure 5b shows the relative protein expression of p-DRP1 when normalized to loading control, β-actin. p-DRP1 expression decreased by 35% after 10 hours of CoCl2-induced hypoxia (p<0.01), of which B355252 treatment was unable to restore. p-DRP1 continued to decrease at the 24h time point, however here B355252 addition increased p-DRP1 expression by 43%, restoring expression to the control's level (p<0.05).
B355252 protects against CoCl2-induced alterations in mitochondrial fission proteins FIS1 and p-DRP1 A) Western blot of mitochondrial fission protein, FIS1, and loading control, β-actin, in HT22 cells. Total protein lysates were obtained as indicated in Figure 4A. Picture is representative of three separate experiments. B) Quantification of average FIS1 expression when normalized to β-actin. C) Western blot of mitochondrial fission protein, p-DRP1, and loading control, β-actin, in HT22 cells. Protein samples were obtained as indicated in Figure 4A and picture is representative of three separate experiments. D) Quantification of average p-DRP1 expression when normalized to β-actin. One-way ANOVA followed by Bonferroni's post-test was used for analysis. ### p<0.001 vs. control. ## p<0.01 vs. control. * p<0.05 vs. CoCl2 alone.
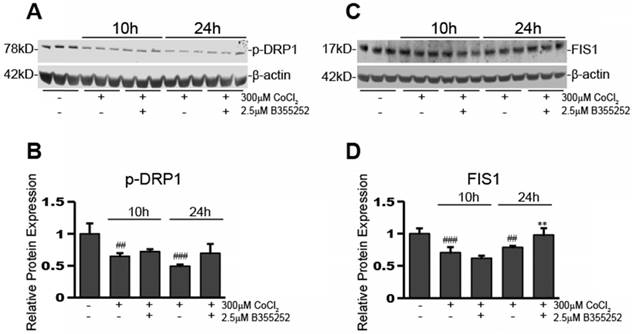
Expression of a second protein involved in mitochondria fission processes, FIS1, was also assessed. Figure 5c shows a Western blot image that is representative of at least three separate experiments. The expression of FIS1 was reduced by 30% after exposure to CoCl2 for 10 hours (p<0.001) (Figure 5d). At 10 hours, the addition of B355252 was unable to rescue FIS1 expression. However, similar to p-DRP1, there was rescue of FIS1 expression with B355252 treatment at 24 hours CoCl2 exposure. CoCl2 alone at 24 hours reduced FIS1 expression by 22% while the B355252 treatment group saw a reduction of only 2% effectively restoring FIS1 expression to that of the control (p<0.01).
Our Western blot analyses have shown that CoCl2 has an overall reductive effect on both mitochondrial fusion and fission. B355252 significantly increased mitochondrial fusion by increasing Mfn2 expression, and reinstalled fission proteins p-DRP1 and FIS1 after 24 hours in CoCl2 treated cells. Alterations in mitochondrial dynamics may be associated with increases in autophagy, therefore our next step was to assess autophagy induction and response to B355252 therapy.
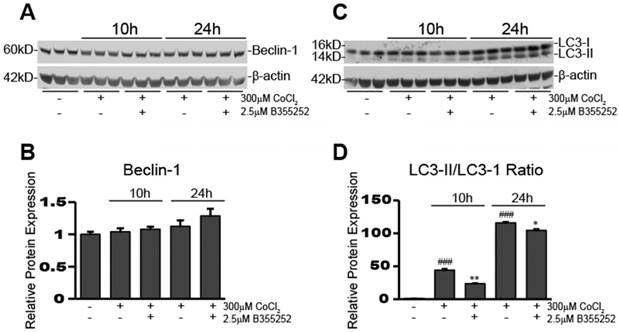
B355252 treatment causes a reduction in CoCl2-induced hypoxia-dependent increases in autophagy
To determine activation of autophagy in our CoCl2-exposed HT22 cells, we used Western blot analysis to examine the expression of autophagy-associated proteins, beclin-1 and LC3-II. Figure 6a&b shows beclin-1 expression remained unaltered after 10 and 24 hours of CoCl2-induced hypoxia and adding B35252 had no effect. Conversely, the ratio of LC3-II/LC3-I expression increased significantly during CoCl2-induced hypoxia at both 10 and 24 hours, as seen in Figure 6c which shows a representative Western blot image from at least three separate experiments. Quantification of the ratio of LC3-II to LC3-I in Figure 6d shows that conversion of LC3-I to LC3-II (an indication of autophagy) increased nearly 50-fold after 10 hours of hypoxia (p<0.001), and over a 100-fold after 24 hours (p<0.001). B355252 treatment significantly reduced the activation of autophagy by roughly 50% at 10 hours (p<0.01). At 24 hours the increase in autophagy from CoCl2 exposure was much more pronounced and B355252's ability to limit it was lessened, effecting a roughly 10% decrease (p<0.05) in the LC3-II/LC3-I ratio.
B355252 reduces CoCl2-induced autophagy A) Western blot of autophagy associated protein, Beclin-1, and loading control, β-actin, in HT22 cells. Protein samples were obtained as indicated in Figure 4A. Picture is representative of three independent experiments. B) Quantification of average Beclin-1 expression when normalized to β-actin. C) Western blot of autophagy associated protein, LC3-I/II in HT22 cells. Protein samples were obtained as indicated in Figure 4A and picture is representative of three separate experiments. D) Quantification of average LC3-II/LC3I ratio. Ratio was obtained by dividing the LC3-II fluorescent signal by the LC3-I fluorescent signal. One-way ANOVA followed by Bonferroni's post-test was used for analysis. ### p<0.001 vs. control. * p<0.05 vs. CoCl2 alone. ** p<0.01 vs. CoCl2 alone.
Discussion
The aim of this study was to understand the effects of hypoxia mimetic, CoCl2, on mitochondrial oxidative stress, mitochondrial dynamics, and the activation of autophagy in HT22 mouse hippocampal cells. Once the damage of our hypoxia model was assessed, we tested out the therapeutic efficacy of a novel small molecule, B355252, in protecting against this damage. CoCl2 itself has been used extensively in the literature to simulate hypoxia, including in HT22 cells, although the doses used and timing have varied. B355252 has also previously been shown to protect against neuronal damage similar to that which occurs during hypoxia, such as in Parkinson's disease and glutamate induced-excitotoxicity, and we anticipated it would effectively confer protection in our chemical hypoxia model.
Before adding B355353, we performed a CoCl2 dose response experiment to verify CoCl2 cytotoxicity and ultimately chose 300 µM CoCl2 as a working concentration. This concentration reduced HT22 cell viability by 30% after 24 hours with viability decreasing by 67% 24 hours after removing the CoCl2. The continued damage that occurs even after CoCl2 is removed, is consistent with the reperfusion phase seen after an ischemic episode in which damage caused by secondary responses, such as inflammation, continue to wreak havoc within the cellular milieu.
We next examined the protective effects of B355252 given two hours prior to adding CoCl2 in cell culture. Our results show that B355252 effectively protects against CoCl2-induced decreases in cell viability with a maximum benefit seen at 5 µM B355252. At this concentration, the deleterious effect of CoCl2 on cell viability is almost entirely reversed. At concentrations higher than 5 µM, however, B355252 becomes toxic to cells and there is a precipitous drop in cell viability during co-incubation that further reduces viability even below that of CoCl2 exposure alone. Because of this seemingly narrow therapeutic index, we explored the protective mechanisms of B355252 using a suboptimal dose of 2.5 µM.
It has previously been shown that CoCl2 increases production of reactive oxygen species (ROS) in HT22 cells [12]. We verified increased ROS levels in our CoCl2-treated cells, using CellRox deep red reagent, and found that B355252 treatment prevents this increase. This is consistent with the observation made by Gilyazova et al., who reported that B355252 treatment decreased glutamate-induced ROS in HT22 cells [9]. We believe this reduction in ROS is a key mechanism of how B355252 increases cell viability against hypoxic insult. Generation of ROS is often associated with changes in the mitochondrial membrane potential which may be more directly related to the demise of the cell through triggering the intrinsic cell death pathway.
The mitochondrial membrane potential (MMP) is typically maintained between 80 and 140 mV in cells [16]. During ischemia there is a depletion of oxygen. This causes negative feedback inhibition of complex IV of the electron transport chain and accumulation of electrons at complexes I and III [16, 24]. The halt of electron flow leads to a loss of the proton gradient across the inner mitochondrial membrane, prevention of ATP production, mitochondrial depolarization, and accumulation of Ca2+ ions in the mitochondria [25]. Ca2+ activates phosphatases, which dephosphorylate Complex IV. This is followed by hyperpolarization of the mitochondrial membrane upon reperfusion, due to loss of allosteric regulation [16]. High MMP leads to the excessive generation of ROS by extending the half-life of reaction intermediates which are involved in the partial reduction of oxygen, and thus production of superoxide at complexes I and III [16].
In our CoCl2 model we found MMP to be increased, indicating hyperpolarization of the membrane. This likely contributed to the excess generation of ROS we observed. This relationship corresponds with the findings of Kumari et al who similarly found that both ROS and MMP were increased in glutamate-exposed HT22 cells [26] and with Fang WL et al who showed the same trends in HT22, Aβ25-35 -exposed cells [27]. The latter group showed that treatment with the drug, Neurotropin, reversed the increase in ROS production and elevation of the MMP while also inhibiting the expression of HIF1-α and increasing cell viability. Since CoCl2 confers its hypoxia-like actions mainly through stabilization of HIF1-α expression, our own reductions in MMP and ROS coupled with the increased cell viability seen with B355252 treatment, are not surprising. However, our observations were contrary to those made by another group, Peng et al., who measured MMP in CoCl2-treated cells using a TMRM probe and confocal laser scanning, and observed depolarization of the mitochondrial membrane [12]. It is possible that this discrepancy stemmed from the different investigative methods used and the times the measurements were conducted. To mitigate this detrimental effect we treated cells with B355252. Just as it did with ROS production, the addition of B355252 significantly decreased the hyperpolarization of our CoCl2-treated cells. Gilyazova et al. also observed protection from MMP depolarization with B355252 treatment in a Parkinson's Disease model [28]. We can surmise from these findings that B355252 neuroprotection involves stabilizing changes in mitochondrial membrane potential.
Neuroprotection may also occur through stabilization of mitochondrial dynamics. Several recent reviews have highlighted how disturbances in mitochondrial dynamics, particularly in fission and fusion proteins, DRP1, Fis1, OPA1, and Mfn2, can contribute to neuropathology [29-31]. In this study, the influence of CoCl2 and B355252 on mitochondrial dynamics was investigated by measuring the expression of proteins involved in mitochondrial fusion and fission. We observed a gradual decrease in the expression of fusion protein, OPA1, following CoCl2-induced toxicity. This suggests that CoCl2 exposure leads to a decrease in mitochondrial fusion. This change in OPA1 was not significantly affected by B355252 treatment however, another prominent fusion protein, Mfn2, was also decreased after 10 hours of hypoxia. However, at 24 hours of hypoxia, its expression increased above that of untreated cells, suggesting that CoCl2 causes a transient decline of mitochondrial fusion. Peng et al. also noticed a decrease in Mfn2 in their in vitro CoCl2 model, but in contrast to our findings, this decrease occurred after 24 hours exposure [12]. This group also used an in vivo middle cerebral artery occlusion (MCAO) model which showed Mfn2 decreases at both 12 and 24 hours [12]. B355252 treatment of CoCl2-exposed cells showed complete Mfn2 restoration after 10 hours and, at 24 hours post treatment, Mfn2 experienced a further increase in expression that was 1.5-fold beyond the control in our study. This confirms that B355252 protection against CoCl2-induced hypoxia involves an increase in Mfn2 expression. Mfn2 overexpression has previously been shown to have a protective role against CoCl2-induced hypoxia [12]. In a study by Peng et al, LV-Mfn2 transfected HT22 cells showed decreased apoptosis after CoCl2-induced hypoxia, compared to LV-control cells [12] Mfn2 has also been shown to have a protective role against glucose-oxygen deprivation in HT22 cells by restoring MMP and reducing apoptosis [32].
It has also been shown in an in vivo MCAO model that ischemia disrupts the equilibrium of mitochondrial dynamics to favor mitochondrial fission [19]. In our current study, expression of fission proteins, p-DRP1 and FIS1, were examined via Western blot to determine the effect of CoCl2 exposure on mitochondrial fission. DRP1 phosphorylated at the Ser616 amino acid facilitates fission, while phosphorylation on Ser637 and 656 inhibits fission [33, 34]. Therefore, Ser 616 p-DRP1 was used in our study. We saw an unexpected decrease in p-DRP1 at 10 hours, which further decreased at 24 hours of CoCl2-induced hypoxia. B355252 treatment only had a minimal effect on the protein after 24 hours. FIS1 expression was also found to be decreased at 10 and 24 hours of hypoxia in our study. At 24 hours, B355252 treatment restored FIS1 to control levels. Overall, we noticed a decrease in mitochondrial fission due to CoCl2, as opposed to an expected increase. However, such a result is not without precedent. Jang and Koh found that DRP1 expression was reduced in both a MCAO rat model and in HT22 cells exposed to glutamate, and they attributed the accompanying neuronal cell death to these injury-induced declines in DRP1 expression [35]. It is possible that in our model, a drop in DRP1-mediated fission due to CoCl2 exposure, could be resulting in activation of the DNA damage response due to genome instability and replication stress, such as that seen in a study monitoring the effects of DRP1 inhibition in breast cancer cells [36]. DRP1 and FIS1 deficiency have also been associated with impaired mitochondrial function due to a decrease in fission-dependent mechanisms of mitophagy that work to remove damaged mitochondria within the cell [33]. Overall, mitochondrial fission is associated with fragmentation of the mitochondria and plays a key role in maintaining homeostasis [19, 37]. To confirm that CoCl2 exposure suppresses mitochondrial fission, further research should be conducted to visualize the mitochondrial structure using confocal microscopy and a mitochondrial staining dye. A true decrease in fission should render a more tubular shape to the mitochondria.
Mitochondria can occupy up to 30% of the volume of mature neuronal cells [38]. It is no surprise that neurodegenerative diseases, such as Parkinson's disease, Alzheimer's disease and stroke, are characterized by mitochondrial dysfunction, indicated by inhibition of the electron transport chain (ETC), reduced ATP production, and deletion of mitochondrial DNA. [15]. The mitochondrion is an essential organelle for cell energy metabolism and homeostasis, due to its role in generation of ATP, regulation of calcium ions, and synthesis of amino acids, lipids, and nucleotides [25]. Taken together, our results show that CoCl2 disrupts mitochondrial dynamics by reducing both fusion and fission and, in both cases, B355252 appears to have some influence on mitigating these changes with the end result being increased cell viability.
Finally, autophagy following CoCl2 exposure was investigated with Beclin-1 and LC3-II. Beclin-1 is involved in the early formation of autophagosomes. In our study there were no significant changes from CoCl2 or B355252 when the level of Beclin-1 was quantified. Beclin-1 induction has been previously reported in HT22 cells after exposure to CoCl2 [39]. It was noted that Beclin-1 expression peaks at 10 hours exposure and then gradually reduces. In our own experiments we saw slight increases at 10 and 24 hours CoCl2 exposure, but the changes were not significant. This could be due to our lower dose of 300 µM CoCl2 instead of the 500 µM utilized by Yang et al. On the other hand, conversion of LC3-I to LC3-II was significantly increased after 10 and 24 hours of CoCl2 exposure. This indicates an increase in autophagy due to CoCl2-induced toxicity. This is consistent with the findings published by Frugier et al showing increased staining of LC3-II and recruitment of autophagic vescicles in human post-mortem brain tissues after stroke [40]. These authors, however, could not determine if the autophagic process was beneficial or deleterious in the context of this study. A review by Xu et al also eludes to the difficulty of determining whether the increase in autophagy seen in neurons following cerebral ischemia serves to enhance or protect against neuronal cell death and because of this, the role of autophagy remains controversial [20]. Immediately following ischemic conditions, autophagy could have a protective function because it enables the removal of damaged mitochondria, which could have resulted in the generation of excess ROS [17]. Conversely, chronic autophagy could result in depletion of mitochondria and other essential organelles, and thus cell death [17]. Such a bimodal response has also been observed after spinal cord ischemia reperfusion injury in rats [41]. In our current study, B355252 treatment was able to significantly reduce the conversion of LC3-I to LC3-II, confirming that the compound limits autophagy induction. At the same time we saw an increase in cell viability with B355252 treatment against CoCl2 toxicity. This would indicate autophagy induction, at the level we see after CoCl2 exposure, is detrimental to the cell's viability and B355252 provides protection by dampening the autophagic process. It does not rule out the possibility that a smaller induction of autophagy would benefit cell survival as B355252 does not completely block autophagy induction. It is possible B355252 exerts its pro-survival functions by preventing cells from crossing the autophagy threshold in which autophagy goes from being protective to catastrophic for the cell.
In this present study we did not examine mitophagy, in which mitochondria as organelles enter the autophagic cascade, however we did note that the mitochondrial protein, COX IV, was itself decreased by CoCl2 treatment (data not shown). COX IV often serves as a marker of mitochondrial mass and has also been used to monitor the last step in the degradation process of mitophagy [42]. Hence, it is possible mitophagy may contribute to the overall changes in autophagy we observed. While beyond the scope of this present study, it would not be surprising to see mitophagy effected by CoCl2/B355252 treatments. Like CoCl2, another stabilizer of Hif-1α, propyl-4-hydroxyalse inhibitor, was shown to increase mitophagy and reduce cell death after experimental retinal detachment in rats [43]. On the other hand, a specific type of mitophagy that is Parkin independent was shown to depend on expression of DRP1 in mouse heart and brain [44]. We have shown here that DRP1 expression is significantly reduced after CoCl2 treatment which could potentially limit this type of mitophagy. However, our results indicate that B355252 improves cell viability after CoCl2 exposure without significantly altering DRP1 expression and therefore this specific type of mitophagy may be unaffected by B355252 or have a limited role in our model. Further studies will be needed to address the potential role of mitophagy in B355252 therapy.
In summary, we confirmed the dose-dependent cytotoxic effect of CoCl2 on HT22 cells, which was ameliorated in a dose-dependent manner by B355252. We observed that B355252 protects HT22 cells from oxidative stress by attenuating ROS generation and decreasing mitochondrial membrane hyperpolarization caused by CoCl2 exposure. CoCl2 had a generally suppressive effect on both mitochondrial fusion and fission. B355252 protected against CoCl2-induced changes in mitochondrial dynamics-regulating proteins, Mfn2, p-DRP1 and FIS1. The increase in autophagy caused by CoCl2 was also lessened with B355252 addition. While these results are promising for the application of B355252 toward mitigating the effects of cerebral ischemia, much about the molecular mechanisms behind its efficacy remain to be discovered. Here we have provided a basic framework in which B355252 functions to increase cell viability, reduce oxidative stress, and rebalance mitochondrial dynamics to suppress damage caused by cerebral ischemia. Further work will need to be done on how to incorporate B355252, whether alone or in combination with other therapeutic options, to provide the most optimal care during cerebral ischemic episodes.
Abbreviations
CoCl2: cobalt chloride; B355252: 4-chloro-N-(naphthalen-1-ylmethyl)-5-(3-(piperazin-1-yl)phenoxy)thiophene-2-sulfonamide; ROS: reactive oxygen species; OPA1: optic atrophy protein 1; Mfn2: mitofusin 2; p-DRP1: phosphorylated dynamin-related protein 1; FIS1: fission protein 1; LC3-I: microtubule-associated protein 1A/1B-light chain three-I; LC3-II: microtubule-associated protein 1A/1B-light chain three-II; rTPA: recombinant tissue plasminogen activator; PD: Parkinson's disease; HIF-1α: hypoxia-inducible factor 1-alpha; DMEM: Dulbecco's Modified Eagles Medium; FBS: fetal bovine serum; TMRM: tetramethylrhodamine methyl ester; MMP: mitochondrial membrane potential; COX IV: cytochrome c oxidase subunit IV.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Mozaffarian D. et al. Heart Disease and Stroke Statistics-2016 Update: A Report From the American Heart Association. Circulation. 2016;133(4):e38-360
2. American Heart Association. Heart disease and stroke statistics update. 2018. http://www.heart.org/HEARTORG/General/Heart-and-Stroke-Association-Statistics_UCM_319064_SubHomePage.jsp
3. World Health Organization. Stroke, Cerebrovascular Accident. 2018. http://www.who.int/topics/cerebrovascular_accident/en/
4. Nikonenko AG. et al. Structural features of ischemic damage in the hippocampus. Anat Rec (Hoboken). 2009;292(12):1914-21
5. Hossmann KA. Pathophysiology and Therapy of Experimental Stroke. Cellular and Molecular Neurobiology. 2006;26(7):1055-1081
6. Moustafa RR, Baron JC. Pathophysiology of ischaemic stroke: insights from imaging, and implications for therapy and drug discovery. Br J Pharmacol. 2008;153(Suppl 1):S44-54
7. Fluri F, Schuhmann MK, Kleinschnitz C. Animal models of ischemic stroke and their application in clinical research. Drug Des Devel Ther. 2015;9:3445-54
8. Williams AL. et al. Microwave-assisted synthesis of 4-chloro-N-(naphthalen-1-ylmethyl)-5-(3-(piperazin-1-yl)phenoxy)thiophene-2-sulfo namide (B-355252): a new potentiator of Nerve Growth Factor (NGF)-induced neurite outgrowth. Tetrahedron. 2010;66(50):9577-9581
9. Gliyazova NS, Huh EY, Ibeanu GC. A novel phenoxy thiophene sulphonamide molecule protects against glutamate evoked oxidative injury in a neuronal cell model. BMC Neurosci. 2013:14 (93). doi: 10.1186/1471-2202-14-93
10. Gallo S. et al. Agonist antibodies activating the Met receptor protect cardiomyoblasts from cobalt chloride-induced apoptosis and autophagy. Cell Death Dis. 2014;5:e1185
11. Chen WY, Chang MS. IL-20 is regulated by hypoxia-inducible factor and up-regulated after experimental ischemic stroke. J Immunol. 2009;182(8):5003-12
12. Peng C. et al. Mitofusin 2 ameliorates hypoxia-induced apoptosis via mitochondrial function and signaling pathways. Int J Biochem Cell Biol. 2015;69:29-40
13. Lu Y. et al. The protective effects of propofol against CoCl2-induced HT22 cell hypoxia injury via PP2A/CAMKIIα/nNOS pathway. BMC Anesthesiol. 2017;17(1):32
14. Siddiq A, Aminova LR, Ratan RR. Hypoxia inducible factor prolyl 4-hydroxylase enzymes: center stage in the battle against hypoxia, metabolic compromise and oxidative stress. Neurochem Res. 2007;32(4-5):931-46
15. Knott AB, Bossy-Wetzel E. Impairing the mitochondrial fission and fusion balance: a new mechanism of neurodegeneration. Ann N Y Acad Sci. 2008;1147:283-92
16. Sanderson TH. et al. Molecular mechanisms of ischemia-reperfusion injury in brain: pivotal role of the mitochondrial membrane potential in reactive oxygen species generation. Mol Neurobiol. 2013;47(1):9-23
17. Marin-Garcia J, Akhmedov AT. Mitochondrial dynamics and cell death in heart failure. Heart Fail Rev. 2016;21(2):123-36
18. Ranieri M. et al. Mitochondrial fusion proteins and human diseases. Neurol Res Int. 2013;2013:293893. doi: 10.1155/2013/293893
19. Kumari S. et al. Hyperglycemia alters mitochondrial fission and fusion proteins in mice subjected to cerebral ischemia and reperfusion. Transl Stroke Res. 2012;3(2):296-304
20. Xu F, Gu JH, Qin ZH. Neuronal autophagy in cerebral ischemia. Neurosci Bull. 2012;28(5):658-66
21. Ginet V. et al. Enhancement of autophagic flux after neonatal cerebral hypoxia-ischemia and its region-specific relationship to apoptotic mechanisms. Am J Pathol. 2009;175(5):1962-74
22. Naves T. et al. Autophagy takes place in mutated p53 neuroblastoma cells in response to hypoxia mimetic CoCl2. Biochem Pharmacol. 2013;85:1153-1161
23. Panee J. et al. The responses of HT22 cells to the blockade of mitochondrial complexes and potential protective effect of selenium supplementation. Int J Biol Sci. 2007;3(5):335-41
24. Borutaite V, Toleikis A, Brown GC. In the eye of the storm: mitochondrial damage during heart and brain ischaemia. FEBS J. 2013;280(20):4999-5014
25. Hagberg H. et al. Mitochondria: hub of injury responses in the developing brain. The Lancet Neurology. 2014;13(2):217-232
26. Kumari S, Mehta SL, Li PA. Glutamate induces mitochondrial dynamic imbalance and autophagy activation: preventive effects of selenium. PLoS One. 2012;7(6):e39382
27. Fang WL. et al. Neurotropin alleviates hippocampal neuron damage through a HIF-1α/MAPK pathway. CNS Neurosci Ther. 2017;23(5):428-437
28. Gliyazova NS, Ibeanu GC. The Chemical Molecule B355252 is Neuroprotective in an In Vitro Model of Parkinson's Disease. Cell Mol Neurobiol. 2016;36(7):1109-22
29. Gao J. et al. Abnormalities of mitochondrial dynamics in neurodegenerative diseases. Antioxidants (Basel). 2017;6(2):doi 10.3390/antiox6020025
30. Burte F. et al. Disturbed mitochondrial dynamics and neurodegenerative disorders. Nat Rev Neurol. 2015;11(1):11-24
31. Golpich M. et al. Mitochondrial dysfunction and biogenesis in neurodegenerative diseases: Pathogensis and treatment. CNS Neurosci Ther. 2017;23(1):5-22
32. Chen XL. et al. Mfn2-mediated preservation of mitochondrial function contributes to the protective effects of BHAPI in response to ischemia. J Mol Neurosci. 2017;63(3-4):267-274
33. Twig G. et al. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J. 2008;27(2):433-46
34. Manczak M, Calkins MJ, Reddy PH. Impaired mitochondrial dynamics and abnormal interaction of amyloid beta with mitochondrial protein Drp1 in neurons from patients with Alzheimer's disease: implications for neuronal damage. Hum Mol Genet. 2011;20(13):2495-509
35. Jang AR, Koh PO. Ischemic brain injury decreases dynamin-like protein 1 expression in a middle artery occlusion animal model and glutamate-exposed HT22 cells. Lab Anim Res. 2016;32(4):194-199
36. Qian W. et al. Mitochondrial hyperfusion induced by loss of the fission protein Drp1 causes ATM-dependent G2/M arrest and aneuploidy through DNA replication stress. J Cell Sci. 2012;125(23):5745-57
37. Hu C, Huang Y, Li L. Drp1-dependent mitochondrial fission plays critical roles in physiological and pathological progresses in mammals. Int J Mol Sci. 2017;18(1):E144
38. Schaper J, Meiser E, Stammler G. Ultrastructural morphometric analysis of myocardium from dogs, rats, hamsters, mice, and from human hearts. Circ Res. 1985;56(3):377-91
39. Yang T. et al. Regulation of Beclin-1 expression by mTORE in CoCl2-induced HT22 cell ischemia-reperfusion injury. Brain Res. 2015;1614:60-66
40. Fugier T. et al. Evidence for the recruitment of autophagic vesicles in human brain after stroke. Neurochem Int. 2016;96(62):62-68
41. Fang B. et al. Role of autophagy in the bimodal stage after spinal cord ischemia reperfusion injury in rats. Neuroscience. 2016;328:107-116
42. Qi Y. et al. ATM mediates spermidine-induced mitophagy via PINK1 and Parkin regulation in human fibroblasts. Sci Rep. 2016;6:24700
43. Liu H. et al. Propyl-4-Hydroxylases inhibitor stabalizes HIF-1α and increases mitophagy to reduce cell death after experimental retinal detachment. Invest Ophthalmol Vis Sci. 2016;57(4):1807-1815
44. Kageyama Y. et al. Parkin-independent mitophagy requires Drp1 and maintains the integrity of mammalian heart and brain. EMBO J. 2014;33:2798-2813
Author contact
![]() Corresponding author: E-Mail address: pliedu; Tel.: +1-919-530-6872; Fax: +1-919-530-6600.
Corresponding author: E-Mail address: pliedu; Tel.: +1-919-530-6872; Fax: +1-919-530-6600.