Impact Factor ISSN: 1449-1907

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2018; 15(7):674-681. doi:10.7150/ijms.23782 This issue Cite
Research Paper
Inhibition of CRL-NEDD8 pathway as a new approach to enhance ATRA-induced differentiation of acute promyelocytic leukemia cells
Shuyuan Liu#, Jinhua Wan#, Yunyuan Kong, Yonglu Zhang, Lagen Wan ![]() , Zhanglin Zhang
, Zhanglin Zhang ![]()
Department of Clinical laboratory, The First Affiliated Hospital of Nanchang University, Nanchang, Jiangxi 330006, China
#These authors contributed equally to this work.
Received 2017-11-10; Accepted 2018-3-2; Published 2018-4-3
Abstract
The cullin-RING ligase (CRL)-NEDD8 pathway maintains essential cellular processes, including cell cycle progression, apoptosis, autophagy, DNA repair, antigen processing and signal transduction. Growing evidence demonstrates that the alteration of the CRL-NEDD8 pathway in some cancers constitutes an attractive target for therapeutic intervention, but the roles of CRL-NEDD8 pathway in acute promyelocytic leukemia (APL) is still unclear. In the present study, we found that ATRA could decrease the expression of NEDD8-activating enzyme E1 (NAE1) and inhibit the neddylation of cullin1 and cullin3 in the APL cell line NB4. Inactivation of cullin neddylation promoted self-degradation of F-box proteins (Skp2, KLHL20, βTrCP) and up-regulated the protein expression of p27kip, DEPTOR and DAPK1. MLN4924, a novel inhibitor of NAE1, significantly suppressed cell growth and enhanced apoptosis of APL cells by blocking cullin neddylation and subsequent accumulation of CRL E3 substrates. Furthermore, MLN4924 effectively enhanced ATRA-induced differentiation of APL cells by promoting autophagy. Our findings not only provide further insights into the mechanism of the CRL-NEDD8 axis, but also provide a better understanding of this pathway as a potential target for therapeutic intervention in APL.
Keywords: ATRA, differentiation, CRL-NEDD8, MLN4924, neddylation
Introduction
The ubiquitin-proteasome system (UPS) plays a critical role in the degradation of most intracellular proteins. As the largest enzyme family of UPS, the cullin-RING ligases (CRLs) are responsible for ubiquitylation of about 20% of cellular proteins for targeted degradation[1]. Increasing reports suggest that CRLs are implicated in the regulation of numerous cellular processes such as cell cycle and apoptosis, and aberrant CRL activity is associated with cancers. CRLs are modular assemblies built around a central cullin scaffold, a substrate receptor module and a RING protein that recruits the E2-conjugating enzyme[2]. Pro-degradative activity of CRLs requires modification of cullin by a small ubiquitin-like protein NEDD8[3]. CRL neddylation involves an ordered transfer of NEDD8 by specific NEDD8-activating enzyme E1 (NAE1), NEDD8-conjugating enzyme E2 (UBE2M or UBE2F) and NEDD8-E3 ligases[4, 5]. The reverse reaction, deneddylation, catalyzed by the COP9 signalosome (CSN), allows subsequent binding of factors to mediate the disassembly and remodeling of CRL complexes[6, 7]. The binding of NEDD8 to cullin family proteins is required for CRL assembly and activation; however, continuous neddylation of cullins leads to the auto-ubiquitination of CRL subunits followed by degradation[8, 9]. Therefore, CRL-NEDD8 controls a high proportion of ubiquitylation events in cells, making this pathway an attractive target for pharmacological manipulation. Recent studies show that retinoic acid-induced gene G (Rig-g), first identified from an APL cell line NB4 treated with ATRA, is able to negatively regulate SCF-E3 ligase activities and largely decrease protein levels of cullin1 and β-TrCP, indicating a significant role for inhibition of CRL-NEDD8 pathway in the ATRA-induced APL differentiation[10, 11].
MLN4924, a specific small molecule inhibitor, specifically blocks the activity of NEDD8 E1-activating enzyme, efficiently inhibits neddylation of all cullins, resulting in inactivation of CRLs and accumulation of their substrates[12, 13]. It has been shown that MLN4924 has anti-tumor activities both in vitro and in vivo. Treatment of tumor cells (lung cancer, pancreatic cancer, AML, B-cell lymphoma, myeloma) with MLN4924 induces cell cycle arrest, apoptosis and senescence[14-19]. These findings suggested the CRL-NEDD8 pathway as a promising therapeutic target and MLN4924 as a potential drug for cancer therapy.
In this study, we found that ATRA inactivated of CRL1 and CRL3-E3 by inhibiting the neddylation of cullin1 and cullin3 in NB4 cells, then up-regulated the substrate proteins p27kip, DEPTOR and DAPK1. Inhibition neddylation of cullins by MLN4924 significantly suppressed cell growth by inducing S phase arrest and promoting apoptosis of NB4 cells. Furthermore, we found that MLN4924 effectively enhanced ATRA-induced differentiation of APL cells via promoting autophagy. These data illustrate the important role of CRL-NEDD8 mediated proteolysis in ATRA-induced differentiation of APL, and provide the basis for MLN4924 combined ATRA in the APL therapeutics.
Materials and methods
Cell culture and reagents
The APL cell line NB4 was cultured in RPMI 1640 (Gibco BRL, Gaithersburg, MD) containing 10% FBS, 2 mmol/L L-glutamine, 10 U/ml penicillin, and 10μg/ml streptomycin at 37°C in a humidified atmosphere containing 5% CO2. ATRA (Sigma, St. Louis, MO) and MLN4924 ( MedChemExpress USA ) were dissolved in DMSO to 100 mmol/L (stock solutions). Protease inhibitors used were PMSF (AMRESCO, Solon, OH) and a cocktail (Roche, Switzerland); they were respectively dissolved in isopropanol and PBS to 100 mmol/L and 50×. All stock solutions were stored at -20℃. Annexin-V-FITC/PI kit was purchased from Bestbio Biotechnology (Bestbio, China). Cell cycle detection kit (COULTER DNA PREP reagent kit) was from Beckman coulter, Inc.
The following primary antibodies were used in this study: rabbit polyclonal anti- Rig-G antibody was described previously[11]; anti-Cul 1 was obtained from Invitrogen (Grand Island, NY); anti-Cul 3 was purchased from BD (Franklin Lakes, NJ); anti-DAPK1 was produced by Sigma (St. Louis, MO); antibodies against LC3, NAE1, p27kip, p-Beclin1 and βTrCP were from Cell Signaling Technologies Inc. (Beverly, MA); anti-NEDD8 was purchased from Abcam (Cambridge, UK); anti-UBE2M and β-actin were from ABclonal (USA); anti-DEPTOR and Skp2 antibodies, anti-mouse IgG, and anti-rabbit IgG were obtained from Santa Cruz Biotechnology Inc. (USA); anti-Beclin1 and KLHL20 were produced by Abgent (USA).
Cell proliferation and morphology assessment
The leukemic cells were treated with ATRA or MLN4924 for 1 to 3 days, harvested, and washed in PBS. Then, viable cells were quantified using Cell Counter (Z2, Beckman Coulter), and 4×104 viable cells were prepared for cytospin onto glass slides (5 min centrifugation at 500 rpm). The cells on glass slides were stained with Giemsa (WG16; Sigma-Aldrich, St. Louis, MO) for 5 minutes, rinsed briefly with distilled water, dried, and observed by microscopy.
Detection of the CD11b antigen
Mouse anti-human CD11b-PC5 antibody (10 μl) was added to a 100 μl cell suspension (∼5×105 cells) and mixed. The samples were stained for 30 min at 25℃, protected from light. After two washes with PBS, cells were fixed with 500 μl 2% paraformaldehyde solution. The expression of the CD11b antigen was detected by flow cytometry (FC500, Beckman Coulter).
Analysis of cell cycle and apoptosis
Cells were treated with or without the drug and cultivated under 37℃ saturated humidity and 5% CO2. 106 cells were harvest in the appropriate manner (centrifuged at 2,000 rpm for 5 min) and removed the supernatant. For cell cycle analysis, added 50 μl DNA PREP LPR reagent for 1 min according to the instructions, and then added 300 μl DNA PREPStain reagent and placed it at room temperature for 30 min. Then detected by flow cytometry (FC500, Beckman Coulter) and analyzed cell cycle by MODFIT2 software. For apoptosis analysis, 5 μl Annexin-V were added after adding 300ml Annexin-V binding solution and placed the mixture at 4℃ for 15min. Added 10 μl PI at indicated time, then analyzed the results by cytometer. Annexin V+ and/or PI+ cells are apoptosis cells.
Western blot analysis
Whole cell lysates were prepared with a lysis buffer containing 1% Triton X-100, 50 mM Tris (pH 8.0), 150 mM NaCl, 1 mM PMSF, 1 mM Na3VO4, and protease inhibitor cocktail. Protein concentrations were determined using Bio-Rad protein assays. Cell lysate proteins (50 μg) were separated on 12% SDS-PAGE, and electro-transferred to nitrocellulose membranes, which were blocked for 30 minutes at room temperature in Tris-buffered saline-0.05% Tween-20 (TTBS) containing 5% non-fat dry milk. After incubation with TTBS containing primary antibodies for 4 h at room temperature, membranes were washed (3×10 min) in TTBS and incubated with peroxidase conjugated secondary antibodies for 1 h. Finally, protein bands were visualized using the enhanced chemiluminescence detection system (Amersham, Piscataway, NJ).
Statistical analysis
Statistical analysis was performed using Student's t-test. P-values <0.05 were considered statistically significant.
Results
ATRA inactivates cullin1- and cullin3-mediated CRL-E3 ligases and results in substrate protein accumulation by inhibiting neddylation in NB4 cells
To investigate the effect of ATRA on the activation of CRL-NEDD8 in cultured APL cells, we treated NB4 cells with ATRA (1μM) for 24, 48 and 72 hours, and determined the expression levels of cullin1 and cullin3 in NB4 cells. As shown in Fig.1A, western blot analysis of NB4 cells with antibodies against cullin1 and cullin3 revealed the dramatic decrease of the neddylated cullin1 and cullin3 band intensity after ATRA treatment for 24h, but no obvious changes for the un-neddylated cullins. Meanwhile, we determined the expression of Rig-g. The results showed that expression of Rig-g protein started at 72h, which is significantly delayed than the decrease of cullins. In addition, we assessed the protein levels of two cullin1 F-box proteins, Skp2 and βTrCP, and the cullin3 adaptor protein KLHL20 in NB4 cells treated with ATRA. The levels of these three F-box proteins were decreased in a time-dependent manner (Fig. 1B). These results showed that ATRA could inactivate cullin1- and cullin3-mediated E3 ligases, which then inhibited the degradation of substrates such as p27kip, DEPTOR and DAPK1 (Fig. 1C).
ATRA inhibits cullin1- and cullin3-mediated CRL-E3 ligases and results in substrate protein accumulation in NB4 cells. Effect on CRL components and substrates after treatment of NB4 cells with 1μM ATRA for 0, 24, 48 and 72 hours. (A) Immunoblotting for cullin1, cullin3 and Rig-G. (B) For two cullin1 F-box proteins, Skp2 and βTrCP, and the cullin3 adaptor protein KLHL20. (C) For the SCFskp2 substrate p27kip, SCFβTrCP substrate DEPTOR and cullin3-CRL substrate DAPK1. (D) For E1-activating enzyme NAE1 and E2-conjugating enzyme UBE2M. The expression of β-actin was used as loading control.
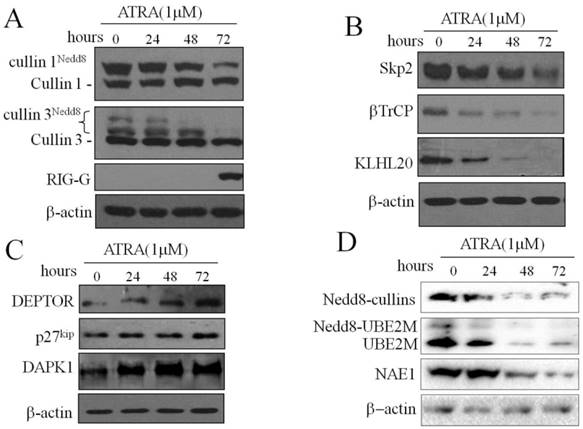
We further hypothesized that inactivation of CRLs may be regulated by neddylation in ATRA treated-NB4 cells. We treated NB4 cells with ATRA and monitored NEDD8-modified cullins, NAE1 and UBE2M by western blot assay. The result showed that the proteins of NEDD8-modified cullins, NAE1 and UBE2M were decreased in ATRA-treated NB4 cells (Fig. 1D). Taken together, our data demonstrated that ATRA potently prevented the neddylation of CRLs and trapped them in an inactive state, the respective CRL substrates could not be ubiquitinated and were protected from degradation by the proteasome. These observations suggest that the important role of CRL-NEDD8 mediated proteolysis in ATRA-induced differentiation of APL cells.
Inhibition of neddylation by MLN4924 induces S phage arrest and promotes apoptosis of NB4 cells
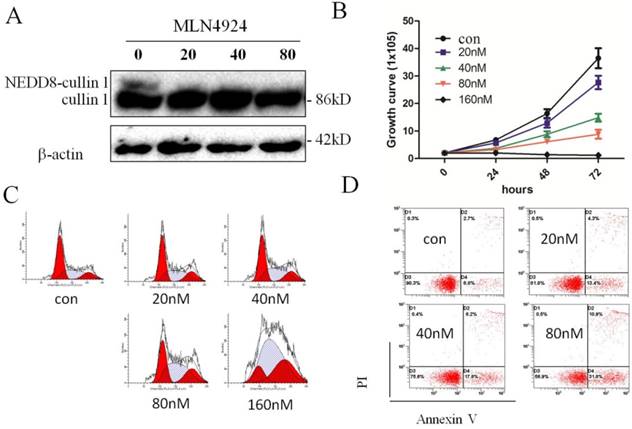
Neddylation contributes to the oncogenic growth of various hematologic malignancies, including acute myeloid leukemia[20]. To investigate effect of CRL-NEDD8 pathway in ATRA-induced differentiation of APL, NB4 cells were treated with varying concentrations of MLN4924 (0, 20, 40, 80 and 160 nM) for 0, 24, 48 and 72 hours. We first determined effect of MLN4924 on cullin neddylation, and the result showed that the neddylated cullin1 was not detectable when treated with 20 nM MLN4924 for 24h (Fig. 2A). Next, we examined the cell proliferation of NB4 cells when treated with MLN4924. As shown in Fig. 2B, MLN4924 inhibited NB4 cells growth in dose- and time-dependent manners. Cell cycle analysis revealed that MLN4924 treatment for 24 hours caused growth arrest at the S phase in a dose-dependent manner (Fig. 2C). Moreover, annexin-V/PI staining assay showed that MLN4924 induced apoptosis of NB4 cells in a dose-dependent manner (Fig. 2D). Collectively, these data demonstrated that MLN4924 potently inhibited cell viability and clonal survival, resulting from induction of S cell cycle arrest and apoptosis.
Inhibition of neddylation by MLN4924 induce S phage arrest and promotes apoptosis of NB4 cells. (A) NB4 cells were treated with MLN4924 (0, 20, 40 and 80 nM) for 0, 24, 48 and 72 hours, and the protein levels of cullin1 were detected by western blotting, with β-actin used as loading control. (B) NB4 cells were exposed to MLN4924 (0, 20, 40, 80 and 160 nM), the growth curve was formed. (C) NB4 cells were treated with MLN4924 for 48 h, stained with PI, and examined with flow cytometry assays. (D) NB4 cells were treated with MLN4924 for 48 h, stained with Annexin-V-FITC and PI, and examined with flow cytometry assays.
Inhibition of neddylation by MLN4924 enhances ATRA-induced differentiation of NB4 cells
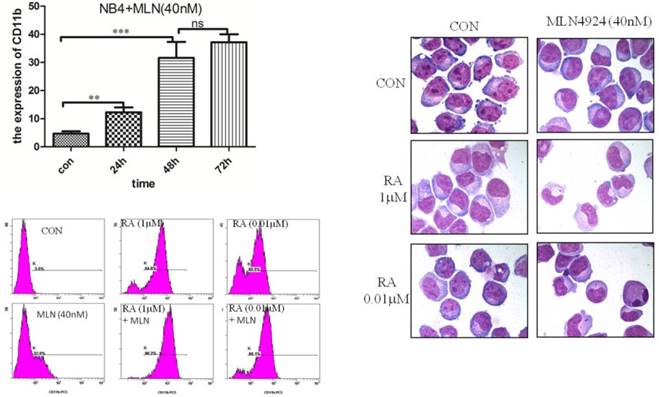
In order to explore the effects of CRL-NEDD8 pathway on ATRA-induced differentiation, we suppressed neddylation by MLN4924 and assessed the myeloid differentiation by measuring the expression of granulo-monocytic differentiation marker CD11b. As the result seen in Fig. 3A, 40 nM MLN4924 induced the CD11b expression in a time-dependent manner. Next, we tested the differentiation of NB4 cells when treated with ATRA combined with MLN4924 (40nM) for 48 hours, we found that the expression of CD11b increased to 84.5% when treated with 1μM ATRA, and 68.5% with 0.01μM ATRA. The percentage of CD11b-positive cells reached to 96.3% when treated with 1μM ATRA combined with MLN4924, and 88.1% for the treatment of 0.01μM ATRA combined with MLN4924 (Fig. 3B). Cell morphology data also demonstrated inhibition of neddylation by MLN4924 could enhance ATRA-induced differentiation of NB4 cells (Fig. 3C).
MLN4924 promotes the expression of CD11b and enhances ATRA induced differentiation of NB4 cells. (A) The myeloid differentiation antigen CD11b was measured by flow cytometry in NB4 cells after treatment with MLN4924 (40 nM) for 0, 24, 48 and 72 hours. (B) NB4 cells were treated with ATRA (0.01 μM) and/or MLN4924 (40 nM) for 48 h, and the expression of CD11b was measured by FCM. (C) Giemsa staining of NB4 cells treated with ATRA (0.01 μM) and/or MLN4924 (40 nM). Graphical data indicates the mean ± S.E.M. ns represent having no statistics, **and *** indicate less than 0.01 and 0.005 of p-values.
Inhibition of neddylation by MLN4924 induced autophagy by up-regulating DAPK1 and Beclin1
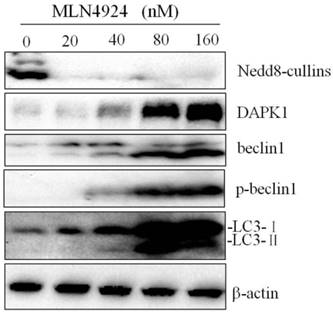
As an important cellular response, autophagy plays a key role in the regulation of cell survival during cellular stresses. Some studies show that MLN4924 effectively induces autophagy in multiple human cancer lines, indicating a general phenomenon. Furthermore, we explored whether MLN4924 affects NB4 cell autophagy. Autophagy in NB4 cells treated with MLN4924 (0, 20, 40, 80 and 160 nM) for 24 hours was detected via testing the conversion of LC3-Ⅰ to LC3-Ⅱ by western blot. As shown in Fig. 4, the conversion of LC3-Ⅰ to LC3-Ⅱ was increased by treatment with MLN4924 and this increase was dose-dependent. Western blot results suggested that MLN4924 could inhibit the neddylation of cullins, and up-regulated DAPK1, Beclin1 and p-Beclin1. Taken together, the data show that MLN4924 could induced autophagy by up-regulating DAPK1 and Beclin1.
Inhibition of neddylation by MLN4924 induced autophagy by up-regulating DAPK1 and Beclin1. NB4 cells were treated with MLN4924 (0, 20, 40, 80 and 160 nM) for 24 h, and the levels of Nedd8, DAPK1, Beclin1, p-Beclin1 and LC3 were examined by Western blot. β-actin was used as loading control.
Inhibition of neddylation by MLN4924 enhances ATRA-induced autophagy
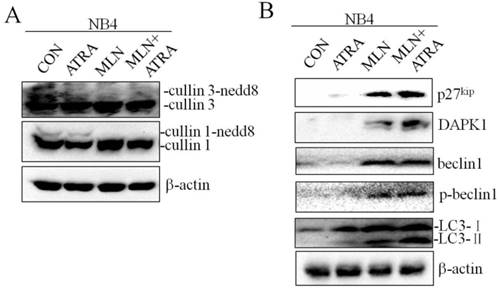
Recent studies have shown that autophagy promotes degradation of the PML/RARa fusion protein and contributes to ATRA induced differentiation of NB4 cells, and CRLs could control autophagy through modification of regulators of autophagy such as DAPK1[21, 22]. We treated NB4 cells with 0.01μM ATRA in combination with 40nM MLN4924, and analyzed the expression of autophagy related proteins by western blot. As shown in Fig. 5A, MLN4924 treatment alone inhibited cullin1 and cullin3 neddylation, demonstrating the inactivation of neddylation pathway. In comparison with MLN4924 alone, MLN4924+ATRA treatment induced significant up-regulation of autophagy related proteins including DAPK1 and p-Beclin1 (Fig. 5B). Interesting, the level of Belin1 in these two group were similar. We also found that MLN4924 combined with ATRA treatment in NB4 cells caused more obvious up-regulation of LC3-II level, indicating that MLN4924 could enhance ATRA-induced autophagy in APL cell line NB4.
Inhibition of neddylation by MLN4924 enhance ATRA induced autophagy. NB4 cells were treated with ATRA (0.01 μM) and/or MLN4924 (40 nM) for 24 h, and the expression of cullin3, cullin1, p27kip, DAPK1, Beclin1, p-Beclin1 and LC3 were analyzed by Western blot. The expression of β-actin was used as loading control.
Discussion
Although great achievements have been made in understanding the mechanistic basis for ATRA-induced differentiation, some other genes may also contribute to the treatment of APL. It is generally considered that Rig-g inhibits the proliferation and propels the ATRA-induced differentiation of NB4 cells, and its expression level is related to the morbid state of APL patients[23, 24]. In the present study, we found that the expression of neddylated cullin1 and cullin3 dramatically decreased at 24h in ATRA-treated NB4 cells, while Rig-G expression decreased at 72h, which is significantly delayed than the decrease of cullins. These results indicated that other mechanisms also contribute to the ATRA-induced differentiation, and cullin-RING ligase may play an important role in the course.
Recent studies have clearly shown that cullin-RING ligase and neddylation pathway are over-activated in various human cancers[17, 25]. CRLs are multi-protein complexes assembled in mammals on seven cullin scaffoles (cullin 1, 2, 3, 4a, 4b, 5 and 7). In the cell, the activities of CRLs can be regulated by the ratio of NEDD8 linkage to the cullin proteins. The binding of NEDD8 to cullin family proteins (neddylation) is required for CRL assembly and activation. Upregulation of CRL-NEDD8 may contribute to tumorigenesis, unrestrained cell proliferation and resistance to apoptosis in cancer. CRL-NEDD8 pathway has emerged as one of the potential cancer targets[17, 25, 26]. In the present study, we found that ATRA inactivated CRL1 and CRL3-E3 by inhibiting the neddylation of cullin1 and cullin3 in NB4 cells, which then up-regulated the substrate proteins p27kip, DEPTOR and DAPK1. To evaluate whether and how CRL-NEDD8 pathway is involved in ATRA-induced differentiation, MLN4924, a novel inhibitor of NAE1, has been used as single agent or in combination with ATRA on APL cell line NB4. The results showed that MLN4924 treatment inhibited the cell proliferation by inducing cell cycle arrest at the S phase in NB4 cells, potently suppressed cell viability and clonal survival. More importantly, the combining MLN4924 with ATRA enhanced cell differentiation. It should be noted that we used a lower dose of ATRA (0.01 μM) than previously reported, and this low dose of ATRA combining with MLN4924 could significantly induce cell differentiation efficiently. To elucidate the underlying mechanism, we focused on potential changes involved in the processes of ATRA treatment.
It has been reported that ATRA stimulates the mTOR-dependent autophagy, which contributes to therapy-induced degradation of the PML-RARα[21]. Inhibiting autophagy blocked PML-RARα degradation and subsequently granulocytic differentiation of human myeloid leukemic cells, demonstrating a role for autophagy in ATRA-induced APL differentiation [21, 27]. In addition, it has been shown that the cul3-KLHL20 E3 ligase regulated autophagy by impacting DAPK1 protein degradation. DAPK phosphorylates Beclin-1 on Thr119 located at a crucial position in its BH3 domain, and thus promoted the dissociation of Beclin-1 from its inhibitor Bcl-XL, resulting in the induction of autophagy[28, 29]. Our results presented above showed MLN4924 combined with ATRA treatment caused more significantly accumulation of DAPK1 and p-Beclin1 and up-regulation of LC3-II level, suggesting that inhibition of neddylation by MLN4924 may enhance ATRA-induced differentiation of NB4 cells, through triggering cell autophagy via accumulated DAPK1 and Beclin1. Meanwhile, Zhao et al showed that MLN4924 induced protective autophagy through inducing accumulation of SCF E3 substrates DEPTOR, a direct inhibitor of mTORC1 and the HIF1-REDD1-TSC1 axis, a negative regulatory pathway of mTORC1[30]. We noted that ATRA could inhibite the degradation of substrates and induce the accumulation of the mTOR-inhibitory protein DEPTOR, revealing that MLN4924-enhanced differentiation may be attributed to blockage of mTOR signals via DEPTOR.
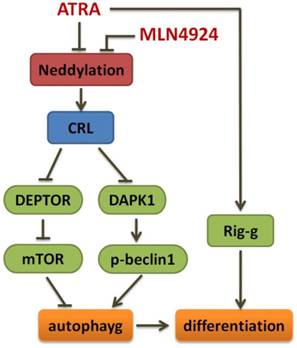
In summary, we demonstrate here that inhibition of neddylation by MLN4924 significantly suppress APL cell growth by blocking cullin neddylation and subsequent accumulation of CRL E3 substrates, which trigger cell cycle regulation and apoptosis, and MLN4924 can induce autophagy by DAPK1 accumulation and effectively enhance ATRA-induced differentiation of APL cells (Fig 6). Our findings not only provide further insights into the mechanism of the CRL-NEDD8 axis, but also contribute to a better understanding of this pathway as a potential target for therapeutic intervention in APL. Furthermore, it is of interest to develop complementary treatment strategies for APL including CRL-NEDD8 inhibitors which increase the sensitivity of APL cell to ATRA action.
Schema of the mechanism for MLN4924 enhancing ATRA-induced differentiation.
Acknowledgements
We thank all members of Department of clinical laboratory of the First Affiliated Hospital of Nanchang University for their support. This work was supported by Natural Science Foundation of China (81760539), Natural Science Foundation of Jiangxi Province (20151BAB205020) and Science and Technology Plan Project of Jiangxi Provincial Health Planning Commission (20171045).
Author Contributions
Zhanglin Zhang contributed to the study design. Shuyuan Liu, Jinhua Wan, Yunyuan Kong, Yonglu Zhang, Lagen Wan, Zhanglin Zhang preformed the research and conducted the data analysis. Zhanglin Zhang and Lagen Wan wrote the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Deshaies RJ, Joazeiro CA. RING domain E3 ubiquitin ligases. Annu Rev Biochem. 2009;78:399-434
2. Zheng N, Schulman BA, Song L, Miller JJ, Jeffrey PD, Wang P, Chu C, Koepp DM, Elledge SJ, Pagano M. et al. Structure of the Cul1-Rbx1-Skp1-F boxSkp2 SCF ubiquitin ligase complex. Nature. 2002;416(6882):703-709
3. Enchev RI, Schulman BA, Peter M. Protein neddylation: beyond cullin-RING ligases. Nat Rev Mol Cell Biol. 2015;16(1):30-44
4. Deshaies RJ, Emberley ED, Saha A. Control of cullin-ring ubiquitin ligase activity by nedd8. Subcell Biochem. 2010;54:41-56
5. Duda DM, Scott DC, Calabrese MF, Zimmerman ES, Zheng N, Schulman BA. Structural regulation of cullin-RING ubiquitin ligase complexes. Curr Opin Struct Biol. 2011;21(2):257-264
6. Pierce NW, Lee JE, Liu X, Sweredoski MJ, Graham RL, Larimore EA, Rome M, Zheng N, Clurman BE, Hess S. et al. Cand1 promotes assembly of new SCF complexes through dynamic exchange of F box proteins. Cell. 2013;153(1):206-215
7. Cope GA, Deshaies RJ. COP9 signalosome: a multifunctional regulator of SCF and other cullin-based ubiquitin ligases. Cell. 2003;114(6):663-671
8. Wee S, Geyer RK, Toda T, Wolf DA. CSN facilitates Cullin-RING ubiquitin ligase function by counteracting autocatalytic adapter instability. Nat Cell Biol. 2005;7(4):387-391
9. Wu JT, Chan YR, Chien CT. Protection of cullin-RING E3 ligases by CSN-UBP12. Trends Cell Biol. 2006;16(7):362-369
10. Xiao S, Li D, Zhu HQ, Song MG, Pan XR, Jia PM, Peng LL, Dou AX, Chen GQ, Chen SJ. et al. RIG-G as a key mediator of the antiproliferative activity of interferon-related pathways through enhancing p21 and p27 proteins. Proc Natl Acad Sci U S A. 2006;103(44):16448-16453
11. Xu GP, Zhang ZL, Xiao S, Zhuang LK, Xia D, Zou QP, Jia PM, Tong JH. Rig-G negatively regulates SCF-E3 ligase activities by disrupting the assembly of COP9 signalosome complex. Biochem Biophys Res Commun. 2013;432(3):425-430
12. Soucy TA, Smith PG, Milhollen MA, Berger AJ, Gavin JM, Adhikari S, Brownell JE, Burke KE, Cardin DP, Critchley S. et al. An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. Nature. 2009;458(7239):732-736
13. Brownell JE, Sintchak MD, Gavin JM, Liao H, Bruzzese FJ, Bump NJ, Soucy TA, Milhollen MA, Yang X, Burkhardt AL. et al. Substrate-assisted inhibition of ubiquitin-like protein-activating enzymes: the NEDD8 E1 inhibitor MLN4924 forms a NEDD8-AMP mimetic in situ. Mol Cell. 2010;37(1):102-111
14. Blank JL, Liu XJ, Cosmopoulos K, Bouck DC, Garcia K, Bernard H, Tayber O, Hather G, Liu R, Narayanan U. et al. Novel DNA damage checkpoints mediating cell death induced by the NEDD8-activating enzyme inhibitor MLN4924. Cancer Res. 2013;73(1):225-234
15. Godbersen JC, Humphries LA, Danilova OV, Kebbekus PE, Brown JR, Eastman A, Danilov AV. The Nedd8-activating enzyme inhibitor MLN4924 thwarts microenvironment-driven NF-kappaB activation and induces apoptosis in chronic lymphocytic leukemia B cells. Clin Cancer Res. 2014;20(6):1576-1589
16. Jia L, Li H, Sun Y. Induction of p21-dependent senescence by an NAE inhibitor, MLN4924, as a mechanism of growth suppression. Neoplasia. 2011;13(6):561-569
17. Li L, Wang M, Yu G, Chen P, Li H, Wei D, Zhu J, Xie L, Jia H, Shi J. et al. Overactivated neddylation pathway as a therapeutic target in lung cancer. J Natl Cancer Inst. 2014;106(6):dju083
18. Lin JJ, Milhollen MA, Smith PG, Narayanan U, Dutta A. NEDD8-targeting drug MLN4924 elicits DNA rereplication by stabilizing Cdt1 in S phase, triggering checkpoint activation, apoptosis, and senescence in cancer cells. Cancer Res. 2010;70(24):10310-10320
19. Luo Z, Yu G, Lee HW, Li L, Wang L, Yang D, Pan Y, Ding C, Qian J, Wu L. et al. The Nedd8-activating enzyme inhibitor MLN4924 induces autophagy and apoptosis to suppress liver cancer cell growth. Cancer Res. 2012;72(13):3360-3371
20. Swords RT, Kelly KR, Smith PG, Garnsey JJ, Mahalingam D, Medina E, Oberheu K, Padmanabhan S, O'Dwyer M, Nawrocki ST. et al. Inhibition of NEDD8-activating enzyme: a novel approach for the treatment of acute myeloid leukemia. Blood. 2010;115(18):3796-3800
21. Isakson P, Bjoras M, Boe SO, Simonsen A. Autophagy contributes to therapy-induced degradation of the PML/RARA oncoprotein. Blood. 2010;116(13):2324-2331
22. Cui D, Xiong X, Zhao Y. Cullin-RING ligases in regulation of autophagy. Cell Div. 2016;11:8
23. Lou YJ, Pan XR, Jia PM, Li D, Xiao S, Zhang ZL, Chen SJ, Chen Z, Tong JH. IRF-9/STAT2 [corrected] functional interaction drives retinoic acid-induced gene G expression independently of STAT1. Cancer Res. 2009;69(8):3673-3680
24. Lou YJ, Pan XR, Jia PM, Jin J, Tong JH. RIG-G inhibits the proliferation of NB4 cells and propels ATRA-induced differentiation of APL cells. Leuk Res. 2016;40:83-89
25. Soucy TA, Dick LR, Smith PG, Milhollen MA, Brownell JE. The NEDD8 Conjugation Pathway and Its Relevance in Cancer Biology and Therapy. Genes Cancer. 2010;1(7):708-716
26. Milhollen MA, Narayanan U, Soucy TA, Veiby PO, Smith PG, Amidon B. Inhibition of NEDD8-activating enzyme induces rereplication and apoptosis in human tumor cells consistent with deregulating CDT1 turnover. Cancer Res. 2011;71(8):3042-3051
27. Orfali N, O'Donovan TR, Nyhan MJ, Britschgi A, Tschan MP, Cahill MR, Mongan NP, Gudas LJ, McKenna SL. Induction of autophagy is a key component of all-trans-retinoic acid-induced differentiation in leukemia cells and a potential target for pharmacologic modulation. Exp Hematol. 2015;43(9):781-793 e782
28. Zalckvar E, Berissi H, Eisenstein M, Kimchi A. Phosphorylation of Beclin 1 by DAP-kinase promotes autophagy by weakening its interactions with Bcl-2 and Bcl-XL. Autophagy. 2009;5(5):720-722
29. Zalckvar E, Berissi H, Mizrachy L, Idelchuk Y, Koren I, Eisenstein M, Sabanay H, Pinkas-Kramarski R, Kimchi A. DAP-kinase-mediated phosphorylation on the BH3 domain of beclin 1 promotes dissociation of beclin 1 from Bcl-XL and induction of autophagy. EMBO Rep. 2009;10(3):285-292
30. Zhao Y, Xiong X, Jia L, Sun Y. Targeting Cullin-RING ligases by MLN4924 induces autophagy via modulating the HIF1-REDD1-TSC1-mTORC1-DEPTOR axis. Cell Death Dis. 2012;3:e386
Author contact
![]() Corresponding authors: Zhanglin Zhang, Email: zhzl1984sjtu.edu.cn and Lagen Wan, Email: wlgme196412com; Tel: +86-0791-88697032, Mail Address: No. 17 Yongwai Street, Donghu District, Nanchang, Jiangxi, 330006, China.
Corresponding authors: Zhanglin Zhang, Email: zhzl1984sjtu.edu.cn and Lagen Wan, Email: wlgme196412com; Tel: +86-0791-88697032, Mail Address: No. 17 Yongwai Street, Donghu District, Nanchang, Jiangxi, 330006, China.