Impact Factor ISSN: 1449-1907

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2017; 14(4):323-332. doi:10.7150/ijms.17834 This issue Cite
Research Paper
TBX20 loss-of-function mutation responsible for familial tetralogy of Fallot or sporadic persistent truncus arteriosus
Ri-Tai Huang1*, Juan Wang2*, Song Xue1, Xing-Biao Qiu3, Hong-Yu Shi3, Ruo-Gu Li3, Xin-Kai Qu3, Xiao-Xiao Yang3, Hua Liu3, Ning Li3, Yan-Jie Li3, Ying-Jia Xu3 ![]() , Yi-Qing Yang3,4,5
, Yi-Qing Yang3,4,5 ![]()
1. Department of Cardiovascular Surgery, Renji Hospital, School of Medicine, Shanghai Jiao Tong University, 1630 Dongfang Road, Shanghai 200127, China;
2. Department of Cardiovascular Medicine, East Hospital, Tongji University School of Medicine, 150 Jimo Road, Shanghai 200120, China;
3. Department of Cardiology, Shanghai Chest Hospital, Shanghai Jiao Tong University, 241 West Huaihai Road, Shanghai 200030, China;
4. Department of Cardiovascular Research Laboratory, Shanghai Chest Hospital, Shanghai Jiao Tong University, 241 West Huaihai Road, Shanghai 200030, China;
5. Department of Central Laboratory, Shanghai Chest Hospital, Shanghai Jiao Tong University, 241 West Huaihai Road, Shanghai 200030, China.
* These two authors contributed equally to this work.
Received 2016-10-6; Accepted 2016-12-20; Published 2017-3-11
Abstract
Congenital heart disease (CHD), the most common form of developmental abnormality in humans, remains a leading cause of morbidity and mortality in neonates. Genetic defects have been recognized as the predominant causes of CHD. Nevertheless, CHD is of substantial genetic heterogeneity and the genetic defects underlying CHD in most cases remain unclear. In the current study, the coding regions and splicing junction sites of the TBX20 gene, which encodes a T-box transcription factor key to cardiovascular morphogenesis, were sequenced in 175 unrelated patients with CHD, and a novel heterozygous TBX20 mutation, p.K274X, was identified in an index patient with tetralogy of Fallot (TOF). Genetic analysis of the proband's available family members showed that his father, elder brother and son had also TOF. In addition, his father and elder brother had also atrial septal defect, and his niece had persistent truncus arteriosus and ventricular septal defect. Analysis of the pedigree revealed that the mutation co-segregated with CHD transmitted in an autosomal dominant fashion, with complete penetrance. The nonsense mutation, which was absent in the 800 control chromosomes, was predicted to produce a truncated protein with only the amino terminus and partial T-box domain left. Functional analyses by using a dual-luciferase reporter assay system showed that the mutant TBX20 lost the ability to transactivate the target gene ANF. Furthermore, the mutation reduced the synergistic activation between TBX20 and NKX2.5 as well as GATA4, two other transcriptional factors previously associated with various CHD, encompassing TOF. This study firstly links TBX20 loss-of-function mutation to familial TOF or sporadic persistent truncus arteriosus, providing novel insight into the molecular pathogenesis of CHD.
Keywords: Congenital heart disease, Tetralogy of Fallot, Genetics, Transcriptional factor, TBX20, Reporter gene assay
Introduction
Congenital heart disease (CHD), which defines a large set of structural defects of the heart and intra-thoracic great vessels that occur during embryogenesis, is the most prevalent form of human birth defect worldwide, affecting approximately 1% of live births, with an estimated prenatal incidence of 10 % [1-3]. According to specific anatomic lesions, CHD is clinically categorized into more than 20 distinct types, including ventricular septal defect, atrial septal defect, patent ductus arteriosus, tetraology of Fallot (TOF) and double outlet right ventricle, of which ventricular septal defect is the most common CHD; while TOF is the most common type of cyanotic CHD [1]. In addition to degraded quality of life and decreased exercise performance, serious CHD may lead to various complications, including retarded brain development or brain injury, thromboembolism, pulmonary hypertension, chronic heart failure, arrhythmias and sudden cardiac demise [4-16]. Although enormous advancement in the diagnosis and treatment of pediatric CHD has been made during the past 50 years, which allows more than 75% of CHD children to survive into adulthood, unfortunately, it has brought about a growing population of adults living with CHD, with the prevalence of CHD in adults being approximately 3000 per million, and this unique CHD population is increasing by almost 5% per year [17,18]. Moreover, these survivors have a significantly increased incidence of late complications and sudden cardiac death [18,19]. Obviously, CHD is still associated with substantial morbidity and mortality, and has imposed a heavy socioeconomic burden on patients' families and societies [18,19]. Therefore, reveal of the molecular mechanisms underpinning CHD is of pronounced clinical importance, especially vital to the healthcare of this growing adult CHD community [17].
Previous studies have demonstrated that CHD are multi-factorial, with both environmental and genetic risk factors involved in the pathogenesis of CHD [17,20,21]. Environmental risk factors for CHD encompass maternal viral infection, drug uses, alcohol consumption, smoking, and exposure to toxic chemicals or ionizing radiation during the first trimester of pregnancy [20]. However, epidemiologic studies convincingly suggest that genetic defects are the predominant causes of CHD [17,21]. To date, mutations in more than 60 genes have been causally linked to CHD [17,21,22-47]. Among these well-established CHD-causing genes, most code for cardiac transcription factors, including homeodomain-containing transcription factors, zinc-finger transcription factors, and T-box transcription factors [17,21,22-48]. Nevertheless, these known genetic causes of CHD can only explain for less than 20% of all CHD [49], and the genetic basis underlying CHD in an overwhelming majority of cases remains unknown.
As a member of the large family of T-box transcription factors, TBX20 have been shown to play a key and dose-sensitive role in cardiovascular genesis. In humans and vertebrate models, TBX20 is amply expressed in the heart throughout cardiac development [50]. In mice, TBX20 is abundantly expressed in the heart progenitor cells, cardiomyocytes of the early heart tube and endothelial cells responsible for endocardial cushions, as well as the precursor structures required for the development of cardiac valves and atrioventricular septum [51]. Targeted disruption of murine Tbx20 led to embryonic death, with unlooped and severely hypoplastic myocardial tubes; while heterozygous Tbx20-null mice suffered from atrial septal anomalies [52-54]. By contrast, Tbx20-knockdown mice exhibited failed septation of the outflow tract, persistent truncus arteriosus and hypoplastic right ventricle [55]. Additionally, in zebrafish and Xenopus, either loss- or gain-of-function of Tbx20 resulted in cardiac development abnormalities [52]. In humans, mutations in TBX20 have been associated with atrial septal defect, ventricular septal defect, TOF and double-outlet right ventricle [30,31,56-60]. These findings make it justifiable to screen TBX20 as a prime candidate gene for CHD in another cohort of patients with CHD.
Materials and methods
Ethics
This study was performed in accordance with the ethical principles of the Declaration of Helsinki. The study protocol was reviewed and approved by the local institutional ethics committee, and written informed consents to the use of the blood samples for genetic analyses were obtained from the study participants or their guardians prior to commencement of this investigation.
Study population
In this study, a cohort of 175 unrelated patients, who fulfilled the criteria of CHD, was recruited from the Han Chinese population. The available family members of the index patient carrying an identified TBX20 mutation were also included. A total of 400 unrelated non-CHD individuals from the same geographic area, who were matched to the CHD cases in age, gender and ethnicity, were registered as control subjects. All the study participants were clinically evaluated in detail, including comprehensive individual and medical histories, detailed physical examination, standard 12-lead electrocardiogram, and transthoracic echocardiography with color flow Doppler. Transesophageal echocardiography, cardiac catheter-based procedure or surgery was performed only when there was a strong clinical indication. Various forms of CHD were diagnosed by cardiac ultrasonic imaging and medical records. The patients who were diagnosed with chromosomal abnormalities or syndromic CHD, such as trisomy 13, trisomy 18, trisomy 21, Turner syndrome, Alagille syndrome, Holt-Oram Syndrome and Noonan syndrome, were excluded from the current study. Clinical research was conducted by professionals without knowledge of genotype.
Mutational analysis of TBX20
Peripheral venous blood samples were drawn from all study subjects. Genomic DNA was isolated from whole blood leukocytes using the Wizard Genomic DNA Purification Kit (Promega, Madison, WI, USA) according to the manufacture's protocol. The primers to amplify the complete exonic sequences as well as small flanking intronic sequences of TBX20 by polymerase chain reaction (PCR) were designed as described previously [30], and was synthesized commercially (Sangon, Shanghai, China). The total volume of a PCR mixture was 25 μL, comprising 11.25 μL of deionized water, 5 μL of 5× Q solution, 2.5 μL of 10× buffer, 2 μL of dNTPs (2.5 mM each), 1 μL of each primer at 20 μM, 0.25 μL of HotStar Taq DNA Polymerase (Qiagen, Hilden, Germany) at 5 U/μL, and 2 μL of genomic DNA at 200 ng/μL. Typical PCR cycling parameters were predenaturation of template and activation of polymerase at 95 ℃ for 15 minutes, followed by 35 cycles of denaturation at 94 ℃ for 30 seconds, annealing at 62 ℃ for 30 seconds and extension at 72 ℃ for 1 minute, with a final extension at 72 ℃ for 10 minutes. Amplification of genomic DNA was performed in 0.2 ml thin-walled PCR tubes on a GeneAmp PCR System 9700 (Applied Biosystems, Foster, CA, USA). The amplified products were fractionated by electrophoresis on 1.5% agarose gels. After purification with the QIAquick Gel Extraction Kit (Qiagen), both strands of each amplicon were PCR-sequenced with the BigDye® Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems) on an ABI PRISM 3130 XL automated DNA analyzer (Applied Biosystems). Resultant sequences were analyzed with a sequence analysis software (Applied Biosystems) and the BLAST program (http://blast.ncbi.nlm.nih.gov/Blast.cgi). Whenever a sequence variant was found, the sample was PCR-sequenced again from both forward and reverse directions to confirm the nucleotide change. For a confirmed mutation, its position was based on the complementary DNA reference sequence for TBX20 (accession no. NM_001077653.2), with the initial codon ATG numbered as residue 1. Additionally, the online databases for human sequence variations, such as the single nucleotide polymorphism (SNP; http://www.ncbi.nlm.nih.gov/SNP) database and the human gene mutation (HGM; http://www.hgmd.org) database, were queried to confirm its novelty.
Alignment of multiple TBX20 protein sequences across species
The amino acid sequences of the TBX20 proteins in human, chimpanzee, monkey, dog, cattle, mouse, fowl, zebrafish and frog were obtained from the GenBank database (http://www.ncbi.nlm.nih.gov/genbank/). By using the Clustal Omega software (http://www.clustal.org/omega/), multiple alignments of amino acid sequences among these species were performed.
Plasmid constructs and site-directed mutagenesis
The recombinant expression vector TBX20-pcDNA3.1, which contains the full-length cDNA of human TBX20, was constructed as previously described [30]. The expression plasmids NKX2.5-pEFSA and GATA4-pSSRa as well as the atrial natriuretic factor-luciferase (ANF-luc) reporter construct, which contains the 2600-bp 5'-flanking region of the ANF gene and encodes the Firefly luciferase, were generously provided by Dr. Ichiro Shiojima, at the Department of Cardiovascular Science and Medicine, Chiba University Graduate School of Medicine, Chiba, Japan. With a complementary pair of primers, the identified mutation was introduced into the wild-type TBX20-pcDNA3.1 construct by PCR-based site-directed mutagenesis using the QuickChange II XL Site-Directed Mutagenesis Kit (Stratagene, La Jolla, CA, USA). The mutant underwent selection by DpnI (TaKaRa, Dalian, China) and was fully sequenced to verify the desired mutation and to exclude any other unwanted sequence variations.
Reporter gene assay of mutant TBX20
Chinese hamster ovary (CHO) cells and COS-7 (a fibroblast-like cell line) cells were cultured in Dulbecco's modified Eagle's medium supplemented with 10% fetal calf serum at 37°C in an incubator supplied with 5% CO2, and maintained in 12-well plates under standard conditions for 48 hours after transfection. One day before transfection, cells were plated at a density of 2×105 cells/well. The PolyFect Transfection Reagent (Qiagen) was used for transfection of cells according to the manufacturer's instructions. The internal control reporter plasmid pGL4.75 (hRluc/CMV, Promega), which expresses Renilla luciferase, was used in transient transfection assays to normalize the trancfection efficiency. Twenty-four hours after plating, CHO cells grown to approximately 90% confluence were transfected with 0.8 μg of wild-type or mutant TBX20-pcDNA3.1 expression vector, 1.0 μg of ANF-luc reporter construct, and 0.04 μg of pGL4.75 control reporter vector. For co-transfection experiments in CHO cells, 0.4 μg of wild-type TBX20-pcDNA3.1 together with 0.4 μg of mutant TBX20-pcDNA3.1 or 0.4 μg of empty pcDNA3.1 vector were used in the presence of 1.0 μg of ANF-luc and 0.04 μg of pGL4.75. For the synergistic activation between TBX20 and NKX2.5 or GATA4 in COS-7 cells, the same amount (0.6 μg) of plasmid (empty pcDNA3.1, wild-type TBX20-pcDNA3.1, NKX2.5-pEFSA, GATA4-pSSRa or K274X-mutant TBX20-pcDNA3.1) was used alone or together, in combination with 1.0 μg of ANF-luc and 0.04 μg of pGL4.75. Transfected cells were washed once on the following day with medium before being refreshed with new medium. Forty-eight hours after transfection, cells were harvested and lysed, and then the Firefly luciferase and Renilla luciferase activities were measured with the Dual-Glo Luciferase Assay Kit (Promega) according to the manufacturer's instructions. For promoter activity assays, all experiments were performed at minimum in triplicate and data are reported as the ratio of Firefly luciferase to Renilla luciferase.
Statistical analysis
All statistical analyses were carried out with the SPSS for Windows software version 19.0 (SPSS, Chicago, IL, USA). Quantitative data are expressed as means ± standard deviations (SD). Student's unpaired t test was used to compare quantitative variables between two groups. Comparison of the qualitative variables between two groups was made by using Pearson's χ2 test or Fisher's exact test when appropriate. All reported p values were two-tailed and statistical significance was defined as p < 0.05.
Results
Clinical findings
A cohort of 175 unrelated patients with CHD underwent a comprehensive clinical evaluation in contrast to a total of 400 unrelated non-CHD control individuals. All the studied patients had echocardiographically or surgically confirmed CHD; while the control subjects had no evidence of congenital defects in cardiovascular structures or positive family history of CHD. None of the study participants had recognized environmental risk factors for CHD, such as maternal viral infection, drug use and long-term exposure to toxic chemicals or ionizing radiation in the first trimester of pregnancy. There is no significant difference in age, gender or ethnicity between patient and control groups. The demographic profiles and clinical features of the CHD patients are summarized in Table 1.
Demographic profiles and clinical characteristics of the patients with congenital heart disease (n = 175).
| Variables | Statistics |
|---|---|
| Male gender (%) | 98 (56) |
| Female gender (%) | 77 (44) |
| Age (years) | 4.3 ± 3.1 |
| Positive family history of congenital heart disease (%) | 42 (24) |
| Clinical classification of congenital heart disease | |
| Isolated congenital heart disease (%) | 75 (43) |
| Ventricular septal defect (%) | 26 (15) |
| Atrial septal defect (%) | 23 (13) |
| Patent ductus arteriosus (%) | 9 (5) |
| Double outlet of right ventricle (%) | 5 (3) |
| Abnormal pulmonary venous connection (%) | 4 (2) |
| Truncus arteriosus (%) | 3 (2) |
| Endocardial cushion defect (%) | 2 (1) |
| Hypoplastic left ventricle (%) | 2 (1) |
| Hypoplastic right ventricle (%) | 1 (1) |
| Complex congenital heart disease (%) | 100 (57) |
| Tetralogy of Fallot (%) | 49 (28) |
| Atrial septal defect + ventricular septal defect (%) | 12 (7) |
| Ventricular septal defect + patent ductus arteriosus (%) | 8 (5) |
| Atrial septal defect + tetralogy of Fallot (%) | 7 (4) |
| Atrial septal defect + patent ductus arteriosus (%) | 7 (4) |
| Double outlet of right ventricle + ventricular septal defect (%) | 6 (3) |
| Ventricular septal defect + transposition of the great arteries (%) | 6 (3) |
| Ventricular septal defect + truncus arteriosus (%) | 5 (3) |
| Incidence of arrhythmias | 16 (9) |
| Atrioventricular block (%) | 10 (6) |
| Atrial fibrillation (%) | 6 (3) |
| Treatment of congenital heart disease | |
| Surgical repair (%) | 121 (69) |
| Catheter-based closure (%) | 43 (25) |
| Follow-up (%) | 11 (6) |
Data are expressed as means and standard deviations, number or percentage.
Identification of a novel TBX20 mutation
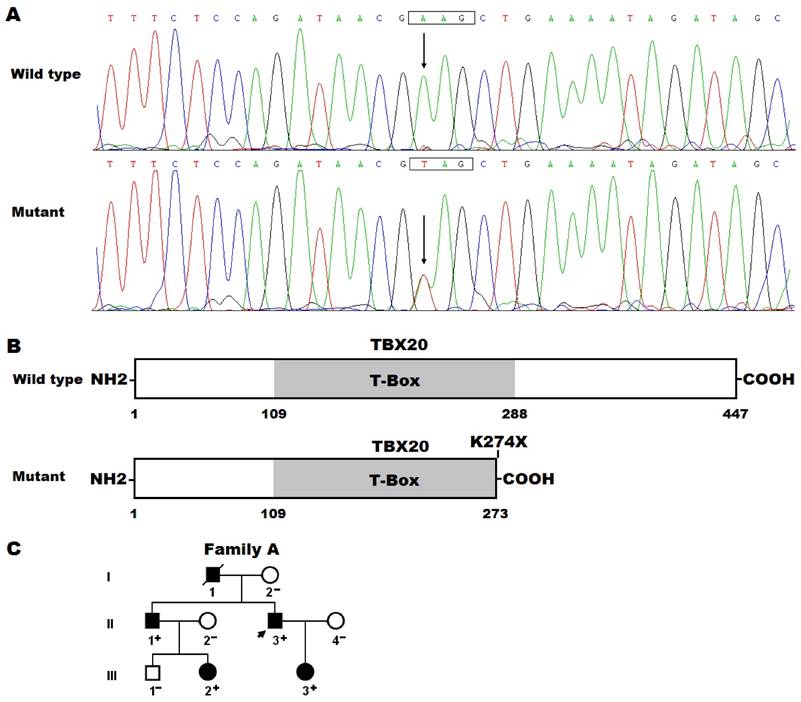
By sequence analysis of the TBX20 gene in 175 unrelated patients with CHD, a novel sequence variation was identified in a patient with TOF, who had a positive family history of CHD. Specifically, a substitution of thymine for adenine in the first nucleotide of codon 274 (c.820A>T), which was predicted to produce a premature stop codon at amino acid position 274 (p.K274X), was identified in the index patient from family A. The sequence chromatograms showing the detected heterozygous TBX20 mutation and its homozygous control sequence are presented in Figure 1A. The schematic diagrams showing the full-length and truncated TBX20 proteins were displayed in Figure 1B. The nonsense mutation was absent in the 800 control chromosomes and not found in the SNP and HGM databases (accessed again October 3, 2016). Genetic analysis of the proband's available family members showed that his father (I-1), elder brother (II-1) and son (III-3) had also TOF. In addition, his father (I-1) and elder brother (II-1) had also congenital atrial septal defect, presenting with pentalogy of Fallot, and his niece (III-2) had persistent truncus arteriosus and ventricular septal defect. Analysis of the pedigree revealed that the mutation co-segregated with CHD transmitted in an autosomal dominant fashion, with complete penetrance (Figure 1C). The phenotypic characteristics and status of TBX20 mutation of the affected family members are reported in Table 2.
Novel TBX20 mutation associated with congenital heart disease. (A) Sequence electropherograms showing the TBX20 mutation and its wild-type control. The arrow points to the heterozygous nucleotides of A/T in the proband (mutant) or the homozygous nucleotides of A/A in a control individual (wild type). The rectangle marks the nucleotides comprising a codon of TBX20. (B) Schematic diagrams showing full-length and truncated TBX20 protein structures with the causative mutation marked. The mutation identified in patients with congenital heart disease is predicted to generate a truncated protein with only amino terminus together with partial T-box structural domain left (mutant). NH2, amino terminus; COOH, carboxyl terminus. (C) Pedigree structure of the family with congenital heart disease. The family was designated as family A and the family members are identified by generations and numbers. Square, male family member; circle, female member; symbol with a slash, deceased member; closed symbol, affected member; open symbol, unaffected member; arrow, proband; +, carrier of the heterozygous mutation; -, non-carrier.
Phenotypic characteristics and TBX20 mutation status of the affected family members.
| Individual | Gender | Age (years) | Cardiac structural defects | TBX20 mutation |
|---|---|---|---|---|
| Family A | K274X | |||
| I-1 | M | 53a | TOF, ASD | NA |
| II-1 | M | 31 | TOF, ASD | +/- |
| II-3 | M | 28 | TOF | +/- |
| III-2 | F | 0 | PTA, VSD | +/- |
| III-3 | F | 2 | TOF | +/- |
M, Male; F, female; TOF, tetralogy of Fallot; ASD, atrial septal defect; PTA, persistent truncus arteriosus; VSD, ventricular septal defect; NA, not available; +/-, heterozygote.
a Age at death.
Evolutionary conservation
As shown in Figure 2, human TBX20 was aligned with orthologous proteins from multiple species, exhibiting that the altered lysine at amino acid position 274 was highly conserved evolutionarily.
Alignment of multiple TBX20 protein sequences across species. The altered lysine at amino acid position 274 (p.K274) of TBX20 is highly conserved evolutionarily among multiple species.

Functional defects of mutant TBX20 in transcriptional activation
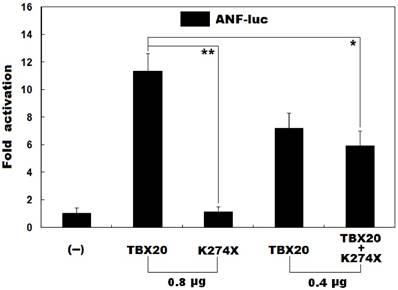
As shown in Figure 3, the same amount (0.8 μg) of wild-type TBX20 and K274X-mutant TBX20 (K274X) activated the ANF promoter by ~11-fold and ~1-fold, respectively. When wild-type TBX20 (0.4 μg) was co-expressed with the same amount (0.4 μg) of K274-mutant TBX20 or empty pcDNA3.1 plasmid, the induced activation of the ANF promoter was ~6-fold or ~7-fold. These results indicate that the K274-mutant TBX20 has no transcriptional activity or significantly dominant-negative effect on its wild-type counterpart.
Failure of the mutant TBX20 to transactivate a target gene. Activation of the ANF-luciferase reporter in cultured CHO cells by wild-type TBX20 or K274X-mutant TBX20 (K274X), alone or together, showed that the K274X-mutant TBX20 had no transcriptional activity or significantly inhibitory effect on its wild-type counterpart. Experiments were carried out in triplicate and data were expressed as mean and standard deviations. * indicates t = 5.7200, p = 0.0046, and ** indicates t = 13.5396, p = 0.0002, when compared with wild-type TBX20 (0.8 μg).
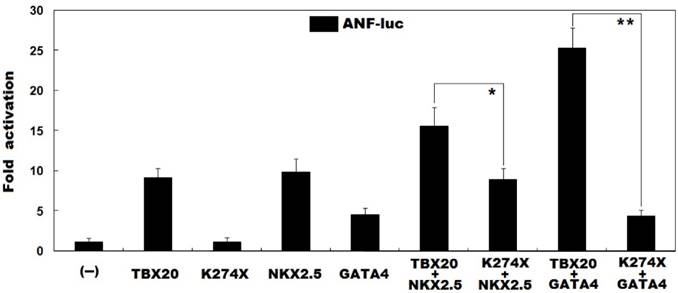
Reduced synergistic activation between TBX20 and NKX2.5 as well as GATA4 by the mutation
As shown in Figure 4, in the presence of NKX2.5, the same amount (0.6 μg) of wild-type TBX20 and K274X-mutant TBX20 (K274X) activated the ANF promoter by ~16-fold and ~9-fold, respectively; while in the presence of GATA4, the same amount (0.6 μg) of wild-type TBX20 and K274X-mutant TBX20 (K274X) activated the ANF promoter by ~25-fold and ~4-fold, respectively. These data indicate that the mutation diminishes the synergistic activation between TBX20 and NKX2.5 as well as GATA4.
Abrogated synergistic transactivation between TBX20 and NKX2.5 as well as GATA4. Activation of the ANF-luciferase reporter in cultured COS-7 cells by wild-type TBX20 or K274X-mutant TBX20 (K274X), alone or in combination with NKX2.5 or GATA4, showed that the mutation disrupted the synergistic transactivation between TBX20 and NKX2.5 as well as GATA4. Experiments were performed in triplicate and data were given as mean and standard deviations. * indicates t = 4.2718, p = 0.0129, and ** indicates t = 14.1437, p = 0.0001, when compared with their wild-type counterparts.
Discussion
In this study, by sequence analysis of the TBX20 gene, a novel heterozygous mutation, c.820A>T, which was predicted to generate a truncated protein (p.K274X), was discovered in a family with CHD, including familial TOF and atrial septal defect as well as sporadic persistent truncus arteriosus and ventricular septal defect. In the family, the mutation co-segregated with CHD with complete penetrance. The nonsense mutation was absent in the 800 referral chromosomes from a control population matched in ethnicity and gender. A cross-species alignment of TBX20 proteins showed that the affected amino acid was highly conserved evolutionarily. Biochemical analyses demonstrated that the mutant TBX20 failed to transactivate a target gene. Furthermore, the mutation reduced the synergistic activation between TBX20 and NKX2.5 or between TBX20 and GATA4. Therefore, it is probable that functional failure of TBX20 confers an enhanced susceptibility to CHD in these mutation carriers.
TBX genes encode a large family of transcription factors that are characterized by a highly conserved DNA-binding motif of 180 amino acid residues, T-box. This T-box recognizes a specific DNA element, the T-half site (the consensus DNA sequence of 5-AGGTGTGA-3), that exists singly, or in multiples with different orientation and spacing in the promoters of target genes, and regulates transcriptional activation or inhibition. Moreover, the T-box is also a conserved interaction domain for other transcription factors, chromatin remodelling complexes and histone-modifying enzymes involved in transcriptional mediation [50]. In this study, the identified mutation is predicted to yield a truncated protein (p.K274X), losing partial T-box along with the carboxyl terminus, thus may be anticipated to impair the transcriptional function of TBX20 probably by interfering with the specific binding to target genes or the interaction with partners of TBX20.
TBX20 has been demonstrated to transactivate multiple target genes that are expressed in the heart during embryogenesis, including ANF, CX40 and CX45, of which ANF is the most extensively investigated one, and its expression in the embryonic heart has been established by synergistic action of TBX20, NKX2.5, GATA4 and other transcriptional factors [50,51]. In this study, the functional roles of a novel TBX20 mutation identified in familial TOF patients were evaluated by transcriptional activity assays, and the results revealed no transcriptional activation of the ANF promoter by the mutant TBX20 protein, alone or together with NKX2.5 or GATA4. These functional data indicate that haploinsufficiency of TBX20 is potentially an alternative molecular mechanism underpinning TOF.
Previous studies have validated that TBX20 physically interacts with multiple transcription factors, including homeobox transcription factor NKX2.5 and zinc-finger transcription factors GATA4, GATA5 and GATA6, cooperating to synergistically activate the expression of several target genes key to cardiogenesis [50,51], and loss-of-function mutations in such transcriptionally cooperative partners as NKX2.5, GATA4, GATA5 and GATA6 have been associated with a wide spectrum of CHD, including atrial septal defect, ventricular septal defect and TOF [34,36,37,50-52,56], which suggest that genetically compromised TBX20 contributes to CHD probably by diminishing synergistic activation of cardiac core genes with these cooperative partners.
Previously, TBX20 mutations have been associated with CHD. In fact, so far at least 13 mutations in the coding region of TBX20 have been identified to be responsible for a great variety of CHD. Kirk et al. [56] sequenced the whole coding exons of TBX20 in 353 index patients with CHD, and identified two heterozygous mutations (p.I152M and p.Q195X) in two index patients with familial CHD. Specifically, in family 1 harboring the missense mutation p.I152M, the proband had atrial septal defect; her grandmother had ventricular septal defect, and her mother had patent foramen ovale. In family 2 carrying the nonsense mutation p.Q195X, the proband had atrial septal defect and coarctation of the aorta, and his mother had mitral valve prolapse with mild regurgitation, dilated cardiomyopathy, and apicolateral hypertrophy. Functional deciphers unveiled that I152M-mutant TBX20 had decreased function while Q195X-mutant TBX20 was effectively null. By PCR-sequencing, Qian et al. [57] screened TBX20 in 192 unrelated children with CHD, and discovered two mutations in four children, including p.H186D in two children (one child had an atrioventicular canal with secundum and primum atrial septal defects, as well as a cleft mitral valve with moderate mitral regurgitation, and the other child had pentalogy of Fallot) and p.L197P in other two children (one had pentalogy of Fallot and the other had atrial septal defect). However, the functional characteristics of the two mutations were not delineated. Liu et al. [58] made a sequence analysis of the exons 2 to 6 of TBX20 in 203 unrelated patients with CHD, and found three non-synonymous mutations of p.A63T, p.I121F and p.T262M in three patients with atrial septal defect, total anomalous pulmonary venous connection and TOF, respectively. However, functional effects of these sequence variants remain to be assayed. Posch et al. [59] analyzed the entire coding sequences of TBX20 in 170 unrelated patients with atrial septal defect, and found a novel mutation of p.I121M in a patient with positive family history of CHD. Genetic analysis of the index patient's pedigree showed that the mutation co-segregated with CHD in the three-generation kindred. Functional assays showed that the mutation p.I121M resulted in a significantly increased transcriptional activity, which was further increased in the presence of transcription factors GATA4/5 and NKX2.5. By whole-exome sequencing, Liu and colleagues [60] analyzed a three-generation family with atrial septal defect, and identified a novel TBX20 mutation, p.D176N. However, functional analysis of the mutation was not performed. Pan and co-workers [30] sequenced the coding exons and flanking introns of the TBX20 gene in 146 unrelated patients with CHD, and identified a novel heterozygous TBX20 mutation, p.R143W in an index patient with double outlet right ventricle. Genetic analyses of the proband's pedigree showed that the mutation co-segregated with the disease. Biological analysis revealed that mutant TBX20 had a significantly diminished transcriptional activity. By using the high resolution melting and DNA sequencing methods, Monroy-Muñoz and partners [31] made a search for TBX20 mutations in a group of 38 patients with atrial or ventricular septal defect, and found three missense mutations (p.Y309D, p.T370O, and p.M395R) in two patients with atrial septal defect, though functional characterizations of these mutations were not carried out. In the present study, a novel TBX20 loss-of-function mutation was identified in patients with familial TOF alone or together with familial atrial septal defect, or sporadic persistent truncus arteriosus and ventricular septal defect. Taken collectively, these findings expand the phenotypic spectrum linked to TBX20 mutations and imply that genetically defective TBX20 is responsible for CHD in a subset of patients.
In conclusion, this study associates TBX20 loss-of-function mutation with increased vulnerability to TOF or persistent truncus arteriosus in humans for the first time, which provides novel insight into the molecular mechanism of CHD and suggests potential implications for genetic counseling and personalized treatment of the patients with CHD.
Acknowledgements
The authors thank the study subjects for their participation in this study. This work was supported by the grants from the National Natural Science Fund of China (81470372), the Key Program for Basic Research of Shanghai, China (14JC1405500) and the Major project of Shanghai Chest Hospital, China (grant no. 2014YZDH10102).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Mozaffarian D, Benjamin EJ, Go AS, Arnett DK, Blaha MJ, Cushman M, de Ferranti S, Després JP, Fullerton HJ, Howard VJ, Huffman MD, Judd SE, Kissela BM, Lackland DT, Lichtman JH, Lisabeth LD, Liu S, Mackey RH, Matchar DB, McGuire DK, Mohler ER 3rd, Moy CS, Muntner P, Mussolino ME, Nasir K, Neumar RW, Nichol G, Palaniappan L, Pandey DK, Reeves MJ, Rodriguez CJ, Sorlie PD, Stein J, Towfighi A, Turan TN, Virani SS, Willey JZ, Woo D, Yeh RW, Turner MB; American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Heart disease and stroke statistics―2015 update: a report from the American Heart Association. Circulation. 2015;131:e29-322
2. van der Linde D, Konings EE, Slager MA, Witsenburg M, Helbing WA, Takkenberg JJ, Roos-Hesselink JW. Birth prevalence of congenital heart disease worldwide: a systematic review and meta-analysis. J Am Coll Cardiol. 2011;58:2241-7
3. Vecoli C, Pulignani S, Foffa I, Andreassi MG. Congenital heart disease: the crossroads of genetics, epigenetics and environment. Curr Genomics. 2014;15:390-9
4. Kahr PC, Radke RM, Orwat S, Baumgartner H, Diller GP. Analysis of associations between congenital heart defect complexity and health-related quality of life using a meta-analytic strategy. Int J Cardiol. 2015;199:197-203
5. Feltez G, Coronel CC, Pellanda LC, Lukrafka JL. Exercise capacity in children and adolescents with corrected congenital heart disease. Pediatr Cardiol. 2015;36:1075-82
6. Rosenblum O, Katz U, Reuveny R, Williams CA, Dubnov-Raz G. Exercise performance in children and young adults after complete and incomplete repair of congenital heart disease. Pediatr Cardiol. 2015;36:1573-81
7. Marelli A, Miller SP, Marino BS, Jefferson AL, Newburger JW. Brain in congenital heart disease across the lifespan: the cumulative burden of injury. Circulation. 2016;133:1951-62
8. Jensen AS, Idorn L, Thomsen C, von der Recke P, Mortensen J, Sørensen KE, Thilén U, Nagy E, Kofoed KF, Ostrowski SR, Søndergaard L. Prevalence of cerebral and pulmonary thrombosis in patients with cyanotic congenital heart disease. Heart. 2015;101:1540-6
9. Galiè N, Humbert M, Vachiery JL, Gibbs S, Lang I, Torbicki A, Simonneau G, Peacock A, Vonk Noordegraaf A, Beghetti M, Ghofrani A, Gomez Sanchez MA, Hansmann G, Klepetko W, Lancellotti P, Matucci M, McDonagh T, Pierard LA, Trindade PT, Zompatori M, Hoeper M, Aboyans V, Vaz Carneiro A, Achenbach S, Agewall S, Allanore Y, Asteggiano R, Paolo Badano L, Albert Barberà J, Bouvaist H, Bueno H, Byrne RA, Carerj S, Castro G, Erol Ç, Falk V, Funck-Brentano C, Gorenflo M, Granton J, Iung B, Kiely DG, Kirchhof P, Kjellstrom B, Landmesser U, Lekakis J, Lionis C, Lip GY, Orfanos SE, Park MH, Piepoli MF, Ponikowski P, Revel MP, Rigau D, Rosenkranz S, Völler H, Luis Zamorano J. 2015 ESC/ERS Guidelines for the diagnosis and treatment of pulmonary hypertension: The Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT). Eur Heart J. 2016;37:67-119
10. Lüscher TF. Frontiers in congenital heart disease: pulmonary hypertension, heart failure, and arrhythmias. Eur Heart J. 2016;37:1407-9
11. Budts W, Roos-Hesselink J, Rädle-Hurst T, Eicken A, McDonagh TA, Lambrinou E, Crespo-Leiro MG, Walker F, Frogoudaki AA. Treatment of heart failure in adult congenital heart disease: a position paper of the Working Group of Grown-Up Congenital Heart Disease and the Heart Failure Association of the European Society of Cardiology. Eur Heart J. 2016;37:1419-27
12. Stout KK, Broberg CS, Book WM, Cecchin F, Chen JM, Dimopoulos K, Everitt MD, Gatzoulis M, Harris L, Hsu DT, Kuvin JT, Law Y, Martin CM, Murphy AM, Ross HJ, Singh G, Spray TL; American Heart Association Council on Clinical Cardiology, Council on Functional Genomics and Translational Biology, and Council on Cardiovascular Radiology and Imaging. Chronic heart failure in congenital heart disease: a scientific statement from the American Heart Association. Circulation. 2016;133:770-801
13. McLeod CJ, Warnes C. Recognition and management of arrhythmias in adult congenital heart disease. Curr Opin Cardiol. 2016;31:117-23
14. Diller GP, Baumgartner H. Sudden cardiac death during exercise in patients with congenital heart disease: the exercise paradox and the challenge of appropriate counseling. Eur Heart J. 2016;37:627-9
15. Jortveit J, Eskedal L, Hirth A, Fomina T, Døhlen G, Hagemo P, Tell GS, Birkeland S, Øyen N, Holmstrøm H. Sudden unexpected death in children with congenital heart defects. Eur Heart J. 2016;37:621-6
16. Williams RG. Late causes of death after congenital heart defects: a population-based study from Finland. J Am Coll Cardiol. 2016;68:499-501
17. Fahed AC, Gelb BD, Seidman JG, Seidman CE. Genetics of congenital heart disease: the glass half empty. Circ Res. 2013;112:707-20
18. Ntiloudi D, Giannakoulas G, Parcharidou D, Panagiotidis T, Gatzoulis MA, Karvounis H. Adult congenital heart disease: a paradigm of epidemiological change. Int J Cardiol. 2016;218:269-74
19. Khairy P, Ionescu-Ittu R, Mackie AS, Abrahamowicz M, Pilote L, Marelli AJ. Changing mortality in congenital heart disease. J Am Coll Cardiol. 2010;56:1149-57
20. Patel SS, Burns TL. Nongenetic risk factors and congenital heart defects. Pediatr Cardiol. 2013;34:1535-55
21. Andersen TA, Troelsen Kde L, Larsen LA. Of mice and men: molecular genetics of congenital heart disease. Cell Mol Life Sci. 2014;71:1327-52
22. Li Y, Klena NT, Gabriel GC, Liu X, Kim AJ, Lemke K, Chen Y, Chatterjee B, Devine W, Damerla RR, Chang C, Yagi H, San Agustin JT, Thahir M, Anderton S, Lawhead C, Vescovi A, Pratt H, Morgan J, Haynes L, Smith CL, Eppig JT, Reinholdt L, Francis R, Leatherbury L, Ganapathiraju MK, Tobita K, Pazour GJ, Lo CW. Global genetic analysis in mice unveils central role for cilia in congenital heart disease. Nature. 2015;521:520-4
23. Homsy J, Zaidi S, Shen Y, Ware JS, Samocha KE, Karczewski KJ, DePalma SR, McKean D, Wakimoto H, Gorham J, Jin SC, Deanfield J, Giardini A, Porter GA Jr, Kim R, Bilguvar K, López-Giráldez F, Tikhonova I, Mane S, Romano-Adesman A, Qi H, Vardarajan B, Ma L, Daly M, Roberts AE, Russell MW, Mital S, Newburger JW, Gaynor JW, Breitbart RE, Iossifov I, Ronemus M, Sanders SJ, Kaltman JR, Seidman JG, Brueckner M, Gelb BD, Goldmuntz E, Lifton RP, Seidman CE, Chung WK. De novo mutations in congenital heart disease with neurodevelopmental and other congenital anomalies. Science. 2015;350:1262-6
24. Quintero-Rivera F, Xi QJ, Keppler-Noreuil KM, Lee JH, Higgins AW, Anchan RM, Roberts AE, Seong IS, Fan X, Lage K, Lu LY, Tao J, Hu X, Berezney R, Gelb BD, Kamp A, Moskowitz IP, Lacro RV, Lu W, Morton CC, Gusella JF, Maas RL. MATR3 disruption in human and mouse associated with bicuspid aortic valve, aortic coarctation and patent ductus arteriosus. Hum Mol Genet. 2015;24:2375-89
25. Abou Hassan OK, Fahed AC, Batrawi M, Arabi M, Refaat MM, DePalma SR, Seidman JG, Seidman CE, Bitar FF, Nemer GM. NKX2-5 mutations in an inbred consanguineous population: genetic and phenotypic diversity. Sci Rep. 2015;5:8848
26. Zheng J, Li F, Liu J, Xu Z, Zhang H, Fu Q, Wang J, Sun K. Investigation of somatic NKX2-5 mutations in Chinese children with congenital heart disease. Int J Med Sci. 2015;12:538-43
27. Wang J, Mao JH, Ding KK, Xu WJ, Liu XY, Qiu XB, Li RG, Qu XK, Xu YJ, Huang RT, Xue S, Yang YQ. A novel NKX2.6 mutation associated with congenital ventricular septal defect. Pediatr Cardiol. 2015;36:646-56
28. Kassab K, Hariri H, Gharibeh L, Fahed AC, Zein M, El-Rassy I, Nemer M, El-Rassi I, Bitar F, Nemer G. GATA5 mutation homozygosity linked to a double outlet right ventricle phenotype in a Lebanese patient. Mol Genet Genomic Med. 2015;4:160-71
29. Chao CS, McKnight KD, Cox KL, Chang AL, Kim SK, Feldman BJ. Novel GATA6 mutations in patients with pancreatic agenesis and congenital heart malformations. PLoS One. 2015;10:e0118449
30. Pan Y, Geng R, Zhou N, Zheng GF, Zhao H, Wang J, Zhao CM, Qiu XB, Yang YQ, Liu XY. TBX20 loss-of-function mutation contributes to double outlet right ventricle. Int J Mol Med. 2015;35:1058-66
31. Monroy-Muñoz IE, Pérez-Hernández N, Rodríguez-Pérez JM, Muñoz-Medina JE, Angeles-Martínez J, García-Trejo JJ, Morales-Ríos E, Massó F, Sandoval-Jones JP, Cervantes-Salazar J, García-Montes JA, Calderón-Colmenero J, Vargas-Alarcón G. Novel mutations in the transcriptional activator domain of the human TBX20 in patients with atrial septal defect. Biomed Res Int. 2015;2015:718786
32. Pan Y, Wang ZG, Liu XY, Zhao H, Zhou N, Zheng GF, Qiu XB, Li RG, Yuan F, Shi HY, Hou XM, Yang YQ. A novel TBX1 loss-of-function mutation associated with congenital heart disease. Pediatr Cardiol. 2015;36:1400-10
33. Perrot A, Schmitt KR, Roth EM, Stiller B, Posch MG, Browne EN, Timmann C, Horstmann RD, Berger F, Özcelik C. CCN1 mutation is associated with atrial septal defect. Pediatr Cardiol. 2015;36:295-9
34. Yang J, Zhu M, Wang Y, Hou X, Wu H, Wang D, Shen H, Hu Z, Zou J. Whole-exome sequencing identifies a new mutation of MYH7 in a Chinese family with left ventricular noncompaction. Gene. 2015;558:138-42
35. Li N, Subrahmanyan L, Smith E, Yu X, Zaidi S, Choi M, Mane S, Nelson-Williams C, Bahjati M, Kazemi M, Hashemi M, Fathzadeh M, Narayanan A, Tian L, Montazeri F, Mani M, Begleiter ML, Coon BG, Lynch HT, Olson EN, Zhao H, Ruland J, Lifton RP, Mani A. Mutations in the histone modifier PRDM6 are associated with isolated nonsyndromic patent ductus arteriosus. Am J Hum Genet. 2016;98:1082-91
36. Boyle L, Wamelink MM, Salomons GS, Roos B, Pop A, Dauber A, Hwa V, Andrew M, Douglas J, Feingold M, Kramer N, Saitta S, Retterer K, Cho MT, Begtrup A, Monaghan KG, Wynn J, Chung WK. Mutations in TKT are the cause of a syndrome including short stature, developmental delay, and congenital heart defects. Am J Hum Genet. 2016;98:1235-42
37. LaHaye S, Corsmeier D, Basu M, Bowman JL, Fitzgerald-Butt S, Zender G, Bosse K, McBride KL, White P, Garg V. Utilization of whole exome sequencing to identify causative mutations in familial congenital heart disease. Circ Cardiovasc Genet. 2016;9:320-9
38. Priest JR, Osoegawa K, Mohammed N, Nanda V, Kundu R, Schultz K, Lammer EJ, Girirajan S, Scheetz T, Waggott D, Haddad F, Reddy S, Bernstein D, Burns T, Steimle JD, Yang XH, Moskowitz IP, Hurles M, Lifton RP, Nickerson D, Bamshad M, Eichler EE, Mital S, Sheffield V, Quertermous T, Gelb BD, Portman M, Ashley EA. De novo and rare variants at multiple loci support the oligogenic origins of atrioventricular septal heart defects. PLoS Genet. 2016;12:e1005963
39. Werner P, Latney B, Deardorff MA, Goldmuntz E. MESP1 mutations in patients with congenital heart defects. Hum Mutat. 2016;37:308-14
40. Yoshida A, Morisaki H, Nakaji M, Kitano M, Kim KS, Sagawa K, Ishikawa S, Satokata I, Mitani Y, Kato H, Hamaoka K, Echigo S, Shiraishi I, Morisaki T. Genetic mutation analysis in Japanese patients with non-syndromic congenital heart disease. J Hum Genet. 2016;61:157-62
41. Kelle AM, Bentley SJ, Rohena LO, Cabalka AK, Olson TM. Ebstein anomaly, left ventricular non-compaction, and early onset heart failure associated with a de novo α-tropomyosin gene mutation. Am J Med Genet A. 2016;170:2186-90
42. Cao Y, Wang J, Wei C, Hou Z, Li Y, Zou H, Meng M, Wang W, Jiang L. Genetic variations of NKX2-5 in sporadic atrial septal defect and ventricular septal defect in Chinese Yunnan population. Gene. 2016;575:29-33
43. Tong YF. Mutations of NKX2.5 and GATA4 genes in the development of congenital heart disease. Gene. 2016;588:86-94
44. Chen J, Qi B, Zhao J, Liu W, Duan R, Zhang M. A novel mutation of GATA4 (K300T) associated with familial atrial septal defect. Gene. 2016;575:473-7
45. Lu CX, Gong HR, Liu XY, Wang J, Zhao CM, Huang RT, Xue S, Yang YQ. A novel HAND2 loss-of-function mutation responsible for tetralogy of Fallot. Int J Mol Med. 2016;37:445-51
46. S Sun YM, Wang J, Qiu XB, Yuan F, Li RG, Xu YJ, Qu XK, Shi HY, Hou XM, Huang RT, Xue S, Yang YQ. A HAND2 loss-of-function mutation causes familial ventricular septal defect and pulmonary stenosis. G3 (Bethesda). 2016;6:987-92
47. Sun YM, Wang J, Qiu XB, Yuan F, Xu YJ, Li RG, Qu XK, Huang RT, Xue S, Yang YQ. PITX2 loss-of-function mutation contributes to tetralogy of Fallot. Gene. 2016;577:258-64
48. McCulley DJ, Black BL. Transcription factor pathways and congenital heart disease. Curr Top Dev Biol. 2012;100:253-77
49. Gelb BD, Chung WK. Complex genetics and the etiology of human congenital heart disease. Cold Spring Harb Perspect Med. 2014;4:a013953
50. Greulich F, Rudat C, Kispert A. Mechanisms of T-box gene function in the developing heart. Cardiovasc Res. 2011;91:212-22
51. Stennard FA, Costa MW, Elliott DA, Rankin S, Haast SJ, Lai D, McDonald LP, Niederreither K, Dolle P, Bruneau BG, Zorn AM, Harvey RP. Cardiac T-box factor Tbx20 directly interacts with Nkx2-5, GATA4 and GATA5 in regulation of gene expression in the developing heart. Dev Biol. 2003;262:206-24
52. Cai CL, Zhou W, Yang L, Bu L, Qyang Y, Zhang X, Li X, Rosenfeld MG, Chen J, Evans S. T-box genes coordinate regional rates of proliferation and regional specification during cardiogenesis. Development. 2005;132:2475-87
53. Singh MK, Christoffels VM, Dias JM, Trowe MO, Petry M, Schuster-Gossler K, Bürger A, Ericson J, Kispert A. Tbx20 is essential for cardiac chamber differentiation and repression of Tbx2. Development. 2005;132:2697-707
54. Stennard FA, Costa MW, Lai D, Biben C, Furtado MB, Solloway MJ, McCulley DJ, Leimena C, Preis JI, Dunwoodie SL, Elliott DE, Prall OW, Black BL, Fatkin D, Harvey RP. Murine T-box transcription factor Tbx20 acts as a repressor during heart development, and is essential for adult heart integrity, function and adaptation. Development. 2005;132:2451-62
55. Takeuchi JK, Mileikovskaia M, Koshiba-Takeuchi K, Heidt AB, Mori AD, Arruda EP, Gertsenstein M, Georges R, Davidson L, Mo R, Hui CC, Henkelman RM, Nemer M, Black BL, Nagy A, Bruneau BG. Tbx20 dose-dependently regulates transcription factor networks required for mouse heart and motorneuron development. Development. 2005;132:2463-474
56. Kirk EP, Sunde M, Costa MW, Rankin SA, Wolstein O, Castro ML, Butler TL, Hyun C, Guo G, Otway R, Mackay JP, Waddell LB, Cole AD, Hayward C, Keogh A, Macdonald P, Griffiths L, Fatkin D, Sholler GF, Zorn AM, Feneley MP, Winlaw DS, Harvey RP. Mutations in cardiac T-box factor gene TBX20 are associated with diverse cardiac pathologies, including defects of septation and valvulogenesis and cardiomyopathy. Am J Hum Genet. 2007;81:280-91
57. Qian L, Mohapatra B, Akasaka T, Liu J, Ocorr K, Towbin JA, Bodmer R. Transcription factor neuromancer/TBX20 is required for cardiac function in Drosophila with implications for human heart disease. Proc Natl Acad Sci U S A. 2008;105:19833-8
58. Liu C, Shen A, Li X, Jiao W, Zhang X, Li Z. T-box transcription factor TBX20 mutations in Chinese patients with congenital heart disease. Eur J Med Genet. 2008;51:580-7
59. Posch MG, Gramlich M, Sunde M, Schmitt KR, Lee SH, Richter S, Kersten A, Perrot A, Panek AN, Al Khatib IH, Nemer G, Mégarbané A, Dietz R, Stiller B, Berger F, Harvey RP, Ozcelik C. A gain-of-function TBX20 mutation causes congenital atrial septal defects, patent foramen ovale and cardiac valve defects. J Med Genet. 2010;47:230-5
60. Liu JJ, Fan LL, Chen JL, Tan ZP, Yang YF. A novel variant in TBX20 (p.D176N) identified by whole-exome sequencing in combination with a congenital heart disease related gene filter is associated with familial atrial septal defect. J Zhejiang Univ Sci B. 2014;15:830-7
Author contact
![]() Corresponding author: Dr. Ying-Jia Xu, Department of Cardiology, Shanghai Chest Hospital, Shanghai Jiao Tong University, 241 West Huaihai Road, Shanghai 200030, China. E-mail: 18616889888com; Prof. Yi-Qing Yang, Department of Cardiovascular Research Laboratory, Shanghai Chest Hospital, Shanghai Jiao Tong University, 241 West Huaihai Road, Shanghai 200030, China. E-mail: dryyqedu.cn.
Corresponding author: Dr. Ying-Jia Xu, Department of Cardiology, Shanghai Chest Hospital, Shanghai Jiao Tong University, 241 West Huaihai Road, Shanghai 200030, China. E-mail: 18616889888com; Prof. Yi-Qing Yang, Department of Cardiovascular Research Laboratory, Shanghai Chest Hospital, Shanghai Jiao Tong University, 241 West Huaihai Road, Shanghai 200030, China. E-mail: dryyqedu.cn.