Impact Factor ISSN: 1449-1907

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2017; 14(5):452-461. doi:10.7150/ijms.18329 This issue Cite
Research Paper
Stabilization of 4E-BP1 by PI3K kinase and its involvement in CHK2 phosphorylation in the cellular response to radiation
Zi-Jian Yu1, Hui-Hui Luo2, 3, Zeng-Fu Shang4, Hua Guan3, Bei-Bei Xiao4, Xiao-Dan Liu3, Yu Wang3, Bo Huang2 ![]() , Ping-Kun Zhou2, 3
, Ping-Kun Zhou2, 3 ![]()
1. Department of Hepatobiliary Surgery, the First Affiliated Hospital, University of South China, Hengyang, Hunan Province 421001, P.R. China;
2. Institute for Environmental Medicine and Radiation Health, the College of Public Health, University of South China, Hengyang, Hunan Province 421001, P.R. China;
3. Department of Radiation Toxicology and Oncology, Beijing Key Laboratory for Radiobiology (BKLRB), Beijing Institute of Radiation Medicine, 100850 Beijing, P.R. China;
4. School of Radiation Medicine and Protection, Medical College of Soochow University, Collaborative Innovation Center of Radiation Medicine of Jiangsu Higher Education Institutions, Suzhou, Jiangsu Province 215123, P.R. China.
Received 2016-11-13; Accepted 2017-3-1; Published 2017-4-9
Abstract
Objectives: 4E-BP1 is a family member of eIF4E binding proteins (4E-BPs) which act as the suppressors of cap-dependent translation of RNA via competitively associating with cap-bound eIF4E. RNA translation regulation is an important manner to control the cellular responses to a series of stress conditions such as ionizing radiation (IR)-induced DNA damage response and cell cycle controlling. This study aimed to determine the mechanism of 4E-BP1 stabilization and its potential downstream target(s) in the response to IR.
Methods: PI3Ks kinase inhibitors were used to determine the signaling control of 4E-BP1 phosphorylation and protein stability. shRNA strategy was employed to silence the expression of 4E-BP1 in HeLa and HepG2 cells, and determine its effect on the irradiation-induced CHK2 phosphorylation. The protein degradation/stability was investigated by western blotting on the condition of blocking novel protein synthesis by cycloheximide (CHX).
Results: The phosphorylation of 4E-BP1 at Thr37/46 was significantly increased in both HepG2 and HeLa cells by ionizing radiation. Depression of 4E-BP1 by shRNA strategy resulted in an incomplete G2 arrest at the early stage of 2 hours post-irradiation, as well as a higher accumulation of mitotic cells at 10 and 12 hours post-irradiation as compared to the control cells. Consistently, the CHK2 phosphorylation at Thr68 induced by IR was also attenuated by silencing 4E-BP1 expression. Both PI3K and DNA-PKcs kinase inhibitors significantly decreased the protein level of 4E-BP1, which was associated with the accelerated degradation mediated by ubiquitination-proteasome pathway.
Conclusion: PI3K kinase activity is necessary for maintaining 4E-BP1 stability. Our results also suggest 4E-BP1 a novel biological role of regulating cell cycle G2 checkpoint in responding to IR stress in association with controlling CHK2 phosphorylation.
Keywords: 4E-BP1, PI3K kinase
Introduction
The mRNA translation is strictly regulated in various normal physiological processes and stress conditions to maintain cellular homeostasis [1]. Cap-dependent mRNA translation is a master mode for protein synthesis in eukaryotic cells. Typically, the rate of cap-dependent mRNA translation is primarily determined by the formation of eukaryotic translation initiation factor 4F (eIF4F) at the origin step, which recruits a ribosome onto the 7-methyl-guanosine (m7GTP) 5'-cap of an mRNA [2]. The eIF4F complex consists of 3 core members: eIF4G, eIF4A and eIF3, and its assembly on 5' cap of mRNA relies on the binding of scaffold factor-eIF4G with eIF4E [2]. A family of proteins termed as eIF4E binding proteins (4E-BPs) acts as suppressors of cap-dependent translation via competitively associating with cap-bound eIF4E [3]. Among them, 4E-BP1 is the most thoroughly characterized and is generally accepted as one of the principle downstream targets of mTOR signaling [4]. The hyperphosphorylated 4E-BP1 mediating by mTOR loses its affinity with eIF4E and therefore facilitates cap-dependent translation initiation [5]. Due to its pivotal role in mRNA translation inhibition, 4E-BP1 is widely recognized as a growth suppressor in mammalian cells. However, overexpression of 4E-BP1 is identified in various human cancers, and has been associated with poor prognosis in several cases [6-8]. Our and collaborators' recent study indicates that a prostate-specific PC-1/PrLZ protein binds directly with 4E-BP1 and the PC-1/PrLZ-4E-BP1 interaction blocks ubiquitin/proteasome pathway (UPP) mediated 4E-BP1 degradation [9]. 4E-BP1 up-regulation is also associated with increased expression of PC-1/PrLZ in patients' prostate tumor tissues [9]. The precise molecular mechanism of 4E-BP1 protein stability regulation and its important significance in tumor progression is still obscure.
The protein synthesis is tightly controlled throughout the cell cycle progression [10, 11]. Cap-dependent translation initiation has been reported decreased during mitosis in comparison with interphase [11]. Wilker and colleagues revealed that cap-dependent translation is markedly inhibited by tumor suppressor 14-3-3δ when cells enter mitosis [12]. However, accumulating evidences show that a hyperphosphorylated 4E-BP1 exists in mitotic cells and some mitotic specific kinases, such as CDK1 and Plk1 are responsible for 4E-BP1 phosphorylation [13-16]. Chang's group identified a noncanonical CDK1-mediated phosphorylation site of 4E-BP1 at its Ser83. Mutation of this site has no impact on cap-dependent translation but at least partially restore the viral protein-induced cell transformation, suggesting an uncovered translation-independent role of 4E-BP1 in mitosis [14]. In accord with this report, the phosphorylated 4E-BP1 seems to associate with mitotic progression regulation, and loss of 4E-BP1 disrupts mitotic spindle structure seriously and leads to chromosomal DNA misaligned [16].
In addition to normal cell cycle progression, several studies also revealed the selective regulation of protein translation in response to a variety of stress condition, including irradiation (IR) exposure [17]. EIF4E has been identified as a potent target of radiotherapy for tumor cells [18]. Interestingly, Dubois and colleagues also demonstrated that 4E-BP1 is a logical target to improve radiosensitivity of glioblastoma engraft tumors via decreasing hypoxia tolerance of tumors tissues [19]. Braunstein et al. found that IR increases protein synthesis early after IR treatment and subsequently inhibits mRNA translation at late time points post IR exposure. Both of these effects rely on the assembly of MRN (Mre11-Rad50-Nbs1) complex and the function of DNA damage response kinase-ATM. At late time following IR, UPP-mediated degradation of 4E-BP1 is inhibited, which consequently promotes the sequestration of eIF4E and blocks cap-dependent protein synthesis [20]. The cell cycle checkpoint is an essential response induced by IR and provides adequate time to repair the damaged DNA before cells entering mitosis [21]. Based on the reports that 4E-BP1 participates in both cell cycle and DNA damage response regulation, we ask the question whether 4E-BP1 will involve in IR-induced cell cycle checkpoint regulation. Our present study shows that increased 4E-BP1 phosphorylation presents in IR-induced G2 phase arrest. Loss of 4E-BP1 decreases the IR-induced phosphorylation of check-point protein Chk2 and thereby attenuates G2 checkpoint maintenance. Interestingly, our work suggests that PI3K and DNA-PKcs kinase activation could prolong the stability of 4E-BP1 through blocking its ubiquitination-mediated degradation.
Materials and Methods
Cells cultures and Irradiation
HeLa cells and HepG2 cells was maintained in DMEM (HyClone) supplemented with 10% fetal bovine serum (HyClone), 100 unites per ml of penicillin and 100 μg/ml of streptomycin in a humidified incubator at 37º C with 5% CO2. A 60Co γ-rays source was used to irradiate the cells at the dose rate of 110.4 cGy/min at room temperature. After irradiation of 4 Gy, the cells were harvested either immediately or at an indicated time of post-irradiation culture, and then subjected to further cell cycle analysis and western blotting detection of proteins expression.
Antibodies and Chemicals
All the antibodies used in present study are commercially available: 4E-BP1 (53H11) polyclone antibody (#9644), phosphor-4E-BP1 (Thr37/46) antibody (#9459), Chk2 antibody and phosphor-Chk2(Thr68) antibody were purchased from Cell Signaling Technology (Danvers, MA, USA). Phosphor-H3 (Ser10p) was purchased from Bethyl Laboratories Inc (Montgomery, TX, USA). Ub (P4D1) antibody, HA antibody and β-actin antibody (C4) were purchased from Santa Cruz Biotechnology Inc (CA, USA). IgG (HRP labelled) second antibody was purchased from Beijing Zhongshan Biotechnology CO. LTD (Beijing, China). DNA-PKcs activity inhibitor NU7026 and cycloheximide (CHX) were purchased from Sigma (St. Louis, MO, USA). PI3K kinase inhibitor LY294002 and rapamycin were purchased form Cell Signaling Technology (Danvers, MA, USA).
Construction of vector expressing 4E-BP1 targeting shRNA and plasmids transfection
The pSico vector, which was obtained from Dr. Andrea Ventura in Jacks laboratory, MIT Center for Cancer Research, Cambridge, MA, USA, was used to construct 4E-BP1 specific shRNA expressing vector pSicoR-shR-4E-BP1. The 4E-BP1 shRNA targeting sequence is 5'- gtttgagatggacatttaa -3'. 4E-BP1 specific shRNA coding oligos was. According to the methodology of shRNA design, following 4E-BP1 shRNA coding oligos were synthesized, annealed and cloned into the position between HpaI and XhoI restriction endonuclease sites of pSicoR vectors. 4E-BP1 specific shRNA coding oligos: sense-strand as 5'-TGTTTGAGATGGACATTTAATTCAAGAGATTAAATGTCCATCTCAAACTTTTTTC-3'; antisense-strand as 3'-ACAAACTCTACCTGTAAATTAAGTTCTCTAATTTACAGGTAGAGTTTGAAAAAAGAGCT-5'. HepG2 cells were transfected with 4E-BP1 shRNA expressing vectors pSicoR-shR-4E-BP1 or control shRNA vectors using lipofectamine 2000 (Invitrogen Corp., Carlsbad, CA, USA) according to the manufacturer's instructions. The transfected cells were harvested at given times for further experiments.
Cell growth analysis
1×104 cells per well were seeded in 24-well culture plates. The cell numbers from three wells were counted every day after plating for each group. Three independent experiments were performed, and the means were used to depict the growth curve.
Cell cycle and G2 arrest analyses
After 4 Gy γ-rays irradiation (IR), the cells were harvested either immediately or at the indicated times post-irradiation, and fixed with 75% ethanol. The cells were resuspended in PBS plus 0.1% saponin and 1μg/ml RNase A (Sigma), incubated for 20 min at 37°C, and stained with 25μg/ml propidium iodide (PI) (Sigma). The cell cycle distribution was evaluated by flow cytometry, counting more than 10,000 cells per sample. To analyze the radiation-induced G2 arrest, the mitotic cells were counted by flow cytometry detection of the marker protein phosphorylated histone H3 (Ser10) positive cells based on the immunofluorescence staining. The fixed cells were treated with 0.25% Triton X-100 in PBS for 15 min, stained with 10 μg/ml of FITC-conjugated phospho-histone H3 (Ser10) antibody for 1 h at room temperature in the dark, and then stained with propidium iodide (20 μg/ml). Cellular fluorescence was measured using a FACSCalibur flow cytometer (BD Pharmingen, USA). Two‐dimensional dot plots were generated using ModFit LT software.
Western blotting analysis
The cells were harvested and washed twice in ice-cold phosphate buffered saline. Cell pellets were treated with lysis buffer (50 mmol/L Tris-HCL, pH 7.5, 1% Noridet P40, 0.5% Sodium deoxycholate, 150 mmol/L NaCl, 1 piece of protease inhibitor cocktail tablet in 50 ml solution), and the total protein was isolated. Protein (50 μg) was resolved on SDS/PAGE (8%), and then transferred onto the polyvinylidene fluoride (PVDF) membrane for western blotting detection.
Determination of 4E-BP1 protein stability and ubiquitination
The stock solution of PI3K kinase inhibitor LY294002 and DNA-PKcs activity inhibitor NU7026 (Sigma) was prepared in DMSO. HepG2 cells were co-treated with 10 μM ly294002 or 20 μM NU7026 with 100 μg/ml cycloheximide (CHX). After the treated for a given time, the cells harvested for western blotting analysis of 4E-BP1 protein level. For analysis of 4E-BP1 ubiquitination, HepG2 cells were transfected with HA-tagged 4E-BP1 vectors mediated by lipofectamine 2000 (Invitrogen Corp., Carlsbad, CA, USA). 24 h after the transfections, the cells were treated with 10 μM Ly294002 with or without the co-treatment of MG123 for 4 h. Cells lysates were immunoprecipitated (IP) using the anti-HA Affinity Matrix. The ubiquitination levels and the amount of HA-4E-BP1 protein in the IP product were detected by western blotting using the ubiquitin and HA antibodies.
Results
Ionizing radiation induced phosphorylation of 4E-BP1
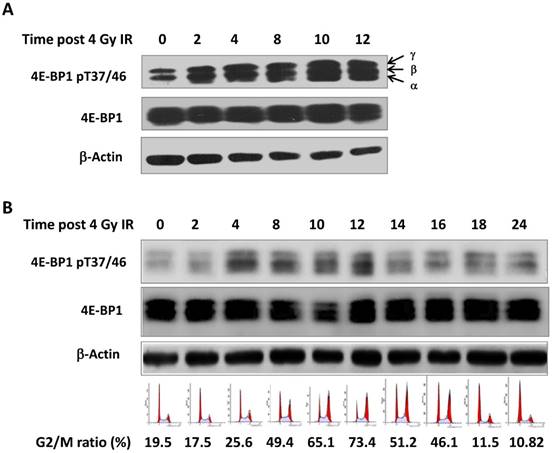
Seven phosphorylation sites (Thr 37, Thr 46, Ser 65 Thr 70, Ser 83, Ser 101, Ser 112) have been identified in 4E-BP1 protein [22]. Among them, the phosphorylation of 4E-BP1 on Thr37 and Thr46 sites is recognized as a priming event to allow the subsequent phosphorylation of other sites [23]. We irradiated HepG2 cells with 4 Gy γ-ray and harvested cells at different times post treatment (0, 2, 4, 8, 12 and 24 hours). As shown in Figure 1A, phosphorylation of 4E-BP1 at its Thr37/46 was significantly increased after irradiation. Interestingly, we also observed a band shift of 4E-BP1 to the upper position of the electrophoretic gels, suggesting phosphorylation on other sites might be also triggered by IR (Figure 1A). The phosphorylation of 4E-BP1 at Thr37/46 was also induced by IR in HeLa cells (Figure 1B). The cell cycle distribution of HeLa cells at indicated times after IR was monitored by flow cytometry (Figure 1B). As shown in Figure 1B, the phosphorylation levels of 4E-BP1 is highly correlated with IR-induced G2/M arrest.
Phosphorylation of 4E-BP1 protein induced by γ-ray irradiation. A, Increased level of phosphorylated 4E-BP1/T37/T46 in HepG2 cells after 4 Gy of γ-ray irradiation; B, Increased level of phosphorylated 4E-BP1/T37/T46 in HeLa cells after 4 Gy of γ-ray irradiation and its association with accumulation of G2/M population.
Loss of 4E-BP1 disrupted IR-induced G2 arrest and impairs G2 checkpoint related CHK2 activation
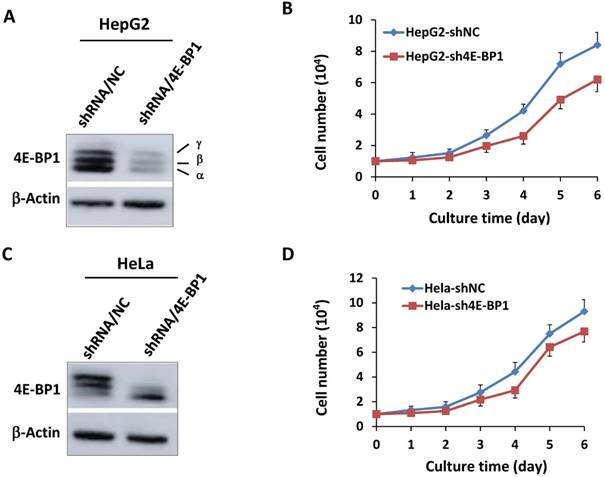
To further determine the role 4E-BP1 in IR-induced G2 checkpoint, we constructed stable 4E-BP1-depleted HeLa and HepG2 cells using specific 4E-BP1 shRNA strategy. The shRNA can efficiently inhibit expression of 4E-BP1 in both HepG2 cells (Figure 2A) and HeLa cells (Figure 2C). Consistent with its function in mitosis progression, Silencing of 4E-BP1 expression markedly suppressed the tumor cells growth (Figure 2B and D).
Depression of 4E-BP1 by specific shRNA and its effect on cell proliferation. A, Decreased level of 4E-BP1 protein in HepG2 cells transfected with 4E-BP1 specific shRNA vectors (shRNA/4E-BP1) as compared with the cells transfected with the control vector (shRNA/NC); B, Decreased proliferation activity of HepG2-sh4E-BP1 cells transfected with 4E-BP1 specific shRNA vectors as compared with HepG2-shNC cells transfected with the control vector (shRNA/NC); C, Decreased level of 4E-BP1 protein in HeLa cells transfected with 4E-BP1 specific shRNA vectors (shRNA/4E-BP1) as compared with the cells transfected with the control vector (shRNA/NC); D, Decreased proliferation activity of HeLa-sh4E-BP1 cells transfected with 4E-BP1 specific shRNA vectors as compared with HeLa-shNC cells transfected with the control vector.
Due to the association between the increased level of 4E-BP1 phosphorylation and IR-induced G2/M arrest, we speculated that 4E-BP1 might play an important role in DNA damage checkpoint of cell cycle. To verify the potent role of 4E-BP1 on IR-induced G2 arrest and mitotic blockage, 4E-BP1-deficient and control HeLa cells were expose to 4-Gy irradiation and then analyzed the mitotic cells population by flow cytometry after staining with an antibody against phospho-H3/Ser10, which is widely accepted as a molecular marker of mitotic cells. As shown in Figure 3A and B, IR dramatically decreased the percentage of pH3 histone-positive cells (mitotic index) in both 4E-BP1 depletion (HeLa-sh4E-BP1) and control cells from 2-8 hours, indicating a robust G2 arrest in both of these cell lines. However, the frequency of pH3 was relative higher in 4E-BP1-deficient HeLa cells when compared with control cells at 2 and 8 hours following 4 Gy irradiation, suggesting that the control of G2 checkpoint in 4E-BP1-deficient HeLa cells might be not as strictly as which in control cells. The persistent higher ratio of mitotic cells at 10 and 12 hours post-irradiation in 4E-BP1 cells group (as compared with control cells) implied a mitotic delay occurred in 4E-BP1 cells.
The effect of 4E-BP1 depression on G2/M checkpoint in response to γ-ray irradiation. A, Representative flow cytometry histograms of HeLa-sh4E-BP1 cells and the control HeLa-shNC cells. The cells were immunostained with phospho-histone H3 (Ser10) antibody to measure the proportion of mitotic cells. The cells were collected at the indicated times post 4 Gy irradiation and immunostained for flow cytometry analysis. The “control” means the control cells without irradiation; B, Quantitative measurement of pH3 (Ser10)-positive mitotic cells. The data are presented as the mean and standard deviation of three independent experiments. *p < 0.05, # p < 0.01: 4E-BP1 depressed HeLa-sh4e-BP1 cells compared with control HeLa-shNC cells at the same time point; C, Effect of 4E-BP1 depression on the cell cycle progression of 4 Gy irradiated HeLa cells. The dynamic distributions of each phase populations were measured by flow cytometry; D. The effect of 4E-BP1 depression on Chk2 phosphorylation in response to γ-ray irradiation.
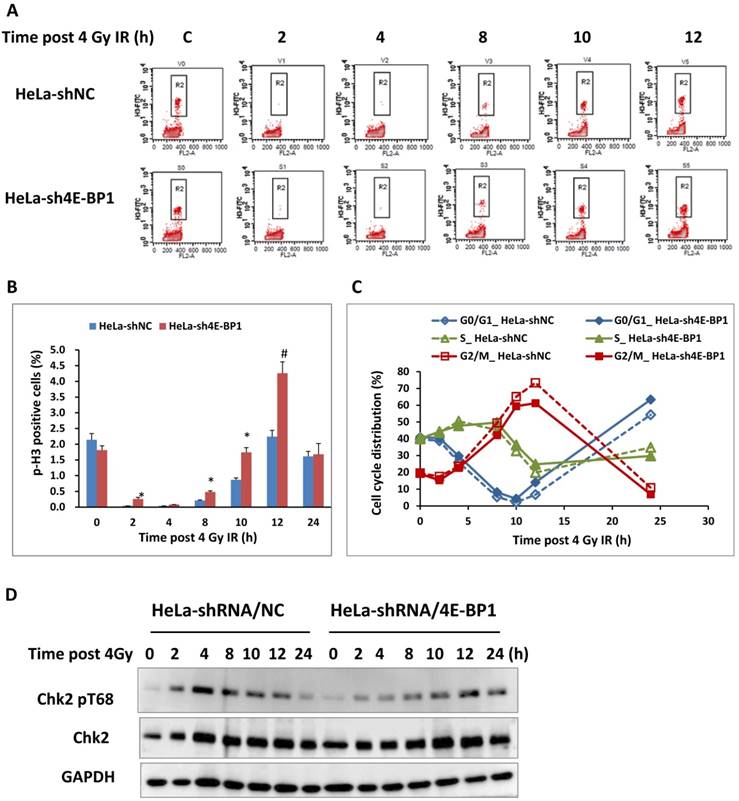
The G2 checkpoint is in the late G2 phase and provides adequate time to repair the damage DNA [21]. The activation of checkpoint kinase 2 (CHK2) plays the dominant role in DNA damage induced G2 checkpoint through phosphorylating a series of downstream targets, including Cdc25C and p53 [24]. Our previous studies also proved that CHK2 was activated during normal mitosis progression. Activation CHK2 relies on its phosphorylation at Thr68 site by ATM and DNA-PKcs, respectively in G2 arrest and mitotic phase [24-28]. As shown in Figure 3D, the phosphorylation of CHK2 at T68 increased significantly 2 hours after irradiation and reach the peak 4 hours following IR in control HeLa cells, when the cells exhibits most tight G2 arrest based on the flow cytometry data. The phosphorylated form of CHK2 reduced 8 hours after IR accompanied by the releasing of cells from G2 arrest. In contrast, loss of 4E-BP1 impaired IR-induced CHK2 phosphorylation at the early stage (2-8 hours) post-irradiation, but a weak enhancement of CHK2 phosphorylation was observed at 12 -24 hours following IR. Thus, it could be attributed to the failure of CHK2 activation that the G2 arrest was not thorough induced at 2 hours post-irradiation. Based on these data, 4E-BP1 might play important role in facilitating the activation of ATM-Chk2 signal or other pathways in response to IR.
4E-BP1 protein stability was associated with PI3K and DNA-PKcs kinase activity
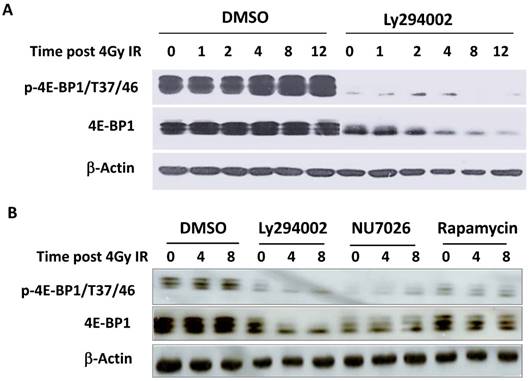
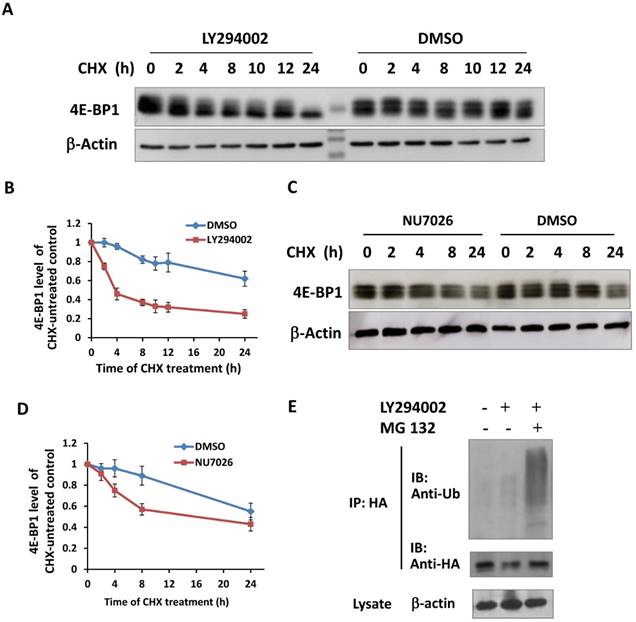
It has been reported that the PI3K-Akt-mTOR signal pathway is mainly responsible for the phosphorylation of 4E-BP1 at Thr37/46 in response to upstream stimuli [29]. To investigate whether PI3K deficiency disrupts IR-induced phosphorylation of 4E-BP1, HepG2 cells were treated with PI3K inhibitor Ly294002 for 2 hours before irradiation and then were harvested at different time post-IR. Interestingly, Ly294002 not only blocked the phosphorylation of 4E-BP1, also markedly reduced 4E-BP1's protein levels (Figure 4A). The PI3K-Akt is the upstream pathway of mTOR and also has been shown to confer the activity of DNA-PKcs. To determine the potent mechanism in which PI3K maintains 4E-BP1 protein level, HeLa cells were treated with inhibitors of PI3K (Ly294002), mTOR (rapamycin) and DNA-PKcs (NU7026). As shown in Figure 4B, both Ly294002 and NU7026 inhibited 4E-BP1 proteins expression level in HeLa cells. However, mTOR inhibitor rapamycin suppressed phosphorylation of 4E-BP1, but did not affect the protein level as strict as by Ly294002 and NU7026. We then exposed HeLa cells to protein synthesis inhibitor cycloheximide (CHX) for different times to investigate 4E-BP1 stability. The combined treatment of CHX with eitherLy294002 (Figure 5A & 5B) or NU7026 (Figure 5C & 5D) dramatically reduced 4E-BP1's protein stability. Our and other's studies demonstrated that 4E-BP1 can be degraded through ubiquitin-proteasome pathway. Therefore, HA-tagged 4E-BP1 vectors were transfected into HepG2 cells, the cells were treated with Ly294002 at 24 hours after the transfection. Cells were harvested and cells lysates were immunoprecipitated (IP) using the anti-HA Affinity Matrix. As estimated, PI3K inhibitor increased 4E-BP1 ubiquitination (Figure 5E). When the protein degradation was inhibited by MG132, a much more significantly increased level of the ubiquitinated 4E-BP1 was detected.
Decrease of 4E-BP1 protein level by PI3K kinase inhibitors. A, PI3K kinase inhibitor Ly294002 resulted in a sharp decrease of 4E-BP1 protein in HepG2 cells. Cells were treated with 10 μM ly294002 for 2h, then irradiated with 4 Gy and harvested 0, 1, 2, 4, 8, 12 h later for the western blotting analysis of proteins expression; B, The effects of PI3K kinase inhibitor Ly294002, DNA-PKcs inhibitor NU7026 and mTOR inhibitor rapamycin on the expression of 4E-BP1 protein in HeLa cells. Cells were treated with 10 μM ly294002, or 10 μM NU7026, or 100 nM rapamycin for 2h, then irradiated with 4 Gy and harvested 0, 4, 8 h later for western blotting analysis.
Inactivation of PI3K destabilized 4E-BP1 protein. A, Ly294002 treatment resulted in a rapid degradation of 4E-BP1 protein in HepG2 cells. Cells were co-treated with 10 μM ly294002 and cycloheximide (CHX), and harvested at 0, 2, 4, 8, 10, 12 and 24 h after the treatments for western blotting analysis; B, Quantification of 4E-BP1 protein level alterations induced by ly294002 based on densitometric scanning of the western blotting signals of the 4E-BP1 protein; C, NU7026 treatment resulted in a rapid degradation of 4E-BP1 protein in HepG2 cells. Cells were co-treated with 20 μM NU7026 and cycloheximide (CHX), and harvested at 0, 2, 4, 8 and 24 h after the treatments for western blotting analysis; D, Quantification of 4E-BP1 protein level alterations induced by NU7026 based on densitometric scanning of the western blotting signals of the 4E-BP1 protein; E, Ly294002 treatment likely increased the ubiquitination of 4E-BP1. HA tagged 4E-BP1 vectors were transfected and expressed in HepG2 cells, 24 h after the transfections, the cells were treated with 10 μM Ly294002 with or without the co-treatment of MG123 for 4 h. Cells lysates were immunoprecipitated (IP) using the anti-HA Affinity Matrix. The ubiquitination levels and the amount of HA-4E-BP1 protein in the IP product were detected by western blotting using the ubiquitin and HA antibodies, respectively.
Discussion
The hyperphosphorylated form of 4E-BP1 is correlated with the accumulation of mitotic cells and has been reported to be mediated by the cardinal mitotic kinase Cdk1 [13-15]. Moore's group revealed an association between the phosphorylation of 4E-BP1 at its T37/46 sites and mitotic marker (pH3-Ser10) in single cell level using flow cytometry [15]. Mitotic-specific hyperphosphorylation of 4E-BP1 was abolished by Cdk1 inhibitor RO-3306, but was resistant to mTOR inhibitor, suggesting that Cdk1 might substitute for the translational regulation function of mTOR during mitosis progression [15]. Our previous study demonstrated that another important mitotic kinase Plk1 also directly phosphorylates purified 4E-BP1 protein in vitro. 4E-BP1interacted with Plk1, and phosphorylated 4E-BP1 colocalized with Plk1 at the mitotic spindle apparatus [16]. Plk1 plays central role in facilitating multiple mitotic processes from G2-M transition to final cytokinesis[30]. Consistent with mitotic function of Plk1, loss of 4E-BP1 also disrupted mitotic progression and led to aberrant chromosome segregation [16]. In the present study, we found that 4E-BP1 exhibits hyperphosphorylation in IR-induced G2 phase arrest. mTOR inhibitor rapamycin significantly impaired IR-induced hyperphosphorylation of 4E-BP1, indicating mTOR might be one of the upstream kinases of 4E-BP1 during G2 checkpoint (Figure 4). The canonical Ser2481 site phosphorylated mTOR has been demonstrated to localize at mitotic spindle during prometaphase and midbody at the cytokinesis stage [31]. However, the exact role of mTOR in G2/M checkpoint warrants further investigation.
The increased phosphorylation of 4E-BP1 in IR-induced G2 arrest indicated its potential function in DNA damage checkpoint. The cell cycle checkpoint kinase Chk2 is the key regulator in the DNA damage induced G2 checkpoint pathway [24, 25]. In response to irradiation, the PI3K-like kinase ATM participates in activation of checkpoint signal through phosphorylating the Chk2's N-terminal T68 site [24, 25]. This phosphorylation event subsequently promotes formation of homodimer and autophosphorylation of Chk2 [24, 25]. Here, we observed that depletion of 4E-BP1 led to an incomplete G2 arrest at the early time (2 hours) post-IR, suggesting 4E-BP1 involves in IR-induced G2 checkpoint maintenance. Consistent with this phenomenon, loss of 4E-BP1 dramatically impairs phosphorylation of Chk2 T68 in early stage after IR exposure (Figure 3). However, knocking down of 4E-BP1, a weak increase of Chk2 phosphorylation occurred at 12 hours post-IR when 4E-BP1-depleted cells exhibit relative higher levels of mitotic index. Our previous works demonstrated that another DNA damage response kinase DNA-PKcs plays dominant role in phosphorylating Chk2 during mitotic progression and the DNA-PKcs-mediated activated Chk2 in turn phosphorylates other substrates such as Brca1 and γH2AX specifically in mitosis[26, 28]. Based on the observations, it is rational to assume that 4E-BP1 contributes to DNA damage-induced G2 checkpoint via assisting ATM-Chk2 activation but seems to have no effect on mitotic DNA-PKcs-Chk2 signal pathway. In addition to its essential role in regulating multiple processes of cell division, Plk1 is also crucial to restart cell cycle progression after DNA damage induced G2 arrest [32-34]. Furthermore, Plk1 has been revealed to directly interact and phosphorylate Chk2 protein [35]. Mps1 is another important mitotic kinase which helps to maintain a robust DNA damage checkpoint [36]. The kinase-dead form of Mps1 disrupts IR or UV induced G2/M arrest and impaired Chk2 T68 phosphorylation [36]. The Cdk1-mediated hyperphosphorylation of 4E-BP1 during mitosis is considered to regulate the translation of some important mRNAs in the short time-scale mitosis [37]. Actually, the expression of many mitotic regulators have been increased in G2 phase of the cell cycle [38, 39]. It is still unclear whether the hyperphosphorylation of 4E-BP1 in DNA damage-induced G2 arrest also facilitates these important mitotic or checkpoint related protein translation.
Interestingly, our study showed that Ly294002, a PI3K inhibitor, markedly reduced protein level of 4E-BP1. An earlier report demonstrated the ubiquitination-mediated degradation of 4E-BP1 and this process is regulated by phosphorylation [40]. The PI3K-Akt-mTOR signal pathway plays the major role in phosphorylating 4E-BP1 [4]. However, we didn't find that mTOR inhibitor reduces 4E-BP1 expression as the PI3K inhibitor does. Previous report has shown that inhibition of PI3K by Ly294002 modulated tumor cells radiosensitivity through blocking irradiation-induced phosphorylation of DNA-PKcs [41]. Our present study proves that DNA-PKcs inhibitor NU7026 also accelerated protein degradation rate of 4E-BP1. Our recent study found DNA-PKcs interacts with anaphase-promoting complex/cyclosome (APC/C) core component APC2 [42]. The APC/C complex together with its co-activator Cdc20 and Cdh1 form a functional E3 ubiquitin ligase to target cell cycle proteins' destruction by proteasome pathway. Loss of DNA-PKcs has impact on several mitotic protein degradation, including cyclin B1 [42], Plk1 [43] and Cdc20 [42] itself. The regulation effects of DNA-PKcs on APC/C complex rely on the kinase activity of DNA-PKcs. It would be interesting to determine whether 4E-BP1 is ubiquitinated by APC/C complex in which DNA-PKcs is involved. PC-1/PrLZ is a prostate specific expressed oncogenic protein and is reported to prevent ubiquitination of 4E-BP1 through direct binding [9]. Interestingly, PC-1/PrLZ also participates in IR-induced G2/M checkpoint [44].
Accumulating evidence showed that 4E-BP1 is overexpressed in variety of tumors [8]. The prolonged G2 checkpoint is thought to be beneficial to repair the DNA lesion [45]. Therefore, our findings provided new insight of 4E-BP1 in IR-induced cell cycle checkpoint regulation, and further indicated that 4E-BP1 might be a potential radiotherapeutic target.
Conclusion
4E-BP1 is a family member of eIF4E binding proteins (4E-BPs) which act as the suppressors of cap-dependent translation of RNA via competitively associating with cap-bound eIF4E. PI3K kinase activity is necessary for maintaining 4E-BP1 stability. Our results also demonstrated 4E-BP1 a novel biological role of controlling cell cycle G2 checkpoint in responding to IR stress, and which is associated with the regulation of CHK2 phosphorylation.
Acknowledgements
This work was supported by the National Key Basic Research Program (973 Program) of MOST, China (Grant No. 2015CB910601), the Chinese National Natural Science Foundation (Grant number: 81272994 and 81530085). The authors thank Dr. Andrea Ventura (Jacks laboratory, MIT Center for Cancer Research, Cambridge, MA, USA) for providing the pSico vectors.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Reid DW, Nicchitta CV. Diversity and selectivity in mRNA translation on the endoplasmic reticulum. Nature reviews Molecular cell biology. 2015;16:221-31
2. Jackson RJ, Hellen CU, Pestova TV. The mechanism of eukaryotic translation initiation and principles of its regulation. Nature reviews Molecular cell biology. 2010;11:113-27
3. Gingras AC, Raught B, Sonenberg N. eIF4 initiation factors: effectors of mRNA recruitment to ribosomes and regulators of translation. Annual review of biochemistry. 1999;68:913-63
4. Mamane Y, Petroulakis E, LeBacquer O, Sonenberg N. mTOR, translation initiation and cancer. Oncogene. 2006;25:6416-22
5. Hsieh AC, Costa M, Zollo O, Davis C, Feldman ME, Testa JR. et al. Genetic dissection of the oncogenic mTOR pathway reveals druggable addiction to translational control via 4EBP-eIF4E. Cancer cell. 2010;17:249-61
6. Braunstein S, Karpisheva K, Pola C, Goldberg J, Hochman T, Yee H. et al. A hypoxia-controlled cap-dependent to cap-independent translation switch in breast cancer. Molecular cell. 2007;28:501-12
7. Martin ME, Perez MI, Redondo C, Alvarez MI, Salinas M, Fando JL. 4E binding protein 1 expression is inversely correlated to the progression of gastrointestinal cancers. The international journal of biochemistry & cell biology. 2000;32:633-42
8. Armengol G, Rojo F, Castellvi J, Iglesias C, Cuatrecasas M, Pons B. et al. 4E-binding protein 1: a key molecular "funnel factor" in human cancer with clinical implications. Cancer research. 2007;67:7551-5
9. Yu L, Shang ZF, Wang J, Wang H, Huang F, Zhang Z. et al. PC-1/PrLZ confers resistance to rapamycin in prostate cancer cells through increased 4E-BP1 stability. Oncotarget. 2015;6:20356-69
10. Pyronnet S, Pradayrol L, Sonenberg N. A cell cycle-dependent internal ribosome entry site. Molecular cell. 2000;5:607-16
11. Prescott DM, Bender MA. Synthesis of RNA and protein during mitosis in mammalian tissue culture cells. Experimental cell research. 1962;26:260-8
12. Wilker EW, van Vugt MA, Artim SA, Huang PH, Petersen CP, Reinhardt HC. et al. 14-3-3sigma controls mitotic translation to facilitate cytokinesis. Nature. 2007;446:329-32
13. Greenberg VL, Zimmer SG. Paclitaxel induces the phosphorylation of the eukaryotic translation initiation factor 4E-binding protein 1 through a Cdk1-dependent mechanism. Oncogene. 2005;24:4851-60
14. Velasquez C, Cheng E, Shuda M, Lee-Oesterreich PJ, Pogge von Strandmann L, Gritsenko MA. et al. Mitotic protein kinase CDK1 phosphorylation of mRNA translation regulator 4E-BP1 Ser83 may contribute to cell transformation. Proceedings of the National Academy of Sciences of the United States of America. 2016;113:8466-71
15. Shuda M, Velasquez C, Cheng E, Cordek DG, Kwun HJ, Chang Y. et al. CDK1 substitutes for mTOR kinase to activate mitotic cap-dependent protein translation. Proceedings of the National Academy of Sciences of the United States of America. 2015;112:5875-82
16. Shang ZF, Yu L, Li B, Tu WZ, Wang Y, Liu XD. et al. 4E-BP1 participates in maintaining spindle integrity and genomic stability via interacting with PLK1. Cell Cycle. 2012;11:3463-71
17. Holcik M, Sonenberg N. Translational control in stress and apoptosis. Nature reviews Molecular cell biology. 2005;6:318-27
18. Hayman TJ, Williams ES, Jamal M, Shankavaram UT, Camphausen K, Tofilon PJ. Translation initiation factor eIF4E is a target for tumor cell radiosensitization. Cancer research. 2012;72:2362-72
19. Dubois L, Magagnin MG, Cleven AH, Weppler SA, Grenacher B, Landuyt W. et al. Inhibition of 4E-BP1 sensitizes U87 glioblastoma xenograft tumors to irradiation by decreasing hypoxia tolerance. International journal of radiation oncology, biology, physics. 2009;73:1219-27
20. Braunstein S, Badura ML, Xi Q, Formenti SC, Schneider RJ. Regulation of protein synthesis by ionizing radiation. Molecular and cellular biology. 2009;29:5645-56
21. Dasika GK, Lin SC, Zhao S, Sung P, Tomkinson A, Lee EY. DNA damage-induced cell cycle checkpoints and DNA strand break repair in development and tumorigenesis. Oncogene. 1999;18:7883-99
22. Qin X, Jiang B, Zhang Y. 4E-BP1, a multifactor regulated multifunctional protein. Cell Cycle. 2016;15:781-6
23. Gingras AC, Gygi SP, Raught B, Polakiewicz RD, Abraham RT, Hoekstra MF. et al. Regulation of 4E-BP1 phosphorylation: a novel two-step mechanism. Genes & development. 1999;13:1422-37
24. Ahn J, Urist M, Prives C. The Chk2 protein kinase. DNA repair. 2004;3:1039-47
25. Bartek J, Lukas J. Chk1 and Chk2 kinases in checkpoint control and cancer. Cancer cell. 2003;3:421-9
26. Shang Z, Yu L, Lin YF, Matsunaga S, Shen CY, Chen BP. DNA-PKcs activates the Chk2-Brca1 pathway during mitosis to ensure chromosomal stability. Oncogenesis. 2014;3:e85
27. Shang ZF, Huang B, Xu QZ, Zhang SM, Fan R, Liu XD. et al. Inactivation of DNA-dependent protein kinase leads to spindle disruption and mitotic catastrophe with attenuated checkpoint protein 2 Phosphorylation in response to DNA damage. Cancer research. 2010;70:3657-66
28. Tu WZ, Li B, Huang B, Wang Y, Liu XD, Guan H. et al. gammaH2AX foci formation in the absence of DNA damage: mitotic H2AX phosphorylation is mediated by the DNA-PKcs/CHK2 pathway. FEBS letters. 2013;587:3437-43
29. Morita M, Gravel SP, Hulea L, Larsson O, Pollak M, St-Pierre J. et al. mTOR coordinates protein synthesis, mitochondrial activity and proliferation. Cell Cycle. 2015;14:473-80
30. Petronczki M, Lenart P, Peters JM. Polo on the Rise-from Mitotic Entry to Cytokinesis with Plk1. Developmental cell. 2008;14:646-59
31. Vazquez-Martin A, Sauri-Nadal T, Menendez OJ, Oliveras-Ferraros C, Cufi S, Corominas-Faja B. et al. Ser2481-autophosphorylated mTOR colocalizes with chromosomal passenger proteins during mammalian cell cytokinesis. Cell Cycle. 2012;11:4211-21
32. Macurek L, Lindqvist A, Lim D, Lampson MA, Klompmaker R, Freire R. et al. Polo-like kinase-1 is activated by aurora A to promote checkpoint recovery. Nature. 2008;455:119-23
33. Mamely I, van Vugt MA, Smits VA, Semple JI, Lemmens B, Perrakis A. et al. Polo-like kinase-1 controls proteasome-dependent degradation of Claspin during checkpoint recovery. Current biology: CB. 2006;16:1950-5
34. van Vugt MA, Bras A, Medema RH. Polo-like kinase-1 controls recovery from a G2 DNA damage-induced arrest in mammalian cells. Molecular cell. 2004;15:799-811
35. Tsvetkov L, Xu X, Li J, Stern DF. Polo-like kinase 1 and Chk2 interact and co-localize to centrosomes and the midbody. The Journal of biological chemistry. 2003;278:8468-75
36. Wei JH, Chou YF, Ou YH, Yeh YH, Tyan SW, Sun TP. et al. TTK/hMps1 participates in the regulation of DNA damage checkpoint response by phosphorylating CHK2 on threonine 68. The Journal of biological chemistry. 2005;280:7748-57
37. Shuda M, Chang Y, Moore PS. Mitotic 4E-BP1 hyperphosphorylation and cap-dependent translation. Cell Cycle. 2015;14:3005-6
38. Winey M, Huneycutt BJ. Centrosomes and checkpoints: the MPS1 family of kinases. Oncogene. 2002;21:6161-9
39. Deeraksa A, Pan J, Sha Y, Liu XD, Eissa NT, Lin SH. et al. Plk1 is upregulated in androgen-insensitive prostate cancer cells and its inhibition leads to necroptosis. Oncogene. 2013;32:2973-83
40. Elia A, Constantinou C, Clemens MJ. Effects of protein phosphorylation on ubiquitination and stability of the translational inhibitor protein 4E-BP1. Oncogene. 2008;27:811-22
41. Toulany M, Kasten-Pisula U, Brammer I, Wang S, Chen J, Dittmann K. et al. Blockage of epidermal growth factor receptor-phosphatidylinositol 3-kinase-AKT signaling increases radiosensitivity of K-RAS mutated human tumor cells in vitro by affecting DNA repair. Clinical cancer research: an official journal of the American Association for Cancer Research. 2006;12:4119-26
42. Shang ZF, Tan W, Liu XD, Yu L, Li B, Li M. et al. DNA-PKcs Negatively Regulates Cyclin B1 Protein Stability through Facilitating Its Ubiquitination Mediated by Cdh1-APC/C Pathway. International journal of biological sciences. 2015;11:1026-35
43. Huang B, Shang ZF, Li B, Wang Y, Liu XD, Zhang SM. et al. DNA-PKcs associates with PLK1 and is involved in proper chromosome segregation and cytokinesis. Journal of cellular biochemistry. 2014;115:1077-88
44. Shang ZF, Wei Q, Yu L, Huang F, Xiao BB, Wang H. et al. Suppression of PC-1/PrLZ sensitizes prostate cancer cells to ionizing radiation by attenuating DNA damage repair and inducing autophagic cell death. Oncotarget. 2016;7:62340-51
45. Begg AC, Stewart FA, Vens C. Strategies to improve radiotherapy with targeted drugs. Nature reviews Cancer. 2011;11:239-53
Author contact
![]() Corresponding authors: Bo Huang, PhD., Institute for Environmental Medicine and Radiation Health, the College of Public Health, University of South China, Hengyang, Hunan Province 421001, P.R. China. Tel: +86-013187205402, E-mail: huangbo0930com; Ping-Kun Zhou, PhD., Department of Radiation Toxicology and Oncology, Beijing Key Laboratory for Radiobiology (BKLRB), Beijing Institute of Radiation Medicine, 100850 Beijing, P. R. China. Tel: +86-10-66931217; Fax: +86-10-68183899, E-mail: zhoupkac.cn.
Corresponding authors: Bo Huang, PhD., Institute for Environmental Medicine and Radiation Health, the College of Public Health, University of South China, Hengyang, Hunan Province 421001, P.R. China. Tel: +86-013187205402, E-mail: huangbo0930com; Ping-Kun Zhou, PhD., Department of Radiation Toxicology and Oncology, Beijing Key Laboratory for Radiobiology (BKLRB), Beijing Institute of Radiation Medicine, 100850 Beijing, P. R. China. Tel: +86-10-66931217; Fax: +86-10-68183899, E-mail: zhoupkac.cn.