Impact Factor ISSN: 1449-1907

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2018; 15(6):603-609. doi:10.7150/ijms.24492 This issue Cite
Research Paper
Glucagon-Like Peptide-1 Receptor Agonist and Glucagon Increase Glucose-Stimulated Insulin Secretion in Beta Cells via Distinct Adenylyl Cyclases
Young-Sun Lee1, Hee-Sook Jun1,2,3 ![]()
1. Lee Gil Ya Cancer and Diabetes Institute, Gachon University, Incheon, 406-840, Republic of Korea
2. College of Pharmacy and Gachon Institute of Pharmaceutical Science, Gachon University, Incheon, 406-840, Republic of Korea
3. Gachon Medical Research Institute, Gil Hospital, Incheon, 405-760, Republic of Korea
Received 2017-12-21; Accepted 2018-3-2; Published 2018-3-14
Abstract
Diabetes mellitus is a chronic disease in which the pancreas no longer produces enough insulin. Pancreatic alpha cell mass increases in response to insufficient insulin secretion. However, the reason for this increase is not clear. It is possible that the increased alpha-cells may stimulate compensatory insulin release in response to the insufficient insulin such as insulin resistance. In this study, we investigated whether glucagon and glucagon-like peptide-1 (GLP-1), hormones produced by alpha cells, contribute to insulin secretion in INS-1 cells, a beta cell line. We confirmed that alpha cell area in the pancreatic islets and glucagon secretion were increased in HFD-induced obese mice. Co-treatment with glucagon and exendin-4 (Ex-4), a GLP-1 receptor agonist, additively increased glucose-stimulated insulin secretion in INS-1 cells. In parallel, cAMP production was also additively increased by co-treatment with these hormones. The increase of insulin secretion by Ex-4 in the presence of high glucose was inhibited by 2'5'-dideoxyadenosine, a transmembrane adenylyl cyclase inhibitor, but not by KH-7, a soluble adenylyl cyclase inhibitor. The increase of insulin secretion by glucagon in INS-1 cells was inhibited by both 2'5'-dideoxyadenosine and KH-7. We suggest that glucagon and GLP-1 produced from alpha cells additively increase cAMP and insulin secretion in the presence of high glucose via distinct adenylyl cyclases in INS-1 cells, and this may contribute to the compensatory increase of insulin secretion by an increase of pancreatic alpha cell mass under conditions of insulin resistance.
Keywords: Glucagon-like peptide-1, glucagon, insulin secretion, beta cell, alpha cell
Introduction
Diabetes is a metabolic disease which is characterized by high blood glucose levels, and type 2 diabetes is associated with both insulin resistance and insulin deficiency. In the early stage of type 2 diabetes, insulin secretion is increased to compensate for insulin resistance [1].
Islet adaptation to insufficient insulin involves compensatory changes in not only beta cells, but also in pancreatic alpha cells [2] [3]. Alpha cells are markedly increased in conditions of lack of beta cells such as injury to beta cells and patients with recent-onset type 1 diabetes [4, 5], and also in the condition of insulin resistance as a result of high fat diet-induced obesity [6, 7]. As a result, increased glucagon secretion is an accompanying phenomenon in type 2 diabetes [3, 6, 8]. However, it is not clear yet the reasons for compensatory increase of pancreatic alpha cells.
In pancreatic alpha cells, glucagon-like peptide-1 (GLP-1) is also produced in addition to glucagon [9, 10]. Glucagon and GLP-1 are hormones derived from the transcriptional product of the proglucagon gene [11]. Post-translational processing of proglucagon by prohormone convertase-2 (PC2) produces glucagon, and further processing of proglucagon by prohormone convertase-1/3 (PC1/3) yields GLP-1 [12-14].
In addition to its classical role as a promoter of gluconeogenesis and glycogenolysis, glucagon, as well as GLP-1, are known to be stimulators of insulin release in beta cell lines and pancreatic islets [15-17]. Binding of glucagon to the glucagon receptor activates adenylyl cyclase and generates cAMP, followed by activation IP3 and increase of calcium, contributing to various biological effects such as gluconeogenesis in liver hepatocytes [18]. Upon GLP-1 receptor activation, adenylyl cyclase is activated and cAMP generated, leading, in turn, to cAMP-dependent activation of second messenger pathways, such as the PKA and Epac pathways, contributing to insulin release in beta cells [9].
We hypothesize that the increased alpha cells in insulin insufficient condition such as insulin resistance may increase the production of glucagon and GLP-1 secretion, contributing to the increase of insulin secretion in beta-cells. Although it is already known that each of glucagon and GLP-1 is stimulator of insulin secretion, we don't know whether glucagon and GLP-1 act additively or synergistically increase insulin secretion in beta cells and the molecular mechanisms. In this study, we investigated the effects of co-treatment of glucagon and Ex-4, a GLP-1 receptor agonist, on insulin secretion in INS-1 cells and found that co-treatment of glucagon and Ex-4 additively increased insulin secretion in the presence of high glucose via a distinct adenylyl cyclase.
Materials and Methods
Materials
The following reagents were purchased: exendin-4 (Ex-4), glucagon, and KH-7 (Sigma, St. Louis, USA) and 2'5'-dideoxyadenosine (Calbiochem, La Jolla, CA).
Animals
C57BL/6 mice were obtained from the Korea Research Institute of Bioscience and Biotechnology (Daejeon, Korea) and were maintained at a facility at Gachon University. Male C57BL/6 mice (4 weeks old) were fed a high fat diet (HFD; 60% kcal from fat) for 8 weeks. All animal experiments were carried out under a protocol approved by the Institutional Animal Care and Use committee at Lee Gil Ya Cancer and Diabetes Institute, Gachon University.
Cell culture
INS-1 cells were cultured in RPMI-1640 medium supplemented with 10% heat-inactivated fetal bovine serum, 11 mM glucose, 2 mM L-glutamine, 100 U/mL penicillin, and 100 µg/mL streptomycin at 37°C in a humidified atmosphere containing 95% air and 5% CO2. The cells were seeded at a density of 2 × 105/well in 24-well plates.
Immunohistochemical analysis
C57BL/6 mice were sacrificed after 8 weeks of HFD feeding. Pancreata were removed, fixed in 10% formalin, and embedded in paraffin. The tissue sections were then incubated with primary antibody solution: guinea-pig anti-insulin (DAKO, 1:100) and rabbit anti-glucagon (DAKO, 1:100). Texas Red-conjugated goat anti-guinea-pig IgG (Santa Cruz Biotechnology, 1:200) and fluorescein isothiocyanate-conjugated goat anti-rabbit IgG (Santa Cruz Biotechnology,1:200) were used as secondary antibodies. Fluorescence was imaged using a laser scanning confocal fluorescent microscope (LSM 700, Carl Zeiss MicroImaging, Jena, Germany). The areas of alpha cells, beta cells, and islets were imaged and analyzed by CQ1spinning disk confocal systems (Yokogawa Electric Corporation) using CellVoyager software.
Glucagon measurement
To investigate the level of glucagon in serum, non-fasting blood samples were obtained from C57BL/6 mice at 8 weeks after HFD feeding prior to sacrifice. We measured the concentration of serum glucagon with a Mercodia glucagon ELISA kit (Mercodia AB, Sweden).
Insulin secretion measurement
INS-1 cells were seeded at a density of 2 × 105/well in 24-well plates with 11 mM glucose RPMI medium. After 24 h, the medium was changed to 5 mM glucose RPMI media and then 24 h later, the medium was changed to 0.2 mM Krebs Ringer Buffer (KRB). After 2 h, the cells were stimulated with 3 mM or 17 mM glucose with or without glucagon, Ex-4, or both for 2 h. At the end of the stimulation, the amount of secreted insulin was quantified using a rat insulin EIA kit (Alpco Diagnostics, Windham, NH, USA) according to the manufacturer's method.
Intracellular cAMP measurement
INS-1 cells were seeded at a density of 1 × 106/well in 6-well plates in 11 mM glucose RPMI medium and cultured. After 24 h, the medium was changed to 17 mM glucose RPMI and then Ex-4 or glucagon was added for 15 min. The medium was removed, and the cells were lysed by incubation with 1 ml of 0.1 M HCl for 20 min at room temperature. The lysate was centrifuged at 3,000 rpm for 5 min to separate the cellular debris, and the supernatant was used for determination of cAMP concentration using a cAMP ELISA kit (Enzo Life Sciences, Farmingdale, NY, USA).
Statistical analysis
All experiments were conducted with a minimum of three different samples, and data are presented as the mean ± SD. Statistical analysis was performed using an unpaired parametric Student's t-test for two groups or ANOVA followed by Fisher's protected least significant difference test for multiple groups. P<0.05 was accepted as significant.
Results
Alpha cell area and glucagon secretion increased in HFD-induced obese mice
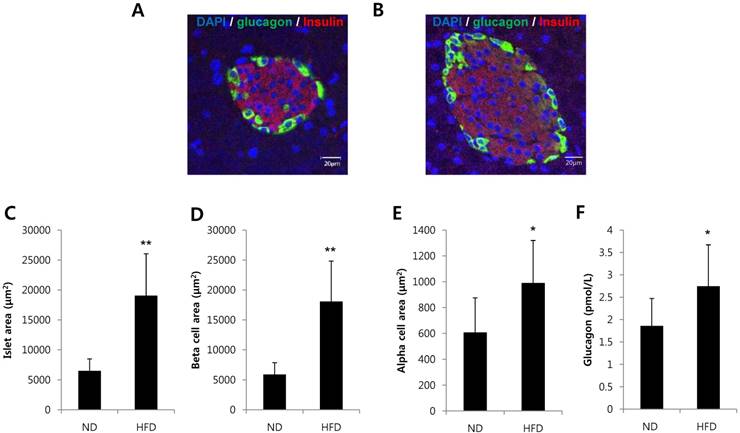
To investigate whether alpha cells are increased in a condition of insulin resistance, we analyzed pancreatic tissues and serum glucagon levels in mice fed a HFD for 8 weeks. The area of islets, beta cell area per islet and alpha cell area per islet were significantly increased in HFD-induced obesity mice (Fig. 1A-E). Serum glucagon levels were significantly increased in HFD-induced obese mice compared with mice fed a normal diet (Fig. 1F). These results suggest that islet adaptation to conditions that would induce insulin resistance increased the alpha cell area and glucagon secretion in vivo.
Alpha cells and glucagon secretion were increased in HFD-induced obese mice. C57BL/6 mice were fed a HFD or normal diet (ND) for 8 weeks. Pancreatic tissues and serum were harvested. Pancreatic sections from (A) ND-fed mice or (B) HFD-fed mice were double-stained with anti-glucagon and anti-insulin antibodies. The area of (C) the islets, (D) beta cells per islet, and (E) alpha cells per islet was measured. (F) The concentration of glucagon in the serum was determined by a glucagon ELISA kit. N=7~10 mice and 54-57 islets. Data are means ± SD, *P<0.05 compared with the ND group.
Glucagon and Ex-4 co-treatment additively increased insulin secretion in high glucose-stimulated INS-1 cells
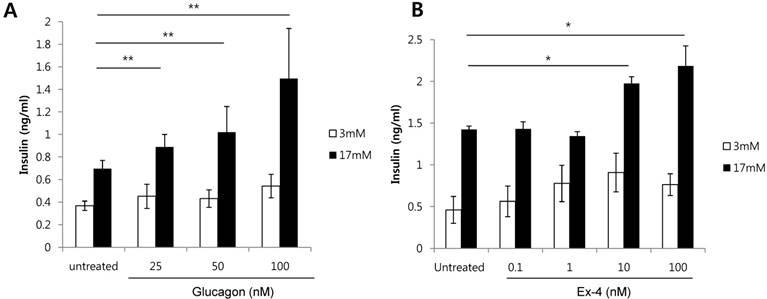
As a HFD increases alpha cells, and both glucagon and GLP-1 are produced from alpha cells, we investigated whether glucagon or GLP-1 increases the secretion of insulin in glucose-stimulated beta cells. Glucagon treatment of INS-1 cells significantly increased insulin secretion in a dose-dependent manner in the presence of high glucose (17 mM) (Fig. 2A). Similarly, Ex-4, GLP-1 receptor agonist, also significantly increased insulin secretion at 10 nM or 100 nM of Ex-4 treatment in the presence of high glucose (17 mM) (Fig. 2B). These results indicate that both glucagon and Ex-4 dose-dependently increased insulin secretion in high glucose-stimulated INS-1 cells.
Glucagon and Ex-4 dose dependently increased glucose-induced insulin secretion in INS-1 cells. INS-1 cells were incubated in media with 11 mM glucose for 24 h, 5 mM glucose for 12 h, and in KRB for 2 h. Cells were then treated with the indicated concentrations of (A) glucagon or (B) Ex-4 in 3 mM or 17 mM glucose KRB for 2 h. Insulin levels in the media were measured with an insulin EIA kit. Results are representative of 3 independent experiments. Data are means ± SD, *P<0.05, **P<0.001 compared with the untreated group.
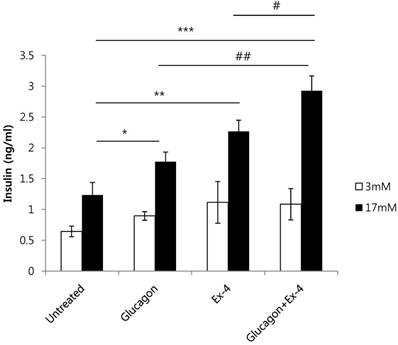
To investigate the effect of glucagon and Ex-4 co-treatment on insulin secretion, INS-1 cells were treated with glucagon (50 nM) and/or Ex-4 (10 nM) in the presence of 17 mM or 3 mM glucose. We found that insulin secretion was increased by glucagon or Ex-4 treatment alone, and glucagon and Ex-4 co-treatment additively and significantly increased insulin secretion over treatment with either compound alone in the presence of 17 mM glucose, but not in the presence of 3 mM glucose (Fig. 3).
Glucagon and Ex-4 co-treatment increased glucose-induced insulin secretion in INS-1 cells. INS-1 cells were incubated in media with 11 mM glucose for 24 h, 5 mM glucose for 12 h, and in KRB for 2 h. Cells were then treated with glucagon (50 nM), Ex-4 (10 nM), or both in 3 mM or 17 mM glucose KRB for 2 h. Insulin levels in the media were measured with an insulin EIA kit. Results are representative of 3 independent experiments. Data are means ± SD, *P<0.05, **P<0.01, ***P<0.001 compared with the untreated group and # P<0.05, ## P<0.01 compared with the glucagon and Ex-4 treated group.
Glucagon and Ex-4 increased insulin secretion via distinct adenylyl cyclase in high glucose-stimulated INS-1 cells
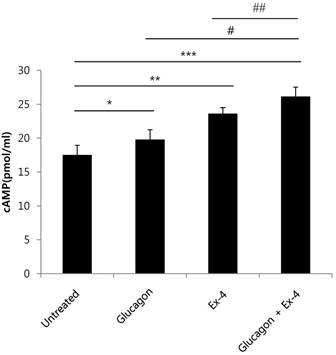
cAMP and its signaling pathways are important for the actions of glucagon and GLP-1[19] [20]. Therefore, we measured cAMP production in INS-1 cells treated with glucagon, Ex-4, or both. cAMP production was significantly increased in glucagon or Ex-4-treated INS-1 cells compared with untreated INS-1 cells in the presence of 17 mM glucose. Co-treatment with glucagon and Ex-4 additively and significantly augmented cAMP levels over either treatment alone (Fig. 4).
Glucagon and Ex-4 co-treatment increased cAMP levels in INS-1 cells. INS-1 cells were incubated in media with 11 mM glucose for 24 h and then treated with glucagon (50 nM), Ex-4 (10 nM), or both in 17 mM glucose media for 15 min. The cells were lysed and the supernatant was assayed for measurement of cAMP concentrations. Results are representative of 5~6 independent experiments. Data are means ± SD. *P<0.05, **P<0.005, *** P<0.001 compared with the untreated group, # p<0.05, ## p<0.01 compared with the glucagon and Ex-4-treated group.
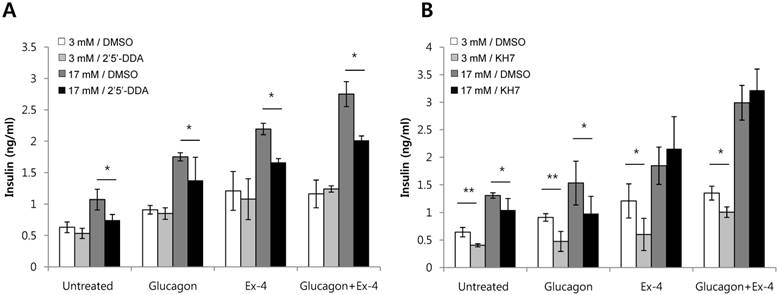
Adenylyl cyclases catalyze the conversion of ATP to cAMP. In mammals, adenylyl cyclases fall into two classes: transmembrane adenylyl cyclase (tmAC) and soluble adenylyl cyclase (sAC) [21]. Antagonists of adenylyl cyclases are 2'5'-dideoxyadenosine, a P-site antagonist of tmAC (IC50 = 3-16 uM), and KH7, a selective inhibitor of sAC (IC50 = 2-5 uM) [22]. To investigate which adenylyl cyclase is involved in insulin secretion by glucagon or Ex-4, INS-1 cells were pretreated with 2'5'-dideoxyadenosine or KH7 and the amount of secreted insulin was measured. Both glucagon-stimulated insulin secretion and Ex-4-induced insulin secretion were inhibited by the tmAC inhibitor, 2'5'-dideoxyadenosine, in the presence of high glucose. As well, co-treatment with glucagon and Ex-4 was inhibited by the tmAC inhibitor in the presence of high glucose (Fig. 5A). Glucagon-stimulated insulin secretion was inhibited by the sAC inhibitor, KH7, in the presence 17 mM glucose, but insulin secretion stimulated by Ex-4 or co-treatment with glucagon and Ex-4 was not inhibited (Fig. 5B).
Glucagon and Ex-4 co-treatment increased glucose-induced insulin secretion via distinct adenylyl cyclases in INS-1 cells. INS-1 cells were incubated in media with 11 mM glucose for 24 h, 5 mM glucose for 12 h, and 0.2 mM KRB for 90 min. Cells were then pre-treated for 30 min with vehicle (DMSO) with or without (A) 2'5'-dideoxyadenosine (2'5'-DDA; 100 μM) or (B) KH7 (30 uM) followed by treatment with glucagon (50 nM), Ex-4 (10 nM), or both for 2 h. Insulin levels in the media were measured with an insulin EIA kit. Results are representative of 4~6 independent experiments. Data are means ± SD, *P<0.05, **P<0.01 compared with 2'5'-dideoxyadenosine or KH7 treatment.
Discussion
HFD-fed mice are associated with insulin resistance and hyperglucagonemia [23, 24]. Pancreatic alpha cells are normally about 15-20 % of the islet mass, but they are increased in conditions of insulin insufficiency or insulin resistance [4, 7, 25]. Their primary role is to secrete glucagon. GLP-1 is also secreted from alpha cells, as well as from intestinal L-cells, and GLP-1 secretion from alpha cells is particularly increased in the presence of high glucose or in damaged islets [25, 26]. Glucagon mainly regulates hepatic glucose production to maintain glucose homeostasis [27], and hyperglucagonemia contributes to increased glucose production and hyperglycemia caused by beta cell failure [28, 29]. GLP-1, is an important insulin secretagogue, and increases beta-cell proliferation and insulin biosynthesis. Both glucagon and GLP-1 are also known to be stimulators of insulin release in beta cell lines and pancreatic islets [15-17], and both glucagon and GLP-1 receptors are expressed in human islets [30] and use cAMP signaling pathways [31, 32]. However, it is not clear whether these hormones, which are both produced from alpha cells, can additively or synergistically stimulate glucose-induced insulin secretion and which adenylyl cyclase is involved. In this study, we found that co-treatment of glucagon and GLP-1 additively potentiates insulin secretion in the presence of high glucose, but not low glucose, in INS-1 cells.
Next, we investigated the downstream mechanisms of this additive effect. GLP-1 and glucagon have distinct receptors, specific G protein-coupled receptors that contain seven transmembrane domains [16, 33] and mediate cAMP production [31, 32]. Adenylyl cyclases catalyze the conversion of ATP to cAMP [19]. In mammals, the family of adenylyl cyclases includes at least ten isozymes which fall into two classes of adenylyl cyclase: tmACs and sACs [21]. TmACs are at the plasma membrane and receive extracellular signals through the G-protein-coupled receptors. sACs are ubiquitously expressed and reside in specific subcellular compartments throughout the cell [34]. Mammalian sAC activity is stimulated by bicarbonate [35], Ca2+ and cellular ATP levels [36], but is insensitive to G protein or forskolin [37]. Calcium-sensitive adenylyl cyclase 8 (ADCY8, one of the tmACs) plays a central role for cAMP-induced amplification of exocytosis in GLP-1 signaling [38]. Although adenylyl cyclases are required for glucagon signaling [39], isozymes of adenylyl cyclases have not been identified as mediator of glucagon-induced insulin secretion.
We found that treatment with KH7, an sAC inhibitor, suppressed glucagon-induced insulin secretion, but not Ex-4-mediated insulin secretion, in the presence of high glucose. However, treatment with 2'5'-dideoxyadenosine, the preferential inhibitor of tmAC, suppressed both glucagon- and Ex-4-induced insulin secretion. This suggests that while glucagon operates through both the action of tmACs and sACs, Ex-4 operates only through the action of tmACs. Thus, co-treatment of glucagon and Ex-4 additively enhanced insulin secretion through the action of different adenylyl cyclases to elevate cAMP.
In summary, we investigated the effect of co-stimulation of GLP-1 and glucagon in beta-cells and found that co-treatment of Ex-4 and glucagon additively increased insulin secretion in high glucose conditions via distinct adenylyl cyclases. In addition, these results support previous findings that pancreatic alpha cells undergo compensatory changes (hyperplasia) during the development of insulin insufficiency or insulin resistance to increase the production of glucagon and GLP-1, which may contribute to reducing hyperglycemia by increasing insulin secretion.
Acknowledgements
We thank Dr. Ann Kyle for editorial assistance. This research was supported by a grant of Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (2015R1D1A1A01060232) and by a grant from the Korea Health Technology R&D Project through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health & Welfare, Republic of Korea (grant number: HI14C1135).
Abbreviations
Ex-4: Exendin-4; GLP-1: glucagon-like peptide-1; HFD: high fat diet; KRB: Krebs Ringer Buffer; sAC: soluble adenylyl cyclase; tmAC: transmembrane adenylyl cyclase.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Czech MP. Insulin action and resistance in obesity and type 2 diabetes. Nat Med. 2017;23:804-14
2. Ahren B, Pacini G. Importance of quantifying insulin secretion in relation to insulin sensitivity to accurately assess beta cell function in clinical studies. Eur J Endocrinol. 2004;150:97-104
3. Larsson H, Ahren B. Islet dysfunction in insulin resistance involves impaired insulin secretion and increased glucagon secretion in postmenopausal women with impaired glucose tolerance. Diabetes Care. 2000;23:650-7
4. Thorel F, Nepote V, Avril I, Kohno K, Desgraz R, Chera S. et al. Conversion of adult pancreatic alpha-cells to beta-cells after extreme beta-cell loss. Nature. 2010;464:1149-54
5. Willcox A, Richardson SJ, Bone AJ, Foulis AK, Morgan NG. Evidence of increased islet cell proliferation in patients with recent-onset type 1 diabetes. Diabetologia. 2010;53:2020-8
6. Ahren B. Glucagon secretion in relation to insulin sensitivity in healthy subjects. Diabetologia. 2006;49:117-22
7. Ellingsgaard H, Ehses JA, Hammar EB, Van Lommel L, Quintens R, Martens G. et al. Interleukin-6 regulates pancreatic alpha-cell mass expansion. P Natl Acad Sci USA. 2008;105:13163-8
8. Li XC, Zhuo JL. Current Insights and New Perspectives on the Roles of Hyperglucagonemia in Non-Insulin-Dependent Type 2 Diabetes. Curr Hypertens Rep. 2013;15:522-30
9. Lee YS, Jun HS. Anti-diabetic actions of glucagon-like peptide-1 on pancreatic beta-cells. Metabolism. 2014;63:9-19
10. Marchetti P, Lupi R, Bugliani M, Kirkpatrick CL, Sebastiani G, Grieco FA. et al. A local glucagon-like peptide 1 (GLP-1) system in human pancreatic islets. Diabetologia. 2012;55:3262-72
11. Layer P, Holst JJ, Grandt D, Goebell H. Ileal release of glucagon-like peptide-1 (GLP-1). Association with inhibition of gastric acid secretion in humans. Dig Dis Sci. 1995;40:1074-82
12. Lee YS, Jun HS. Anti-diabetic actions of glucagon-like peptide-1 on pancreatic beta-cells. Metabolism: clinical and experimental. 2014;63:9-19
13. Heller RS, Aponte GW. Intra-islet regulation of hormone secretion by glucagon-like peptide-1-(7-36) amide. The American journal of physiology. 1995;269:G852-60
14. Yabe D, Rokutan M, Miura Y, Komoto I, Usui R, Kuwata H. et al. Enhanced glucagon-like peptide-1 secretion in a patient with glucagonoma: implications for glucagon-like peptide-1 secretion from pancreatic alpha cells in vivo. Diabetes research and clinical practice. 2013;102:e1-4
15. Dalle S, Smith P, Blache P, Le-Nguyen D, Le Brigand L, Bergeron F. et al. Miniglucagon (glucagon 19-29), a potent and efficient inhibitor of secretagogue-induced insulin release through a Ca2+ pathway. J Biol Chem. 1999;274:10869-76
16. Kawai K, Yokota C, Ohashi S, Watanabe Y, Yamashita K. Evidence That Glucagon Stimulates Insulin-Secretion through Its Own Receptor in Rats. Diabetologia. 1995;38:274-6
17. Thorens B, Waeber G. Glucagon-Like Peptide-I and the Control of Insulin-Secretion in the Normal-State and in Niddm. Diabetes. 1993;42:1219-25
18. Yang HH, Yang LH. Targeting cAMP/PKA pathway for glycemic control and type 2 diabetes therapy. J Mol Endocrinol. 2016;57:R93-R108
19. Moens K, Heimberg H, Flamez D, Huypens P, Quartier E, Ling ZD. et al. Expression and functional activity of glucagon, glucagon-like peptide I, and glucose-dependent insulinotropic peptide receptors in rat pancreatic islet cells. Diabetes. 1996;45:257-61
20. Tian G, Sandler S, Gylfe E, Tengholm A. Glucose- and Hormone-Induced cAMP Oscillations in alpha- and beta-Cells Within Intact Pancreatic Islets. Diabetes. 2011;60:1535-43
21. Zippin JH, Farrell J, Huron D, Kamenetsky M, Hess KC, Fischman DA. et al. Bicarbonate-responsive "soluble" adenylyl cyclase defines a nuclear cAMP microdomain. J Cell Biol. 2004;164:527-34
22. Ramos LS, Zippin JH, Kamenetsky M, Buck J, Levin LR. Glucose and GLP-1 stimulate cAMP production via distinct adenylyl cyclases in INS-1E insulinoma cells. J Gen Physiol. 2008;132:329-38
23. Pacini G, Thomaseth K, Ahren B. Contribution to glucose tolerance of insulin-independent vs. insulin-dependent mechanisms in mice. Am J Physiol-Endoc M. 2001;281:E693-E703
24. Ahren B, Simonsson E, Scheurink AJW, Mulder H, Myrsen U, Sundler F. Dissociated insulinotropic sensitivity to glucose and carbachol in high-fat diet-induced insulin resistance in C57BL/6J mice. Metabolism. 1997;46:97-106
25. Thyssen S, Arany E, Hill DJ. Ontogeny of regeneration of beta-cells in the neonatal rat after treatment with streptozotocin. Endocrinology. 2006;147:2346-56
26. Nie Y, Nakashima M, Brubaker PL, Li QL, Perfetti R, Jansen E. et al. Regulation of pancreatic PC1 and PC2 associated with increased glucagon-like peptide 1 in diabetic rats. J Clin Invest. 2000;105:955-65
27. Lv SH, Qiu XC, Li J, Liang JY, Li WD, Zhang C. et al. Glucagon-induced extracellular cAMP regulates hepatic lipid metabolism. J Endocrinol. 2017;234:73-87
28. Basu R, Chandramouli V, Dicke B, Landau B, Rizza R. Obesity and type 2 diabetes impair insulin-induced suppression of glycogenolysis as well as gluconeogenesis. Diabetes. 2005;54:1942-8
29. Mang Y, Osborne MC, Monia BP, Bhanot S, Gaarde WA, Reed C. et al. Reduction in glucagon receptor expression by an antisense oligonucleotide ameliorates diabetic syndrome in db/db mice. Diabetes. 2004;53:410-7
30. Huypens P, Ling Z, Pipeleers D, Schuit F. Glucagon receptors on human islet cells contribute to glucose competence of insulin release. Diabetologia. 2000;43:1012-9
31. Luo GC, Kong XC, Lu LN, Xu XH, Wang H, Ma XS. Glucagon-like peptide 1 potentiates glucotoxicity-diminished insulin secretion via stimulation of cAMP-PKA signaling in INS-1E cells and mouse islets. Int J Biochem Cell B. 2013;45:483-90
32. Li SY, Li J, Cao GL, Zhang Z, Wang YW, Sun K. Effect of glucagon on insulin secretion through cAMP signaling pathway in MIN6 cells. Int J Clin Exp Patho. 2015;8:5974-80
33. Thorens B. Expression Cloning of the Pancreatic Beta-Cell Receptor for the Gluco-Incretin Hormone Glucagon-Like Peptide-1. P Natl Acad Sci USA. 1992;89:8641-5
34. Zippin JH, Chen YQ, Nahirney P, Kamenetsky M, Wuttke MS, Fischman DA. et al. Compartmentalization of bicarbonate-sensitive adenylyl cyclase in distinct signaling microdomains. Faseb J. 2002;16:82
35. Chen YQ, Cann MJ, Litvin TN, Iourgenko V, Sinclair ML, Levin LR. et al. Soluble adenylyl cyclase as an evolutionarily conserved bicarbonate sensor. Science. 2000;289:625-8
36. Zippin JH, Chen YQ, Straub SG, Hess KC, Diaz A, Lee D. et al. CO2/HCO3- and calcium-regulated soluble adenylyl cyclase as a physiological ATP sensor (vol 288, pg 33283, 2013). J Biol Chem. 2014;289:12679
37. Buck J, Sinclair ML, Schapal L, Cann MJ, Levin LR. Cytosolic adenylyl cyclase defines a unique signaling molecule in mammals. P Natl Acad Sci USA. 1999;96:79-84
38. Roger B, Papin J, Vacher P, Raoux M, Mulot A, Dubois M. et al. Adenylyl cyclase 8 is central to glucagon-like peptide 1 signalling and effects of chronically elevated glucose in rat and human pancreatic beta cells. Diabetologia. 2011;54:390-402
39. Quesada I, Tuduri E, Ripoll C, Nadal A. Physiology of the pancreatic alpha-ce glucose secretion: role in glucose homeostasis and diabetes. J Endocrinol. 2008;199:5-19
Author contact
![]() Corresponding author: Hee-Sook Jun, Lee Gil Ya Cancer and Diabetes Institute, Gachon University, 7-45 Songdo-dong, Yeonsu-ku, Incheon, 21999, Republic of Korea. Email: hsjunac.kr
Corresponding author: Hee-Sook Jun, Lee Gil Ya Cancer and Diabetes Institute, Gachon University, 7-45 Songdo-dong, Yeonsu-ku, Incheon, 21999, Republic of Korea. Email: hsjunac.kr