Impact Factor ISSN: 1449-1907

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2017; 14(13):1335-1341. doi:10.7150/ijms.20653 This issue Cite
Research Paper
HSPB8 Promotes the Fusion of Autophagosome and Lysosome during Autophagy in Diabetic Neurons
Xiao-Cheng Li1,2,5, Qi-Kuan Hu2,3, Ling Chen1,2, Si-yang Liu1,2, Shi Su1,2, Hong Tao2,3, Lin-Na Zhang1,2,3, Tao Sun2, Lan-Jie He1,4 ![]()
1. General Hospital of Ningxia Medical University, Yinchuan, 750000, China;
2. Ningxia Key Laboratory of Cerebrocranial Diseases, the National Key Laboratory Incubation Base, Yinchuan, 750000, China;
3. Department of Physiology, Ningxia Medical University, Yinchuan, 750000, China;
4. Department of Endocrinology, Qilu Hospital of Shandong University (Qingdao), Qingdao, 266000, China;
5. Department of Endocrinology, the First Affiliated Hospital of Xi'an Medical University, Xi'an, 710000, China.
Received 2017-4-19; Accepted 2017-9-12; Published 2017-10-15
Abstract
Although autophagy has been proposed to play an emerging role in diabetic neuropathy, autophagy and its possible role remains unclear. Moreover, only few studies about diabetes have explored the autophagy mediated by heat shock protein beta-8 (HSPB8) and Bcl-2 associated athanogene 3 (BAG3). In the present study, we examined the autophagy induced by high glucose levels in an in vivo rat model of diabetes induced by streptozotocin (STZ) and an in vitro model of retinal ganglion cell-5 (RGC5) cells under high glucose conditions. In the spinal cord tissues of the STZ-induced diabetic rats, the levels of light chain 3 (LC3) and Beclin-1-marked autophagy rose with increasing HSPB8 and BAG3 levels. By confocal immunofluorescence, HSPB8 and LC3 were observed to be co-localized in the spinal cord tissues. In the RGC5 cells, high-glucose stimulation upregulated the expression of LC3-Ⅱ, Beclin-1, and HSPB8 in a dose-dependent manner. When the RGC5 cells were subjected to high-glucose conditions, HSPB8 overexpression, along with upregulated LC3-Ⅱ and Beclin-1 expression, increased the autophagic rate, whereas siRNA-silenced HSPB8 decreased the autophagic rate. Furthermore, in GFP-mRFP-LC3 probe experiments, HSPB8 overexpression promoted autophagosome-lysosome fusion, whereas HSPB8 silencing disrupted this process. In the cells treated with HSPB8 and siRNA, the fusion was impaired, as indicated by the elevated p62 expression. HSPB8 overexpression can partly rescue the blocking of the autophagy flux with chloroquine through the reduction of p62 expression level. Our study demonstrated that HSPB8 is involved in the high glucose-induced autophagy under the in vivo and in vitro conditions and critically participated in the autophagosome-lysosome fusion during the autophagy flux.
Keywords: high glucose, autophagy, HSPB8, fusion.
Introduction
Autophagy is emerging as an important pathway in diabetic neuropathy. Hyperglycemia associated with diabetes impairs cellular autophagic pathways by driving the quality control of proteins and cellular organelles. Autophagy undergoes the following two steps: autophagosome formation, autophagosome-lysosome fusion [1]. Autophagy is regulated by a set of conserved genes named as the autophagy-related genes (Atg) [2]. Beclin 1, the first identified mammalian Atg, is an essential autophagy inducer [3]. Beclin 1 forms a regulatory complex with class III phosphatidylinositol-3-kinase which is important in initiating autophagy [4]. Meanwhile, microtubule-associated protein light chain 3 (LC3), which exists on autophagosomes, is a mammalian homolog of yeast Atg 8. Autophagosome formation involves the conversion of LC3-I to the lipidated form LC3-II. This conversion from LC3-I to LC3-II is a common indicator of autophagy [5]. Therefore, LC3 serves as a widely used marker for autophagosomes [6]. The p62 proteins can directly bind to LC3 proteins via a specific sequence motif and can be degraded by autophagy in the lysosomes. If autophagy is inhibited, the p62 proteins will accumulate. Therefore, the increasing of p62 proteins can be used as the marker of autophagy flux inhibition [7].
An emerging role of autophagy in diabetic neuropathy has been recently proposed [8]. However, the role of autophagy in diabetic neuropathy is unclear and extremely complicated. Diabetic rats induced by streptozotocin (STZ) exhibits increased levels of autophagy in their dorsal root ganglia [9]. Notably, a clinical study on young patients with type 1 diabetes mellitus who died from diabetic ketoacidosis and brain edema reported that the pathogenesis of the neuronal deficits was associated with increased macro-autophagy levels [10].
Heat shock proteins (HSPs), also called stress proteins, contain singular conserved structures. HSPs maintain chaperone and kinase activities, and protect against apoptosis and cell impairment [11, 12]. HSPB8, which belongs to a superfamily of small HSPs, functions as a chaperone interacting with Bcl-2 associated athanogene 3 (BAG3) during autophagy [13]. The BAG family of proteins functions as co-chaperones by assisting molecular chaperones to recruit target proteins [14]. The BAG3 protein belongs to the BAG family, which interacts with the ATPase domain of the HSPB8 to regulate autophagy [15]. HSPB8 has been reported to be closely associated with motor neuron diseases, such as Huntington's and Parkinson's diseases and distal hereditary motor neuropathy [16]. HSPB8 is also mutated in clinical diseases, such as hereditary nerve ending disease and Charcot-Marie-Tooth disease [17, 18]. These findings indicate the important role of HSPB8 in neuropathy. Recent studies have reported the involvement of HSPB8 in diabetes. HSPB8 expression levels were decreased in the gastrocnemius muscles of diabetic rats upon fluoride exposure [19]. Therefore, HSPB8 may be involved in the pathogenesis of diabetic neuropathy through the effects on autophagy.
The current study aims to determine the role and mechanism of HSPB8 in autophagy and flux through in vivo and in vitro experiments. We used retinal ganglion cell-5 (RGC5) cells which is a human neuroblastoma cell line in the in vitro experiment.
Materials and Methods
Animals
Male Sprague-Dawley rats (200-250 g) were provided by the Laboratory Animal Center of Ningxia Medical University. Diabetes was induced by intraperitoneal injection of STZ (55 mg/kg body weight) in sodium citrate buffer. Meanwhile, control rats were injected with sodium citrate buffer alone. All the rats were raised for 4 weeks in accordance with the guidelines of the Institutional Animal Care and Use Committee procedures of Ningxia University. The investigation conformed to the United States National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Reagents, cell culture, and treatment
Retinal ganglion cell-5 (RGC5) cells (human neuroblastoma cell line) were cultured in a RPMI-1640 medium (Invitrogen, Carlsbad, CA, USA) containing 10% fetal bovine serum (Invitrogen, Carlsbad, CA, USA). Chloroquine was purchased from Sigma-Aldrich (St. Louis, MO, USA). The HSPB8-pcDNA3.1 plasmids and small interfering RNAs (siRNAs) were constructed and designed by the Transheep Bio Company (Shanghai, China). The siRNA sequences were as follows: 5′-GGAUUGUCUCCAAGAACUUTT-3′ (sense-HSPB8), 5′-AAGUUCUUGGAGACAAUCCTT-3′ (antisense-HSPB8), 5′-UUCUCCGAACGUGUCACGUTT-3′ (nonsilenced-sense), and 5′-ACGUGACACGUUCGGAGAATT-3′ (nonsilenced-antisense). Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) was used to transfect siRNAs into the RGC5 cells, and a final concentration of 50 nM was achieved.
Western blot analysis
The spinal cord tissues of the rats or the cell samples were then lysed with lysis reagent (Beyotime, Shanghai, China). A BAC protein assay kit (Thermo Scientific, Rockford, IL, USA) was used to quantify the proteins. Western blot was then routinely performed by 10% sodium dodecyl sulfate (SDS) gels and polyvinylidene difluoride (PVDF) membranes (Millipore Corporation, Bedford, MA, USA). An Odyssey Infrared Imaging system (Biosciences, Lincoln, NE, USA) was used to detect the blotted membrane. Antibodies against HSPB8 (R&D Systems, Minneapolis, MN, USA), LC3B (Cell Signaling Technology, Beverly, MA, USA), BAG3, Beclin-1, p62, and GADPH (ProteinTech Group, Chicago, IL, USA) were adopted. Secondary antibodies were obtained from LI-COR Biosciences (Lincoln, NE, USA).
Immunofluorescence
The rats were anesthetized and perfused through the ascending aorta with 0.9% NaCl and 4% paraformaldehyde for 1.5 h. After the perfusion, spinal cords were dissected and fixed in 4% paraformaldehyde for 4 h. Then, sucrose (20%-30%) was used to dehydrate the spinal cords. The dehydrated samples were cut into 8-μm-thick frozen slices (cross-sections) in a microtome-cryostat. The cross-sections were subjected to double immunofluorescent staining. The primary antibodies utilized were as followed: HSPB8 (1:150; mouse monoclonal), LC3B (1:200; rabbit monoclonal), and BAG3 (1:200; rabbit monoclonal). FITC- and TRITC-conjugated secondary antibodies (1:100) were obtained from ZhongshanGoldbridge Biotechnology (Beijing, China). Immunofluorescence was observed through confocal microscopy (Olympus, Tokyo, Japan).
Co-immunoprecipitation assays
Spinal cord tissues from the diabetic rats were homogenized, lysed, and then mixed with 50 μl of protein A sepharose beads. Next, the mixture was incubated on a rotary mixer for 30 min at 4 °C with pre-clearing lysates. HSPB8 antibody (2 μg) (R&D Systems, Minneapolis, MN, USA) or control immunoglobulin M (ProteinTech Group, Chicago, IL, USA) was used for co-immunoprecipitation. The resulting solutions were incubated at 4 °C overnight. The solutions were then centrifuged, and the resulting beads were washed with TBST containing a protease inhibitor for three to four times. Approximately 80 μl of supernatant was obtained after the last centrifugation. The pellet was resuspended with 20 μl SDS sample buffer, and then heated at 95-100 °C for 5 min. Western blot was subsequently performed using anti-BAG3 (ProteinTech Group ,Chicago, IL, USA) and anti-HSPB8 (R&D Systems, Minneapolis, MN, USA) antibodies.
Transmission electron microscopy
The transmission electron microscope of the electron microscopy center of Ningxia Medical University was used. Before the cells were observed, they were fixed in phosphate-buffered 2.5% glutaraldehyde for 24 h, and then post-fixed in phosphate-buffered 2% osmium tetroxide for 2 h. Subsequently, the cells were dehydrated in ascending acetone concentrations, and then infiltrated with Spurr's resin for over 24 h. The cells were subsequently observed under a Hitachi H-600 transmission electron microscope (Ibaraki, Japan).
GFP-mRFP-LC3 probe tandem observation
pcDNA3.1-mRFP-GFP-LC3 plasmids were transfected into the RGC5 cells. The HSPB8-pcDNA3.1 plasmid and the siRNA-HSPB8 were transfected simultaneously. The cells were then fixed in 4% paraformaldehyde for 30 min and observed under a confocal microscope.
Statistical evaluation
All data were presented as means ± standard deviations. Student's t-test was used when two groups were compared, and multivariate ANOVA was used to compare more than two groups. A p value of less than 0.05 was considered statistically significant. The statistical analysis was performed on SPSS 17.0 software (IBM SPSS, Inc., Chicago, IL, USA).
Results
Autophagy and HSPB8 were both upregulated by high glucose in vivo and vitro
To determine how autophagic processes are altered during the pathological processes of diabetes, we first examined the autophagy marker proteins in the spinal cord tissues of the STZ-induced diabetic rats. As shown in Fig. 1A, the expression levels of LC3II and Beclin-1 increased considerably, implying that autophagy occurred in the spinal cord. Then, we examined the role of HSPB8 in the autophagic process in the spinal cord. HSPB8 expression increased in the spinal cords of the diabetic rats. Moreover, BAG3 expression increased only slightly in the spinal cord. Through immuofluorescent double staining, we found that HSPB8 and LC3 co-localized in the cytoplasm of the spinal cord cells (Fig. 1B). To further demonstrate the involvement of the HSPB8-BAG3 complex in the autophagy process of the spinal cord, we performed a co-immunoprecipitation experiment using the anti-HSPB8 antibody. The anti-HSPB8 antibody pulled down the BAG3 proteins and showed bands when tested by the BAG3 antibody (Fig. 1C). These results showed that the HSPB8-BAG3 complex was involved in the spinal cord autophagy. In vitro experiments were conducted on the RGC5 cell lines which are retinal ganglion neuron cell lines. The HSPB8 expression increased in a dose-dependent manner when the glucose concentration increased (Fig. 1D). Consistent with the changes observed in the in vivo studies, the expression levels of LC3II and Beclin-1 in the RGC5 cells increased after the cells were subjected to high-glucose stimulus for 32 h. BAG3 expression also increased along with the upregulated HSPB8 expression (Fig. 1D), indicating that the HSPB8-BAG3 complex was involved in the autophagy.
Intensified autophagy and increased HSPB8 expression by high glucose level in vivo and in vitro. (A) Relative protein levels of HSPB8, BAG3, LC3, and Beclin-1 in the spinal cord of STZ-induced diabetic rats. Column represents the grayscale value (n = 9). Values represented as mean ± SD. * p< 0.05 vs control, ** p < 0.01 vs control. (B) Representative confocal microscopic images, thereby showing the co-localization of HSPB8 with LC3 in the spinal cord of diabetic rats. Scale bar = 10 μm. (C) Co-immunoprecipitation experiments by anti-HSPB8 antibody on the spinal cord tissues of diabetic rats. The test showed that HSPB8 binds to BAG3 protein. (D) Representative Western blot and summarized data presenting the protein levels of HSPB8, BAG3, LC3, and Beclin-1 in the RGC5 cells treated with different high glucose concentrations (final concentrations of 18, 25, and 33 mmol/L in medium). The grayscale values (n = 3) are expressed as mean ± S.D. * p< 0.05 vs control, ** p < 0.01 vs control.
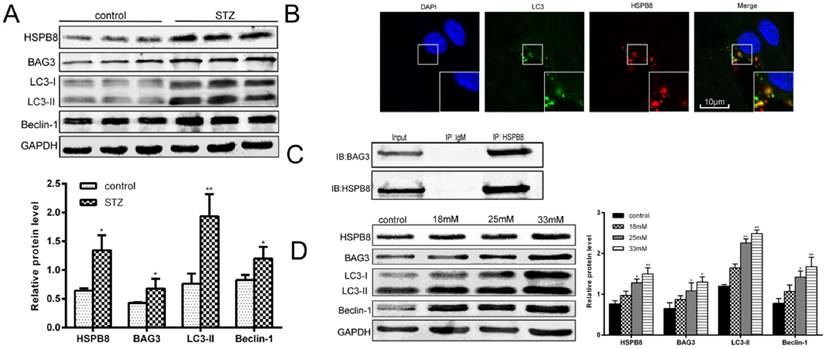
Overall, our data revealed a significant correlation between HSPB8 and Atgs LC3 and Beclin-1.
RGC5 cell autophagy induced by high glucose levels was impaired by HSPB8
To confirm whether HSPB8 is involved in the autophagic process, we performed gain-of-function and loss-of-function experiments on the overexpressed HSPB8 and siRNA vectors. We observed that HSPB8 overexpression increased, whereas the siRNA silencing of HSPB8 decreased the Beclin-1 expression levels (Fig. 2A). Notably, BAG3 exhibited the same results as those of Beclin-1. Thus, HSPB8 can promote the autophagy induced by high-glucose stimulation, whereas HSPB8 silencing can at least partly inhibit the autophagic process.
Gain-and-loss function of HSPB8 in RGC5 cells treated with high glucose concentrations (33 mmol/L). HSPB8 was overexpressed using pcDNA3.1-HSPB8, whereas HSPB8 was silenced by siRNA. (A) Representative Western blot and summarized data (n = 3). * p< 0.05 vs vector, ** p < 0.01 vs vector. (B) Representative electronic microscope graphs showing the autophagosomes in RGC5 cells. Scale bars = 1 µm.
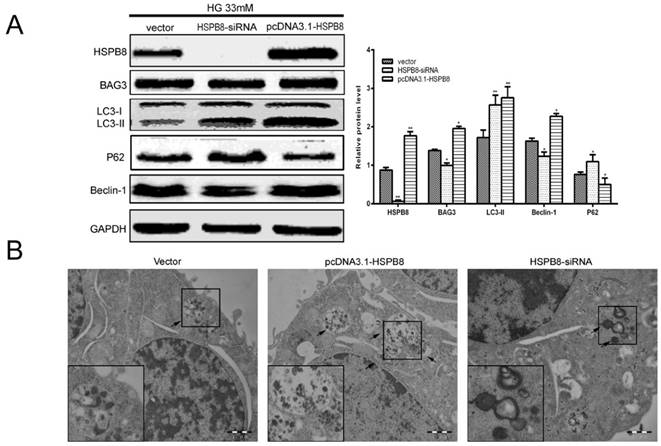
Electron micrograph images showed that typical autolysosomes were present in the cells overexpressing HSPB8 (Fig. 2B). Meanwhile, enlarged lysosomes (indicated by arrows) were observed in the siRNA-HSPB8 cells, suggesting the fusion between the autophagosomes and lysosomes was inhibited. Overall, HSPB8 was a key regulator for the autophagosome-lysosome fusion. The fusion was retarded or blocked without HSPB8.
The most interesting finding in the present study were the expression levels of the LC3 and p62 as markers of the autophagy flux (Fig. 2A). As we know, increased LC3-II level and decreased P62 level indicates that the autophagy flux is smooth and autophagy is activated. In the present study, HSPB8 silencing inhibited the LC3II expression and promoted the p62 expression, indicating autophagy flux was inhibited by silencing HSPB8. Conversely, HSPB8 overexpression increased the LC3II expression and decreased the p62 expression, indicating that the autophagy flux proceeded smoothly.
HSPB8 participated in the autophagosome-lysosome fusion in the RGC5 cells
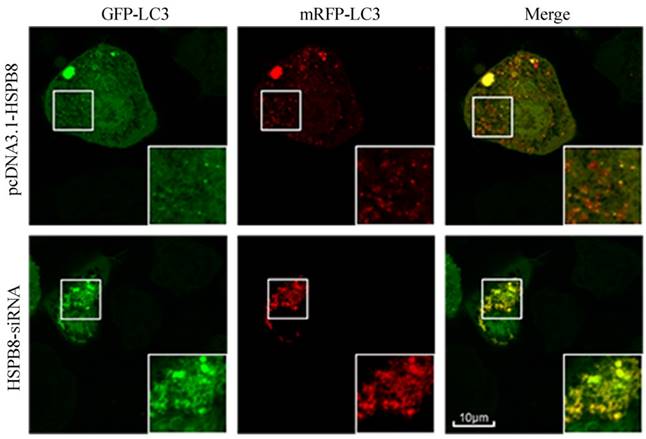
To verify whether HSPB8 is critical during autophagosome-lysosome fusion, we designed and applied a GFP-mRFP-LC3 probe. When the autophagosome-lysosome fusion occurs, the probe comes red. When the fusion is impaired or blocked, the probe comes less reddish and more yellowish [20].
HSPB8 overexpression contributed to prevalent red dots in the RGC5 cells with; while HSPB8 silencing resulted in reduced red dots (Fig. 3). Therefore, HSPB8 silencing exhibited blocking effects on the autophagosome-lysosome fusion and impaired the autolysosome formation.
Tandem GFP-mRFP-LC3 probe analysis of the autophagosome-lysosome fusion. Yellow dots represent the unfused autophagosome, whereas red dots denote the fusion, wherein autolysosomes are formed. The RGC5 cells had been transfected with GFP-RFP-LC3 probes for 32 h. In the HSPB8 siRNA treated cells, an elevated number of yellow dots were observed.
HSPB8 affected the autophagy flux in the chloroquine-treated RGC5 cells
Chloroquine is a compound acting as an autophagosome-lysosome fusion and autophagy flux inhibitor. We designed an experiment to determine whether HSPB8 exerts some effects on chloroquine inhibition. p62 expression increased by chloroquine treatment, indicating autophagy flux was blocked (Fig. 4A). When HSPB8 was overexpressed, the p62 levels decreased considerably after chloroquine treatment. The p62 proteins then generated an evident band with HSPB8 silencing. This band increased to a greater extent than the chloroquine band. Therefore, without HSPB8, the inhibitory effect of chloroquine on the autophagy flux grew considerably. However, when the HSPB8 was overexpressed, the autophagy-flux-blocking effect of chloroquine was partly alleviated.
Effects of HSPB8 on autophagy flux. (A) Rescue of chloroquine effects by HSPB8. The high glucose-(33 mM)-induced autophagy flux was blocked by chloroquine(50µM) in RGC cell. P62 was used as marker of the autophagy flux and examined by western blot. (B) Schematic illustration of HSPB8 function during autophagy. * p< 0.05 vs vector, ** p < 0.01 vs vector, # p< 0.05 vs vector + CQ, ## p < 0.01 vs vector + CQ.
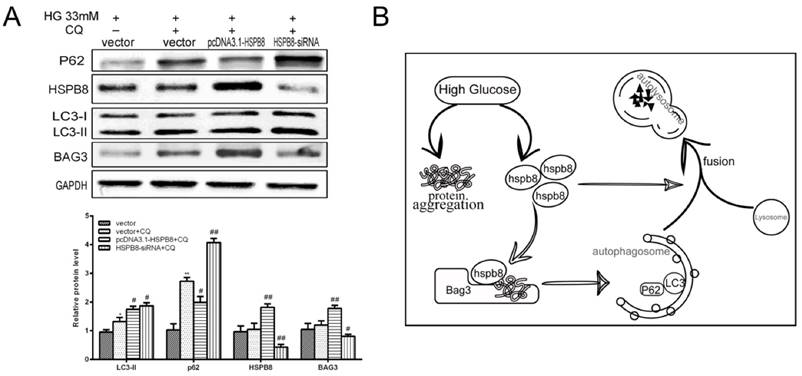
Therefore, HSPB8 overexpression lessened the chloroquine-induced blocking of autophagy flux, while HSPB8 silencing achieved the opposite effect. HSPB8 plays an important role in autophagy flux.
Discussion
In this current study, autophagy occurred intensively in the spinal cord of STZ-induced diabetic rats. The in vitro studies on the RGC5 cells supported the hypothesis that autophagy responds to high glucose. Persistently high glucose caused imbalances on the cell antioxidant capacity and thus promoted oxidative stress-mediated injury and the accumulation of dysfunctional organelles or aggregated proteins within the cells [21]. Hyperglycemia and high-glucose stimulus may activate autophagy through protein aggregation. Studies in Zucker diabetic fatty rats demonstrated that high blood glucose levels stimulate protein aggregation in the brain. The brains extracted from these rats displayed considerably increased levels of protein aggregation, fibronectin expression, protein glycosylation, and mTOR and S6 ribosomal protein phosphorylation [22].
The autophagy mediated by the HSPB8-BAG3 complex can recognize and bind misfolded proteins. As such, the complex can prevent protein aggregation and distinguish the substrates of renaturation from those of proteasomal degradation. HSPB8 is responsible for recognizing the misfolded proteins, whereas BAG3 recruits and activates the macro-autophagy machinery near the chaperone-loaded substrates [23]. Our data demonstrated that the high glucose-induced upregulation of HSPB8 and BAG3 expression was dose-dependent. Along with the autophagy marker LC3 and Beclin-1, the HSPB8-BAG3 complex levels increased in vitro and in vivo. These data suggest that autophagy occurred in the diabetic spinal cord and was mainly caused by the misfolded protein aggregates. The HSPB8-BAG3-mediated autophagy possibly acted as a protective mechanism in response to the high glucose levels. This result was consistent with those of other studies, where autophagy protected against the stress caused by protein aggregation in other nerve-related diseases, such as amyotrophic lateral sclerosis and Parkinson's disease [24].
Autophagy is a dynamic process that involves three steps: autophagosome formation, autophagosome-lysosome fusion, and degradation. Some studies have established the role of HSPB8 as co-chaperon during autophagy flux, where HSPB8 binds with protein aggregates and promotes pre-autophagosome formation [25]. The results of our gain-or-loss-of-function experiments supported this hypothesis. When HSPB8 was overexpressed, high-glucose-induced autophagy intensified, the LC3 expression levels increased, whereas p62 expression levels decreased. However, when HSPB8 expression was decreased by the siRNA, the autophagy flux was impaired, and p62 levels increased evidently, suggesting blocked autophagosome-lysosome fusion. The increased p62 level could not be cleared immediately by the lysosome. Meanwhile, the confocal images of LC3 showed numerous red dots in the HSPB8-overexpressed cells. Notably, HSPB8 overexpression partly rescued the chloroquine's blocking effect on autophagy. Overall, HSPB8 is necessary during the autophagosome-lysosome fusion step in the autophagy flux.
In conclusion, hyperglycemia can augment autophagy levels and upregulate HSPB8 and BAG3 expression in vivo and in vitro. Moreover, HSPB8 plays a critical role in autophagosome-lysosome fusion. Autophagy is particularly important in protecting against diabetic complications including diabetic neuropathy [21], We hypothesize that increasing autophagy in neuronal cells may help these cells ameliorate the bioenergetic crisis, necrosis, and apoptosis associated with diabetic neuropathy. In our study, HSPB8 enhanced the autophagy flux, which indicates that HSPB8 may have protective effects. This study provided a potential target for therapeutic intervention in diabetic neuropathy, or at the least a mean to impede the progress of diabetic neuropathy.
Acknowledgements
This study was supported by grants from the National Natural Science Foundation of China (81360130, 31460300, 31260246), the Ningxia 13th Plan of five- year Major Scientific Program (grant no. 2016BZ 07), Natural Science Foundation of Shaanxi Provincial Department of Education (2013JK0781) and the Ningxia Natural Science Foundation of China (NZ13135).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Park YE, Hayashi YK, Bonne G. et al. Autophagic degradation of nuclear components in mammalian cells. Autophagy. 2009;5:795-804
2. Jiang P, Mizushima N. Autophagy and human diseases. Cell Res. 2014;24:69-79
3. Liang XH, Jackson S, Seaman M. et al. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature. 1999;402:672-6
4. Wirawan E, Lippens S, Vanden Berghe T. et al. Beclin1: a role in membrane dynamics and beyond. Autophagy. 2012;8:6-17
5. Kroemer G, Levine B. Autophagic cell death: the story of a misnomer. Nat Rev Mol Cell Biol. 2008;9:1004-10
6. Inomata M, Niida S, Shibata K. et al. Regulation of Toll-like receptor signaling by NDP52-mediated selective autophagy is normallyinactivated by A20. Cell Mol Life Sci. 2012;69:963-79
7. Bjørkøy G, Lamark T, Pankiv S. et al. Monitoring autophagic degradation of p62/SQSTM1. Methods Enzymol. 2009;452:181-97
8. Rusten TE, Stenmark H. p62, an autophagy hero or culprit? Nat Cell Biol. 2010;12:207-9
9. Gonzalez CD, Lee MS, Marchetti P. et al. The emerging role of autophagy in the pathophysiology of diabetes mellitus. Autophagy. 2011;7:2-11
10. Hoffman WH, Shacka JJ, Andjelkovic AV. Autophagy in the brains of young patients with poorly controlled T1DM and fatal diabeticketoacidosis. Exp Mol Pathol. 2012;93:273-80
11. Ozacmak VH, Barut F, Ozacmak HS. Melatonin provides neuroprotection by reducing oxidative stress and HSP70 expression duringchronic cerebral hypoperfusion in ovariectomized rats. J Pineal Res. 2009;47:156-63
12. Bidmon HJ, Emde B, Oermann E. et al. Heme oxygenase-1 (HSP-32) and heme oxygenase-2 induction in neurons and glial cells ofcerebral regions and its relation to iron accumulation after focal cortical photothrombosis. Exp Neurol. 2001;168:1-22
13. Mymrikov EV, Seit-Nebi AS, Gusev NB. Large potentials of small heat shock proteins. Physiol Rev. 2011;91:1123-59
14. Gurusamy N, Lekli I, Gherghiceanu M. et al. BAG-1 induces autophagy for cardiac cell survival. Autophagy. 2009;5:120-1
15. Ma Y, Gai Y, Yan J. et al. Puerarin Attenuates Anoxia/Reoxygenation Injury Through Enhancing Bcl-2 Associated Athanogene 3 Expression, a Modulator of Apoptosis and Autophagy. Med Sci Monit. 2016;22:977-83
16. Crippa V, Carra S, Rusmini P. et al. A role of small heat shock protein B8 (HspB8) in the autophagic removal of misfolded proteinsresponsible for neurodegenerative diseases. Autophagy. 2010;6:958-60
17. Kwok AS, Phadwal K, Turner BJ. et al. HspB8 mutation causing hereditary distal motor neuropathy impairs lysosomal delivery ofautophagosomes. J Neurochem. 2011;119:1155-61
18. Tang BS, Zhao GH, Luo W. et al. Small heat-shock protein 22 mutated in autosomal dominant Charcot-Marie-Tooth disease type2L. Hum Genet. 2005;116:222-4
19. Lima Leite A, Gualiume Vaz Madureira Lobo J. et al. Proteomic analysis of gastrocnemius muscle in rats with streptozotocin-induced diabetes andchronically exposed to fluoride. PLoS One. 2014;9:e106646
20. Liu F, Wang XY, Zhou XP. et al. Cadmium disrupts autophagic flux by inhibiting cytosolic Ca2+-dependent autophagosome-lysosome fusion in primary rat proximal tubular cells. Toxicology. 2017;383:13-23
21. Yerra VG, Gundu C, Bachewal P. et al. Autophagy: The missing link in diabetic neuropathy? Med Hypotheses. 2016;86:120-8
22. Talaei F, Van Praag VM, Shishavan MH. et al. Increased protein aggregation in Zucker diabetic fatty rat brain: identification of key mechanistictargets and the therapeutic application of hydrogen sulfide. BMC Cell Biol. 2014;15:1
23. Carra S, Seguin SJ, Landry J. HspB8 and Bag3: a new chaperone complex targeting misfolded proteins to macroautophagy. Autophagy. 2008;4:237-9
24. Kiriyama Y, Nochi H. The Function of Autophagy in Neurodegenerative Diseases. Int J Mol Sci. 2015;16:26797-812
25. Carra S, Sivilotti M, Chávez Zobel AT. et al. HspB8, a small heat shock protein mutated in human neuromuscular disorders, has in vivochaperone activity in cultured cells. Hum Mol Genet. 2005;14:1659-69
Author contact
![]() Corresponding author: Lan-Jie He, Department of Endocrinology, General Hospital of Ningxia Medical University, No 692, Shengli Street, Xingqing District, 710000, Yinchuan, Ningxia province, China E-mail: helanjiecom.cn
Corresponding author: Lan-Jie He, Department of Endocrinology, General Hospital of Ningxia Medical University, No 692, Shengli Street, Xingqing District, 710000, Yinchuan, Ningxia province, China E-mail: helanjiecom.cn