Impact Factor ISSN: 1449-1907

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2018; 15(10):1072-1082. doi:10.7150/ijms.25799 This issue Cite
Research Paper
Clinical and Basic Evaluation of the Prognostic Value of Uric Acid in Traumatic Brain Injury
Han Liu, Junchi He, Jianjun Zhong, Hongrong Zhang, Zhaosi Zhang, Liu Liu, Zhijian Huang, Yue Wu, Li Jiang, Zongduo Guo, Rui Xu, Weina Chai, Gang Huo, Xiaochuan Sun ![]() , Chongjie Cheng
, Chongjie Cheng ![]()
Department of Neurosurgery, the First Affiliated Hospital of Chongqing Medical University, Chongqing, China
Received 2018-2-28; Accepted 2018-6-8; Published 2018-6-23
Abstract
Background: As a major antioxidant in serum, uric acid (UA) was once considered only as the leading cause of gout; however, recent studies have validated its neuroprotective role in ischemic stroke. Because the potential protective effects of UA in traumatic brain injury (TBI) remain largely unknown, this study investigated the role of UA in TBI in both clinical patients and experimental animals.
Methods: In TBI patients, serum UA concentrations were measured within 3 days after injury. Clinical outcomes at discharge were classified according to the Glasgow Outcome Scale: good outcome (4-5) and poor outcome (1-3). Risk factors for good outcome were identified via backward logistic regression analysis. For the animal study, a controlled cortical impact (CCI) injury model was established in mice. These mice were given UA at different doses intraperitoneally, and subsequent UA concentrations in mouse serum and brain tissue were determined. Neurological function, oxidative stress, inflammatory response, neuronal maintenance, cerebral blood flow, and lesion size were also assessed.
Results: The serum UA level was significantly lower in TBI patients who had a good outcome (P<0.01), and low serum UA was an independent predictor of good outcome after TBI (P<0.01; odds ratio, 0.023; 95% confidence interval, 0.006-0.082). Consistently, decreased levels of serum UA were observed in both TBI patients and CCI animals (P<0.05), whereas the UA concentration was increased in CCI brain tissue (P<0.05). Administration of UA further increased the UA level in brain tissue as compared to that in control animals (P<0.05). Among the different doses administered, 16 mg/kg UA improved sensorimotor functional recovery, spatial learning, and memory in CCI mice (P<0.05). Moreover, oxidative stress and the inflammatory response were inhibited by UA treatment (P<0.05). UA treatment also improved neuronal maintenance and cortical blood flow (P<0.05) but not lesion size (P>0.05).
Conclusions: UA acted to attenuate neuronal loss, cerebral perfusion impairment and neurological deficits in TBI mice through suppression of neuronal and vascular oxidative stress. Following TBI, active antioxidant defense in the brain may result in consumption of UA in the serum, and thus, a decreased serum UA level could be predictive of good clinical recovery.
Keywords: UA (uric acid), oxidative stress, inflammation, traumatic brain injury
Introduction
In recent decades, the topic of traumatic brain injury (TBI) has gained increasing attention in both the scientific community and clinical practice. While the mortality of severe TBI has decreased by almost 50% over the past 150 years, it remains as high as 30%, and most survivors suffer varying degrees of sequelae [1]. Thus, research to understand the mechanisms of TBI and develop effective treatments is still needed.
Uric acid (UA) is a waste product of purine metabolism [2,3], and it has been known for a long time that a high UA concentration in serum is strongly associated with gout, which is a risk factor for cardiac and renal diseases. Recently though, studies have reported the antioxidant potential of UA and its potential relevance to neuroscience. For example, UA was found to have protective effects in acute stroke and Alzheimer's disease [4,5] as well as against cerebral vascular injury caused by reperfusion [6].
TBI is caused by a sudden profound mechanical injury to neurons, which triggers a cascade of excitotoxic, inflammatory and oxidative factors that contribute to functional disability. Thus, as a potent antioxidant compound [7,8], UA might also have a protective effect in TBI. To investigate the role of UA in TBI, we analyzed the correlation between clinical prognosis and UA level in patients with TBI and explored the underlying pathological mechanisms in an experimental animal TBI model.
Materials and Methods
Retrospective study of cases in TBI patients
From May 2013 to December 2016, data of 193 patients with TBI were collected. In addition, their baseline characteristics and therapies administered during the hospitalization were obtained. Furthermore, the conscious state of the patients was measured on admission, using the Glasgow Coma Scale (GCS) score whereas the Glasgow outcome scale (GOS) score was used to evaluate the outcome at discharge[9]. Moreover, patients with TBI accompanied with a spontaneous brain hemorrhage, stroke, neoplasm, coagulation disorders, aneurysm, or arteriovenous malformations were excluded.
Blood samples were drawn from all patients within 3 days after the onset of TBI and the levels of UA were measured by standard laboratory procedures with urate oxidase reagent using a Dax analyzer (Bayer-Technichon). In addition. the control group was composed of 143 healthy subjects and their data were obtained from the Medical Examination Center of our hospital. The study protocol was approved by the ethics committee of Chongqing Medical University (Permit Number: 2017-146).
Experimental studies in TBI mouse model
Animals
Male C57BL/6 mice (n=90, 12 weeks) were obtained from the experimental animal center of Chongqing Medical University (Chongqing, China), weighing 25-30 g. The mice were randomly allocated into the sham group (n=16), saline group (n=31), UA1 group (n=31), UA2 group (n=6) and UA3 group (n=6) (The number in each sub-group can be found in figure legends). In addition, they were housed in cages with food and water available ad libitum. Experimental protocols were approved by the Chongqing Medical University Administrative Panel on Laboratory Animal Care. All the surgeries were performed under anesthesia, and all efforts were made to minimize mice suffering.
Drugs
UA (Sigma-Aldrich) was dissolved and heated in saline, adjusted to pH 7-8 with NaOH and HCl, and cooled at room temperature [10]. The hyperuricemic mouse model was established by intraperitoneal injection of a 250 mg/kg dose of UA [11]. In addition, the dose of 16 mg/kg was proven to be effective in treating focal ischemic stroke [6]. Thus, in our experiments, mice were treated with three doses of UA (UA1, 16 mg/kg; UA2, 80 mg/kg; UA3, 160 mg/kg) or saline from day 1 to day 14 after controlled cortical impact (CCI) injury [12].
Induction of CCI
After anesthesia, the operation was performed as described by Jiang et al [13]. Furthermore, mice were mounted on the stereotaxic frame of the contusion device TBI-0310 (Precision Systems and Instrumentation, Fair fax, VA, USA). The diameter of the impactor was 3 mm. Then, the machine was set at velocity: 5.0 m/s, depth: 2.0 mm, and dwelling time: 100 ms, which produced a moderately severe contusion in the right sensorimotor cortex and underlying hippocampus, with pronounced behavioral deficits but no virtual mortality [14]. On the other hand, the sham group only received craniotomy without cortical impact.
Following the injury, the bone wax was applied to fill the hole in the skull and the skin incision was sutured. After, the mice were kept on an electric blanket to maintain normal body temperature until they completely woke up from anesthesia.
Determination of Uric Acid (UA) levels
Biases (selection bias, attrition bias, reporting bias and other biases) may exist in clinical research [15]. In our study and in concordance with the clinical data, the concentrations of UA were measured on day 3 post-CCI. After that, the mice were sacrificed. Furthermore, the concentrations of UA in the serum and brain tissue were evaluated by HPLC with ultraviolet detection. In addition, the samples were obtained from sham, saline- and UA-treated mice under isoflurane anesthesia post-CCI. For the brain tissue, the whole brain was divided into right and left hemispheres, after removing olfactory bulb, brain stem, and cerebellum. Briefly [6], serum (100 μl) and brain tissue (700 mg) were deproteinized with 10% TCA (20 μl serum/150 μl brain) firstly. Moreover, samples were sonicated for 20 s and centrifugated for 5 min (12,000 g). Afterwards, 10 μl supernatant was injected into the HPLC system (PerkinElmer, Madrid, Spain) composed of 200 Pump, 717 plus Auto sampler, and 2487 Dual absorbance detector. Then, the reverse phase ODS2 (4.6·200 mm, 5 μm particle size; Waters, Barcelona, Spain) was used. The mobile phase, consisted of methanol-5 mM ammonium acetate-acetonitrile (1:96:3 vol/vol/vol), was run with an isocratic regular low flow rate of 1.2 ml/min and the wavelength ultraviolet detector was set at 292 nm. Finally, the quantification was performed by external calibration.
Neurological Severity Score (NSS)
According to the protocol of Flierl et al [16], NSS was evaluated before the injury and on days 1, 3, 7, 14, 21 and 28 post-CCI. In addition, the assessment was repeated three times in every testing day. All tests were performed by two investigators blinded to the experimental groups.
Wire grip test
The wire grip test apparatus consisted of a stainless steel bar (50 cm long; 2 mm in diameter) mounted on two vertical supports and it was elevated 37 cm above the flat surface. Mice were placed on the bar midway between the supports and were observed for 60 s. According to Wang et al [17], mice were assessed before the injury and on days 1, 3, 7, 14, 21 and 28 post-CCI. In addition, the evaluation was repeated three times in every testing day. All tests were performed by two investigators blinded to the experimental groups.
Morris water maze test
Morris water maze test was performed to assess the learning and spatial memory abilities of mice. This test was performed between the 15th and 20th-day post-TBI. In addition, a submerged platform was placed 1 cm below the water surface and nontoxic white pigment was mixed into water. According to Huang et al [18], each mouse was released from quadrant 1-4 once per day and was allotted 90 s to search the hidden platform. The trial ended when the mouse either found the platform and stayed on it for 5 s or did not find it within 90 s. The mice that could not find the platform was guided to the platform and allowed to stay 20 s on it.
Four trials per day for five consecutive days were performed with the location of the platform kept constant. On the last testing day, the platform was removed and the swimming track, dwelling time, and path length in every quadrant were recorded by a computer. Additionally, in order to exclude the potential difference of visual ability among groups, extra visible trial was performed by using a labeled platform above the water level. All tests were performed by two investigators blinded to the experimental groups.
Western blot
The brain tissue that was dissected from the contused cortex was homogenized on ice in a modified radioimmunoprecipitation buffer (50 mmol/L Tris-HCl, pH 7.5, 50 mmol/L NaCl, 4 mol/L urea, 0.5% SDS, 0.5% NP-40, 0.5% Na-deoxycholate, 5 mmol/L phenylmethylsulfonyl fluoride, 1 mmol/L ethylenediaminetetraacetic acid, 5 mmol/L ethyleneglycoltetraacetic acid, 10 mmol/L dithiothreitol) containing a protease inhibitor cocktail (1:100, Sigma Aldrich). Furthermore, homogenates were centrifugated at 20,000 g for 20 minutes at 4°C. The protein content of lysates was determined by Bio-Rad protein assay (Hercules). Moreover, an equal amount of proteins (20 μg/lane) were separated in 10% sodium dodecyl sulfate-polyacrylamide gels (Invitrogen) and transferred to polyvinylidene difluoride membranes (Millipore). Next, membranes were blocked for 1 hour in 5% non-fat milk in Tris-buffered saline (pH 7.4) containing 0.1% Tween 20, then incubated with primary antibodies overnight at 4°C, including β-actin (1:5000, Abcam), transferrin (1:500, Abcam), superoxide dismutase 1 (1:1000, Abcam) and peroxiredoxin 2 (1:1000, Abcam). After, membranes were washed in Tris-buffered saline-Tween 20 and incubated for 1 hour with an appropriate horseradish peroxidase-conjugated secondary antibody at room temperature. Then, proteins of interest were detected by using the enhanced chemiluminescence Western blotting detection system kit (ECL Plus) and Hyperfilm (Amersham). The optical densities for protein bands were analyzed and quantified with Quantity One 4.6.2. Finally, β-actin was used as an internal reference. Three independent experiments were carried out to verify proteins expression.
Enzyme-linked immunosorbent assay (ELISA)
After harvesting the injured cortical tissue, the homogenate was centrifuged at 5,000 g for 5 minutes. After, the supernatant was collected and prepared for subsequent assay. Then, tumor necrosis factor (TNF)-α, interleukin (IL)-6, and IL-1β were measured using commercially available ELISA kits (NeoBioscience QuantiCyto ELISA system) according to the protocols accompanying the kits. Briefly, the samples were added to each well of monoclonal anti-mouse TNF-α, IL-6 or IL-1β antibody-coated microtiter plates (ELISA plates) for 30 minutes at 37°C. Unbound material was washed off, and bound antibody was detected by addition of horseradish peroxidase for 15 minutes at 37°C. Furthermore, absorbance was measured 15 minutes after the addition of substrate. Finally, a standard curve was constructed using various dilutions of TNF-α, IL-6, and IL-1β.
Immunofluorescence
Brain slices (10 μm) were dried in the air and fixed in 4% paraformaldehyde, then blocked in 5% fetal bovine serum for 80 minutes. After incubation overnight at 4°C with primary antibody to NeuN (Millipore), slices were analyzed with a fluorescence microscope (ECLIPSE Ti-s, Nikon). Neuronal maintenance was calculated by the number of NeuN-positive cells in CA1 and CA3 of ipsilateral hemispheres. Finally, the number of NeuN-positive cells were counted in 3 randomized 20X fields. All the measurements were performed by ImageJ (NIH).
Two-dimensional laser speckle imaging techniques
Cortical blood flow was monitored using the laser speckle techniques as described previously [19,20]. In addition, laser speckle perfusion images were carried out for the TBI model on day 3 post-TBI with PeriCam PSI (Perimed, Beijing, China).
In our study, each mouse was anesthetized with 4% choral hydrate and the body temperature was maintained at 37±0.5°C. After, the skull was shaved, exposed by a midline skin incision and cleaned. Then, the exposed area was kept clean and dry using a tampon during image collection. Next, the PeriCam PSI head was adjusted to ensure that the red cross point of the indicator laser (660 nm) was located at the center of the brain, and the measurement distance was fixed at 10 cm. Additionally, adjustment of the size of the test area was performed by PIM Software version 1.5. Thereafter, cerebral blood flow signals were collected at 785 nm and transferred into blood perfusion images using PIM Software. Also, perfusion images were acquired using PeriCam high-resolution LSCI (PSI system, Perimed) with a 70 mW built-in laser diode for illumination and a CCD camera (PeriCam PSI System; Perimed) installed 10 cm above the skull to allow penetration of the laser in a diffuse manner through the brain. Finally, the acquired images were analyzed for dynamic changes in CBF using PIMSoft (Perimed) [19, 21-22]. Then, the cerebral blood flow changes were recorded over time and expressed as a percentage of baseline (saline group).
Hematoxylin-eosin staining
Mice were anesthetized and transcardially perfused with saline and 4% paraformaldehyde. Afterward, the brains were removed, stored in fresh 4% paraformaldehyde overnight, protected in 30% sucrose, frozen in O.C.T. media, sectioned (20 μm) and mounted onto slides. Next, sections were dried overnight at room temperature, stained with Gill's hematoxylin, followed by counterstaining in 2.5% eosin and then those sections were mounted on coverslips.
Lesion volumes were assessed based on the Cavalieri method of stereology by using Stereologer software (Systems Planning and Analysis) [23]. Brain slices 500 μm apart were stained with hematoxylin and eosin and then photographed. The target level was between 1 mm anterior and 3 mm posterior to the bregma. So brain slices 500 μm apart (total 9 sections in each animal) were collected to cover the core lesion area. Damaged tissue volume=contralateral hemisphere volume-ipsilateral hemisphere volume.
Statistical analysis
All quantitative data were presented as mean±SD (unless indicated otherwise). The risk factors of prognosis were analyzed by stepwise logistic regression. Functional data in the NSS, wire grip test and water maze test were analyzed by two-way analysis of variance (ANOVA) and repeated-measures [24-26], followed by Tukey's post hoc test across groups. In addition, the clinical characteristics in patients, the determination of UA levels, immunoblotting, ELISA, immunofluorescence, cortical blood flow monitoring and hematoxylin and eosin staining data were analyzed by χ2 test, Student's t-test or randomized one-way ANOVA followed by Tukey-Kramer post-hoc tests. The most conservative multiple-test correction was applied using the Bonferroni method. All statistical analysis were processed with the SPSS software (V. 20.0; IBM, Armonk, New York). For all comparisons, the level of significance was set at P<0.05.
Results
Univariate determinants of clinical outcome
A good outcome (GOS=4-5) at discharge was recorded in 118 patients (61.1%), whereas a poor neurological outcome (GOS<4) was recorded for 75 patients (38.9%), including 20 patients (10.4%) who died during hospitalization after UA measurement. The demographic and clinical characteristics of the study population are shown in Table 1. According to univariate analysis, functional outcome was significantly associated with age, hypertension, chronic pulmonary disease, current smoking, injury severity (GCS), and UA concentration (all P<0.01). Notably, the serum UA concentration was not correlated with injury severity (GCS, P>0.05, data not shown).
Association of clinical characteristics with outcomes in TBI patients. The clinical characteristics included the following: age, sex, arterial hypertension (treated or blood pressure values >160 mm Hg systolic or >90 mm Hg diastolic on repeated measures), coronary heart disease (history of angina, myocardial infarction, or congestive heart failure), diabetes (treated or fasting glucose >110 mg/dl at least in 2 separate analyses), chronic pulmonary diseases (COPD, chronic bronchitis, or emphysema), chronic renal disease (chronic glomerulonephritis, uremia, or chronic renal failure), dyslipidemia (treated or >240 mg/dl), smoking ( >5 cigarettes per day), alcohol intake ( >2 drinks per day), Glasgow Coma Scale (3-8) and UA level [50]. GCS indicates Glasgow Coma Scale. Values are number (%) unless indicated otherwise. Age and UA are expressed as mean±SD. N≥5, Pearson Chi-Square; 1≤N<5, Continuity Correction; N=0, Fisher's Exact Test.
| GOS 4-5 (n = 118) | GOS 1-3 (n = 75) | P | |
|---|---|---|---|
| Age, mean ± SD, y | 48.36 ± 1.71 | 58.85 ± 1.68 | 0.0001 |
| Sex | |||
| Male | 87 (74) | 55 (73) | |
| Female | 31 (26) | 20 (27) | 0.952 |
| Hypertension | 17 (14) | 22 (29) | 0.011 |
| Coronary artery disease | 3 (3) | 7 (9) | 0.082 |
| Diabetes | 9 (8) | 10 (13) | 0.195 |
| Chronic pulmonary disease | 8 (7) | 17 (23) | 0.001 |
| Chronic renal disease | 2 (2) | 3 (4) | 0.605 |
| Dyslipidemia | 6 (5) | 6 (8) | 0.414 |
| Current smoking | 49 (42) | 45 (60) | 0.012 |
| Alcohol intake | 36 (31) | 24 (32) | 0.827 |
| GCS 3-8 | 16 (14) | 30 (40) | 0.0001 |
| UA, mean ± SD, μmol/L | 206.90 ± 8.45 | 264.00 ± 13.54 | 0.0002 |
Values are number (%) unless indicated otherwise.
Independent predictors of clinical outcome
Stepwise logistic regression was performed to identify factors independently associated with a good outcome at discharge. As shown in Table 2, a low serum concentration of UA (males, <208 μmol/L and females <155 μmol/L) was positively associated with a good clinical outcome (P<0.01, odds ratio [OR]=0.023, 95% confidence interval [CI]=0.006-0.082), while GCS levels 3-8 and age <65 years predicted a poor outcome (both P<0.05).
Independent predictors of good outcome at hospital discharge. GOS 4-5, age <65 years, male, hypertension, coronary heart disease, diabetes, chronic pulmonary diseases, chronic renal disease, dyslipidemia, current smoking, alcohol intake, GCS 3-8, low UA level, low creatine level, and low urea level were defined as 1. The opposite of these factors was defined as 0. The risk factors of prognosis were analyzed by stepwise logistic regression. The reference interval of UA is 208-428 μmol/L for male and 155-357 μmol/L for female. The reference interval of creatine is 57-97 μmol/L for male and 41-81 μmol/L for female. The reference interval of urea is 3.1-8.0 mmol/L for male and 3.1-8.8 mmol/L for female.
| Odds Ratio | 95% CI | P | |
|---|---|---|---|
| Uric acid ↓ | 0.023 | 0.006-0.082 | <.0001 |
| GCS (3-8) | 2.769 | 1.215-6.309 | 0.0154 |
| Age (< 65 ys) | 0.097 | 0.025-0.374 | 0.0007 |
UA levels in human serum, mouse serum and mouse brain tissue after TBI
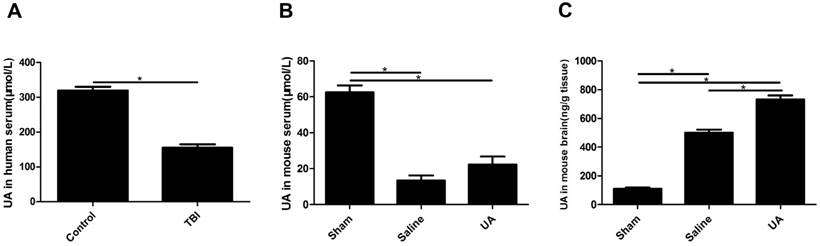
Serum UA levels were significantly lower in TBI patients than in healthy controls (P<0.05, Fig. 1A). To obtain data during the acute phase of TBI, UA levels were detected 3 days post-CCI in the model mice. Serum UA levels were lower in the injury groups than in the sham group (P<0.05, Fig. 1B) following CCI, and when mice were treated with 16 mg/kg UA, no differences were found in serum UA levels between the UA group and saline group (P>0.05, Fig. 1B). The levels of UA in mouse brain tissue were significantly increased after CCI (P<0.05, Fig. 1C), while UA treatment further increased the concentration of UA in brain tissue as compared to the level measured in the saline group (P<0.05, Fig. 1C).
Measurements of UA in human serum, mouse serum and mouse brain tissue after TBI. (A) The measurement of UA in human serum (μmol/L). (B-C) The measurements of UA in mouse serum (μmol/L) and mouse brain tissue (ng/g). Results are presented as mean±SD (n=5 per group), * indicates P<0.05.
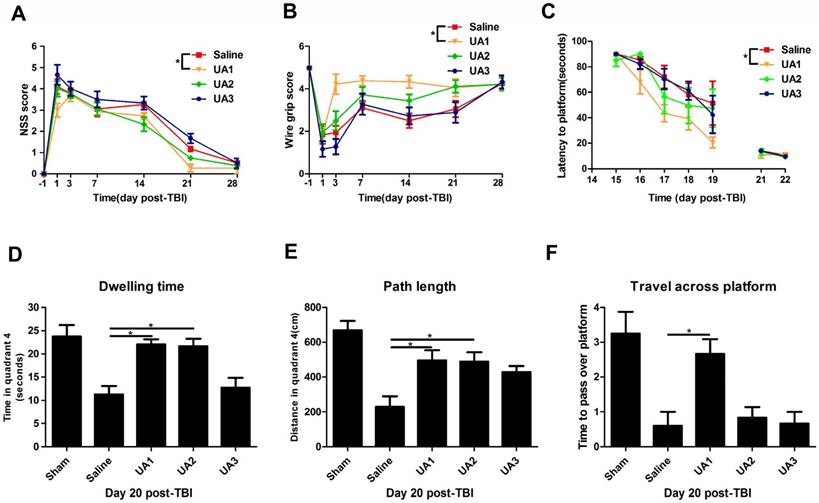
UA treatment improved behavioral performance of mice following CCI
To determine the effects of UA on functional outcomes after CCI, we compared the NSS between the UA-treated and saline-treated groups (Fig. 2A). The mice treated with 16 mg/kg UA exhibited an overall improvement in NSS compared with those treated with saline (P<0.05), whereas treatment with 80 mg/kg UA and 160 mg/kg UA did not show significant benefits (P>0.05). On the wire grip test (Fig. 2B), 16 mg/kg UA improved motor performance compared with saline treatment (P<0.05), whereas 80 mg/kg UA and 160 mg/kg UA did not (P>0.05). Moreover, we examined the four groups in terms of learning ability and spatial memory ability (Fig. 2C-F) in the Morris water maze. After a series of training sessions, animals treated with 16 mg/kg UA took less time to find the hidden platform than those treated with saline (P<0.05, Fig. 2C), whereas no significant advantages were detected after treatment with 80 mg/kg or 160 mg/kg UA (P>0.05, Fig. 2C). On day 20 post-CCI, the dwelling time (Fig. 2D), path length in quadrant 4 (where the platform is located, Fig. 2E) and times to pass over the platform location (Fig. 2F) were recorded with the aid of a computer system. Specifically, as compared to the saline-treated mice, the dwelling time and swimming path length were increased for mice treated with 16 mg/kg UA or 80 mg/kg UA (P<0.05, Fig. 2D&E). In addition, those treated with 16 mg/kg UA passed over the platform location more times (P<0.05, Fig. 2F).
Effects of UA treatment on functional outcomes following CCI. (A) The Neurological Severity Score and (B) the wire grip test. (C-F) The assessment of learning and spatial memory ability in Morris water maze. (C) The latency to locate the hidden platform, (D) dwelling time in quadrant 4, (E) path length in quadrant 4 and (F) the times to pass over the platform. ''Saline” stands for the group that received CCI+Saline, ''UA1” stands for the group that received CCI+UA (16 mg/kg), ''UA2” stands for the group that received CCI+UA (80 mg/kg) and ''UA3” stands for the group that received CCI+UA (160 mg/kg) (n=6 per group). Results are presented as mean±SD. * indicates P<0.05.
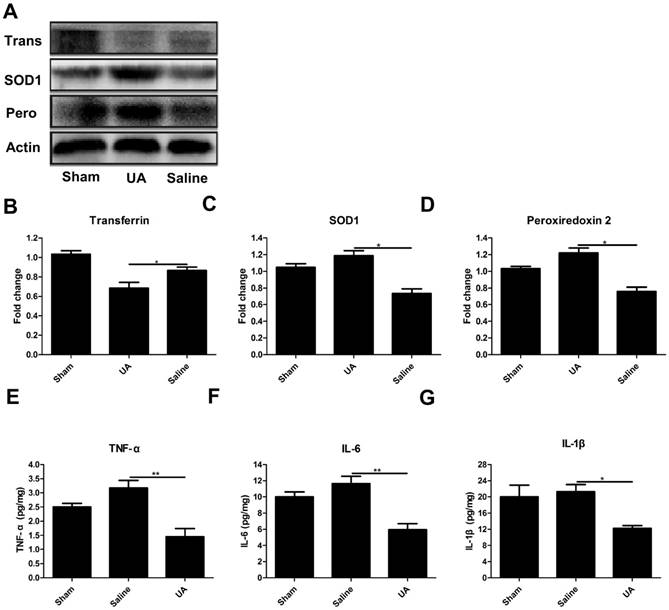
UA inhibited oxidative stress and the inflammatory response following CCI
To determine the effects of UA on oxidative stress after TBI, we measured the protein levels of three oxidative markers: transferrin [27], superoxide dismutase 1 and peroxiredoxin 2 [28] following CCI (Fig. 3A-D). UA (16 mg/kg) treatment significantly promoted the expression of peroxiredoxin 2 and superoxide dismutase 1 while inhibiting transferrin expression (P<0.05). To understand the potential effects of UA on neuroinflammation, we measured the levels of the pro-inflammatory cytokines TNF-α, IL-6 and IL-1β within 3 days post-CCI by ELISA. The levels of TNF-α, IL-1β and IL-6 were significantly lower in the UA group than in the saline group (P<0.05, Fig. 3E-G).
Effects of UA treatment on oxidative stress and the inflammatory response following CCI. (A-D) Representative bands and quantitative analysis of oxidative markers after CCI. (E-G) Quantitative analysis of inflammatory cytokines after CCI by ELISA. Results are presented as mean±SD (n=5 per group). * indicates P<0.05.
UA improved neuronal maintenance and cerebral blood flow but not lesion size following CCI
To evaluate the possible role of UA in acute neuronal degeneration, we assessed hippocampal neuronal maintenance at 7 days post-CCI (Fig. 4A&B). The concept of neuronal maintenance was applied to indicate the survival of neurons following brain injuries [29-31] and was defined according to the number of NeuN-positive cells number [32]. We observed a disrupted hippocampus structure following CCI, and UA (16 mg/kg) treatment significantly attenuated neuronal loss as indicated by an increase in neuron numbers in CA1 (90.25±4.61 vs. 71.00±4.80, P<0.05) and CA3 (95.75±5.14 vs. 69.00±5.79, P<0.05, Fig. 4B).
Effects of UA treatment on neuronal maintenance, cerebral blood flow and lesion size following CCI. (A-B) Representative images and quantitative analysis of neuronal maintenance (NeuN-positive number) in the injured hippocampus after CCI (Scale bar, 200 μm), (n=5 per group). (C-D) Representative laser speckle images and statistical analysis of cortical brain blood flow changes (% CBF in the traumatic hemisphere) in different groups. Color bar shows arbitrary linear perfusion units. Results are expressed as percentage change from baseline (saline group), (n=5 per group). (E-F) Representative images and quantitative analysis of lesion volume in the injured hemisphere after CCI (n=5 per group). * indicates P<0.05.
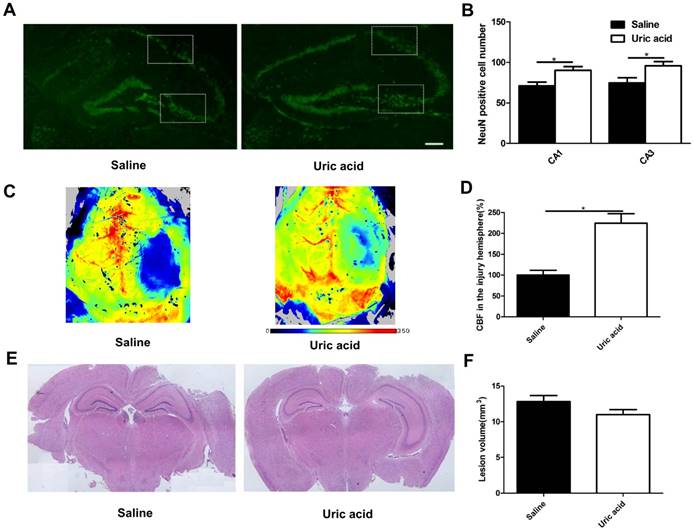
As oxidative stress is also a major cause of endothelial dysfunction in the cerebral circulation [33,34], we evaluated cortical perfusion after UA treatment with laser speckle perfusion images following CCI. Compared with the that observevd in the saline group, UA treatment increased the perfusion efficiency surrounding the lesion areas at 3 days post-CCI (P<0.05, Fig. 4C&D).
The CCI-induced lesion volume was quantified by hematoxylin and eosin staining of coronal brain sections (Fig. 4E) at 28 days post-CCI [35,36]. UA treatment did not induce significant changes in lesion volume (P>0.05, Fig. 4F).
Discussion
To explore the potential protective effects of UA in TBI pathologies, serial experiments were conducted in TBI patients and CCI mice in the present study. Our main findings include that following TBI, the serum UA concentration was significantly lower in patients with a good outcome than in those with a poor outcome. Also, a lower serum UA concentration was an independent predictor of a favorable prognosis. Furthermore, serum UA levels were decreased significantly in TBI patients compared with healthy controls. In the animal experiments, serum UA levels were similarly decreased in CCI model mice compared with those in control mice. In contrast, the UA concentration in brain tissue was increased following CCI and then further enhanced by intraperitoneal injection of UA. Among the different doses of UA tested, 16 mg/kg UA exerted maximal effects in terms of behavioral improvements. The oxidative reaction and inflammatory response were inhibited by UA treatment. Moreover, UA treatment improved hippocampal neuronal maintenance as well as cerebral blood flow after CCI.
The effects of UA represent a double-edged sword in vivo, with UA having long been known as the cause of gout via the deposition of monosodium urate (MSU, UA crystal) in the joints, tendon, kidney and other tissues [37-39]. However, UA is also a major antioxidant, accounting for as much as two-thirds of the total antioxidant capacity in serum [40]. For example, in rats, UA markedly reduces ischemia-induced tyrosine nitration [41], inhibiting vascular oxidative and inflammatory stress [6]. Other researchers reported that UA is involved in the suppression of oxyradical accumulation, stabilization of calcium homeostasis, and preservation of mitochondrial function in rats [42]. Chamorro et al. conducted a phase 2b/3 trial of intravenous urate in acute ischemic stroke and found that urate treatment was not associated with any safety concerns [43]. Another clinical study demonstrated that administration of UA with alteplase reduced infarct growth and led to a better outcome after acute ischemic stroke in women [5]. Amaro et al. demonstrated that UA therapy reduced infarct growth and improved outcomes in patients with hyperglycemia during acute stroke in the URICO-ICTUS trial double-blind study [44]. Furthermore, administration of UA was proven to be protective in experimental allergic encephalomyelitis [45] and to effectively decrease oxidative stress and neuronal loss in animal models of Parkinson's disease [46]. Although the effects of urate itself have not been systematically investigated in TBI models, its precursor inosine was found to improve outcomes in TBI and spinal cord injury [47,48].
Following stroke, an increased UA level in the serum was reported [49,50]. To our surprise, the opposite results were obtained in our study; i.e., the serum UA concentration was reduced in TBI patients and lacked association with injury severity, which was consistent with another study on TBI [51]. Similar results were duplicated in our animal experiments, which avoided the related biases in clinical research [15]. These differences in the results for TBI and stroke may be due to the different pathological properties of the disease models. For instance, the development of TBI is abrupt and robust, whereas stroke is comparatively progressive. Also, there is a rapid decline in serum UA after hospitalization in stroke patients [5]. More intriguingly, the low serum UA level observed in TBI patients in the present study was independently correlated with a favorable outcome, which seems to contradict its neuroprotective role. To our knowledge, UA can penetrate the blood-brain barrier (BBB) [12] or leak from blood into the brain due to BBB disruption [51,52]. In theory, peripheral UA must be recruited to the brain to participate in the oxidative defense post-CCI due to its hepatic source [53], leading to the consumption of serum UA and possibly reflecting a capacity for antioxidant mobilization. Here our results showed that the UA concentration in mouse brain tissue was increased, in concordance with another study of closed head injury [54]. More indirect evidence for this idea is provided by the enhanced UA level observed in brain homogenate after intraperitoneal administration rather than in serum. Data for UA levels in the cerebral spinal fluid of TBI patients should be obtained to further validate our findings.
A 16 mg/kg dose of UA was previously shown to be efficient to attenuate secondary injury and improve neurological function in an experimental stroke model [6]. On the basis of this concentration (16 mg/kg), different doses (1-, 5- and 10-fold increases) were tested to explore the dose associated with maximal neuroprotection in CCI mice. Of course, drug safety was a concern, and the highest dose tested (160 mg/kg) was less than the minimal morbid dose for gout (250 mg/kg) [11]. However, mice treated with 16 mg/kg UA exhibited better improvements than those treated with 80 mg/kg UA, while the highest dose (160 mg/kg) failed to show any benefits in terms of functional outcomes. These observations may be explained by the findings of a recent study showing that excessive UA can also trigger inflammation [12]. Importantly, our results indicated that a proper UA dose exerts its effects via oxidative and inflammatory inhibition following TBI, which extends previous findings of its strong antioxidant activity in acute brain injuries. While several studies have confirmed that endothelial oxidative stress impairs cerebral perfusion in cerebrovascular disease [33,34,55,56], including TBI [57], our study demonstrated that UA treatment increased the perfusion efficiency surrounding lesion areas during the early stage following TBI.
Conclusions
In the current study, we unexpectedly observed reduced serum concentrations of UA in TBI patients, but also found that the UA concentration correlated with injury severity data. We performed an animal study in CCI model mice to explore the mechanism of UA's action in TBI. In summary, our results showed that UA attenuated neuronal loss, cerebral perfusion impairment, and neurological deficits in CCI mice by inhibiting neuronal and vascular oxidative stress. Enhanced antioxidant defense in the brain may result in consumption of UA in the serum, and thus, a reduced serum UA level could be associated with a good clinical outcome following TBI.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 81571159), National Natural Science Foundation for Youth of China (No. 81601072 and No. 81701226), and National Construction Project for Clinical Key Specialty (No. (2011)170). The authors report no conflict of interest concerning the materials or methods used in this study or the findings specified in this paper. In addition, we'd like to thank Rami M. Z. Darwazeh for English proofreading.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Stein SC, Georgoff P, Meghan S, Mizra K, Sonnad SS. 150 years of treating severe traumatic brain injury: a systematic review of progress in mortality. J Neurotrauma. 2010;27:1343-53
2. Johnson RJ, Sautin YY, Oliver WJ, Roncal C, Mu W, Gabriela Sanchez-Lozada L. et al. Lessons from comparative physiology: could uric acid represent a physiologic alarm signal gone awry in western society? Journal of comparative physiology B, Biochemical, systemic, and environmental physiology. 2009;179:67-76
3. Rock KL, Kataoka H, Lai JJ. Uric acid as a danger signal in gout and its comorbidities. Nature reviews Rheumatology. 2013;9:13-23
4. Wu H, Jia Q, Liu G, Liu L, Pu Y, Zhao X. et al. Decreased uric acid levels correlate with poor outcomes in acute ischemic stroke patients, but not in cerebral hemorrhage patients. Journal of stroke and cerebrovascular diseases: the official journal of National Stroke Association. 2014;23:469-75
5. Llull L, Laredo C, Renu A, Perez B, Vila E, Obach V. et al. Uric Acid Therapy Improves Clinical Outcome in Women With Acute Ischemic Stroke. Stroke; a journal of cerebral circulation. 2015;46:2162-7
6. Onetti Y, Dantas AP, Perez B, Cugota R, Chamorro A, Planas AM. et al. Middle cerebral artery remodeling following transient brain ischemia is linked to early postischemic hyperemia: a target of uric acid treatment. American journal of physiology Heart and circulatory physiology. 2015;308:H862-74
7. Guerreiro S, Ponceau A, Toulorge D, Martin E, Alvarez-Fischer D, Hirsch EC. et al. Protection of midbrain dopaminergic neurons by the end-product of purine metabolism uric acid: potentiation by low-level depolarization. Journal of neurochemistry. 2009;109:1118-28
8. Kolz M, Johnson T, Sanna S, Teumer A, Vitart V, Perola M. et al. Meta-analysis of 28,141 individuals identifies common variants within five new loci that influence uric acid concentrations. PLoS genetics. 2009;5:e1000504
9. Petkus V, Preiksaitis A, Krakauskaite S, Zubaviciute E, Rocka S, Rastenyte D. et al. Benefit on optimal cerebral perfusion pressure targeted treatment for traumatic brain injury patients. Journal of critical care. 2017;41:49-55
10. Hoffmeister C, Trevisan G, Rossato MF, de Oliveira SM, Gomez MV, Ferreira J. Role of TRPV1 in nociception and edema induced by monosodium urate crystals in rats. Pain. 2011;152:1777-88
11. Yang Z, Xiaohua W, Lei J, Ruoyun T, Mingxia X, Weichun H. et al. Uric acid increases fibronectin synthesis through upregulation of lysyl oxidase expression in rat renal tubular epithelial cells. American journal of physiology Renal physiology. 2010;299:F336-46
12. Lu W, Xu Y, Shao X, Gao F, Li Y, Hu J. et al. Uric Acid Produces an Inflammatory Response through Activation of NF-kappaB in the Hypothalamus: Implications for the Pathogenesis of Metabolic Disorders. Scientific reports. 2015;5:12144
13. Jiang Y, Brody DL. Administration of COG1410 reduces axonal amyloid precursor protein immunoreactivity and microglial activation after controlled cortical impact in mice. J Neurotrauma. 2012;29:2332-41
14. Zhong J, Jiang L, Huang Z, Zhang H, Cheng C, Liu H. et al. The long non-coding RNA Neat1 is an important mediator of the therapeutic effect of bexarotene on traumatic brain injury in mice. Brain, behavior, and immunity. 2017;65:183-94
15. Mills EJ, Ayers D, Chou R, Thorlund K. Are current standards of reporting quality for clinical trials sufficient in addressing important sources of bias? Contemp Clin Trials. 2015;45:2-7
16. Flierl MA, Stahel PF, Beauchamp KM, Morgan SJ, Smith WR, Shohami E. Mouse closed head injury model induced by a weight-drop device. Nat Protoc. 2009;4:1328-37
17. Wang G, Jiang X, Pu H, Zhang W, An C, Hu X. et al. Scriptaid, a novel histone deacetylase inhibitor, protects against traumatic brain injury via modulation of PTEN and AKT pathway: scriptaid protects against TBI via AKT. Neurotherapeutics. 2013;10:124-42
18. Huang Z, Cheng C, Jiang L, Yu Z, Cao F, Zhong J. et al. Intraventricular apolipoprotein ApoJ infusion acts protectively in Traumatic Brain Injury. Journal of neurochemistry. 2016;136:1017-25
19. Li P, Gan Y, Sun BL, Zhang F, Lu B, Gao Y. et al. Adoptive regulatory T-cell therapy protects against cerebral ischemia. Annals of neurology. 2013;74:458-71
20. Mao L, Li P, Zhu W, Cai W, Liu Z, Wang Y. et al. Regulatory T cells ameliorate tissue plasminogen activator-induced brain haemorrhage after stroke. Brain: a journal of neurology. 2017;140:1914-31
21. Zuo W, Chen J, Zhang S, Tang J, Liu H, Zhang D. et al. IMM-H004 prevents toxicity induced by delayed treatment of tPA in a rat model of focal cerebral ischemia involving PKA-and PI3K-dependent Akt activation. Eur J Neurosci. 2014;39:2107-18
22. Hoda MN, Fagan SC, Khan MB, Vaibhav K, Chaudhary A, Wang P. et al. A 2 x 2 factorial design for the combination therapy of minocycline and remote ischemic perconditioning: efficacy in a preclinical trial in murine thromboembolic stroke model. Exp Transl Stroke Med. 2014;6:10
23. Loane DJ, Pocivavsek A, Moussa CE, Thompson R, Matsuoka Y, Faden AI. et al. Amyloid precursor protein secretases as therapeutic targets for traumatic brain injury. Nature medicine. 2009;15:377-9
24. Tornero D, Wattananit S, Gronning Madsen M, Koch P, Wood J, Tatarishvili J. et al. Human induced pluripotent stem cell-derived cortical neurons integrate in stroke-injured cortex and improve functional recovery. Brain: a journal of neurology. 2013;136:3561-77
25. Zemmar A, Weinmann O, Kellner Y, Yu X, Vicente R, Gullo M. et al. Neutralization of Nogo-A enhances synaptic plasticity in the rodent motor cortex and improves motor learning in vivo. J Neurosci. 2014;34:8685-98
26. Delbary-Gossart S, Lee S, Baroni M, Lamarche I, Arnone M, Canolle B. et al. A novel inhibitor of p75-neurotrophin receptor improves functional outcomes in two models of traumatic brain injury. Brain: a journal of neurology. 2016;139:1762-82
27. Park UJ, Lee YA, Won SM, Lee JH, Kang SH, Springer JE. et al. Blood-derived iron mediates free radical production and neuronal death in the hippocampal CA1 area following transient forebrain ischemia in rat. Acta neuropathologica. 2011;121:459-73
28. Park MH, Jo M, Kim YR, Lee CK, Hong JT. Roles of peroxiredoxins in cancer, neurodegenerative diseases and inflammatory diseases. Pharmacol Ther. 2016;163:1-23
29. Spasic MR, Callaerts P, Norga KK. Drosophila alicorn is a neuronal maintenance factor protecting against activity-induced retinal degeneration. J Neurosci. 2008;28:6419-29
30. Ali YO, Li-Kroeger D, Bellen HJ, Zhai RG, Lu HC. NMNATs, evolutionarily conserved neuronal maintenance factors. Trends in neurosciences. 2013;36:632-40
31. Wang D, Chan CC, Cherry S, Hiesinger PR. Membrane trafficking in neuronal maintenance and degeneration. Cellular and molecular life sciences: CMLS. 2013;70:2919-34
32. Zhuang J, Wei Q, Lin Z, Zhou C. Effects of ADAM10 deletion on Notch-1 signaling pathway and neuronal maintenance in adult mouse brain. Gene. 2015;555:150-8
33. Sochocka M, Koutsouraki ES, Gasiorowski K, Leszek J. Vascular oxidative stress and mitochondrial failure in the pathobiology of Alzheimer's disease: a new approach to therapy. CNS & neurological disorders drug targets. 2013;12:870-81
34. Chrissobolis S, Miller AA, Drummond GR, Kemp-Harper BK, Sobey CG. Oxidative stress and endothelial dysfunction in cerebrovascular disease. Frontiers in bioscience. 2011;16:1733-45
35. Kabadi SV, Stoica BA, Byrnes KR, Hanscom M, Loane DJ, Faden AI. Selective CDK inhibitor limits neuroinflammation and progressive neurodegeneration after brain trauma. Journal of cerebral blood flow and metabolism: official journal of the International Society of Cerebral Blood Flow and Metabolism. 2012;32:137-49
36. Zhao Z, Faden AI, Loane DJ, Lipinski MM, Sabirzhanov B, Stoica BA. Neuroprotective effects of geranylgeranylacetone in experimental traumatic brain injury. Journal of cerebral blood flow and metabolism: official journal of the International Society of Cerebral Blood Flow and Metabolism. 2013;33:1897-908
37. Jin M, Yang F, Yang I, Yin Y, Luo JJ, Wang H. et al. Uric acid, hyperuricemia and vascular diseases. Frontiers in bioscience. 2012;17:656-69
38. Yin Y, Pastrana JL, Li X, Huang X, Mallilankaraman K, Choi ET. et al. Inflammasomes: sensors of metabolic stresses for vascular inflammation. Frontiers in bioscience. 2013;18:638-49
39. Sautin YY, Johnson RJ. Uric acid: the oxidant-antioxidant paradox. Nucleosides Nucleotides Nucleic Acids. 2008;27:608-19
40. Chamorro A, Dirnagl U, Urra X, Planas AM. Neuroprotection in acute stroke: targeting excitotoxicity, oxidative and nitrosative stress, and inflammation. The Lancet Neurology. 2016;15:869-81
41. Romanos E, Planas AM, Amaro S, Chamorro A. Uric acid reduces brain damage and improves the benefits of rt-PA in a rat model of thromboembolic stroke. Journal of cerebral blood flow and metabolism: official journal of the International Society of Cerebral Blood Flow and Metabolism. 2007;27:14-20
42. Yu ZF, Bruce-Keller AJ, Goodman Y, Mattson MP. Uric acid protects neurons against excitotoxic and metabolic insults in cell culture, and against focal ischemic brain injury in vivo. Journal of neuroscience research. 1998;53:613-25
43. Chamorro A, Amaro S, Castellanos M, Segura T, Arenillas J, Marti-Fabregas J. et al. Safety and efficacy of uric acid in patients with acute stroke (URICO-ICTUS): a randomised, double-blind phase 2b/3 trial. The Lancet Neurology. 2014;13:453-60
44. Amaro S, Llull L, Renu A, Laredo C, Perez B, Vila E. et al. Uric acid improves glucose-driven oxidative stress in human ischemic stroke. Annals of neurology. 2015;77:775-83
45. Spitsin SV, Scott GS, Mikheeva T, Zborek A, Kean RB, Brimer CM. et al. Comparison of uric acid and ascorbic acid in protection against EAE. Free radical biology & medicine. 2002;33:1363-71
46. Chen X, Burdett TC, Desjardins CA, Logan R, Cipriani S, Xu Y. et al. Disrupted and transgenic urate oxidase alter urate and dopaminergic neurodegeneration. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:300-5
47. Dachir S, Shabashov D, Trembovler V, Alexandrovich AG, Benowitz LI, Shohami E. Inosine improves functional recovery after experimental traumatic brain injury. Brain research. 2014;1555:78-88
48. Kim D, Zai L, Liang P, Schaffling C, Ahlborn D, Benowitz LI. Inosine enhances axon sprouting and motor recovery after spinal cord injury. PLoS One. 2013;8:e81948
49. Milionis HJ, Kalantzi KJ, Goudevenos JA, Seferiadis K, Mikhailidis DP, Elisaf MS. Serum uric acid levels and risk for acute ischaemic non-embolic stroke in elderly subjects. J Intern Med. 2005;258:435-41
50. Chamorro A, Obach V, Cervera A, Revilla M, Deulofeu R, Aponte JH. Prognostic significance of uric acid serum concentration in patients with acute ischemic stroke. Stroke; a journal of cerebral circulation. 2002;33:1048-52
51. Yang S, Ma Y, Liu Y, Que H, Zhu C, Liu S. Arachidonic acid: a bridge between traumatic brain injury and fracture healing. J Neurotrauma. 2012;29:2696-705
52. Hooper DC, Scott GS, Zborek A, Mikheeva T, Kean RB, Koprowski H. et al. Uric acid, a peroxynitrite scavenger, inhibits CNS inflammation, blood-CNS barrier permeability changes, and tissue damage in a mouse model of multiple sclerosis. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2000;14:691-8
53. Johnson RJ, Nakagawa T, Sanchez-Lozada LG, Shafiu M, Sundaram S, Le M. et al. Sugar, uric acid, and the etiology of diabetes and obesity. Diabetes. 2013;62:3307-15
54. Moor E, Shohami E, Kanevsky E, Grigoriadis N, Symeonidou C, Kohen R. Impairment of the ability of the injured aged brain in elevating urate and ascorbate. Experimental gerontology. 2006;41:303-11
55. Toda N, Okamura T. Cigarette smoking impairs nitric oxide-mediated cerebral blood flow increase: Implications for Alzheimer's disease. J Pharmacol Sci. 2016;131:223-32
56. Park L, Wang G, Zhou P, Zhou J, Pitstick R, Previti ML. et al. Scavenger receptor CD36 is essential for the cerebrovascular oxidative stress and neurovascular dysfunction induced by amyloid-beta. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:5063-8
57. Chrissobolis S, Faraci FM. The role of oxidative stress and NADPH oxidase in cerebrovascular disease. Trends in molecular medicine. 2008;14:495-502
Author contact
![]() Corresponding author: Xiaochuan Sun, Phone: 13883022455; E-mail: sunxch1445com and Chongjie Cheng, Phone: 15334587556; E-mail: 358187887com
Corresponding author: Xiaochuan Sun, Phone: 13883022455; E-mail: sunxch1445com and Chongjie Cheng, Phone: 15334587556; E-mail: 358187887com