Impact Factor ISSN: 1449-1907
- Issue 9; 2026
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Volume 23; 2026
- Past Issues
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
1. Introduction
2. Materials and Methods
3. Results
4. Discussion
5. Conclusion
Acknowledgements
Author Contributions
References

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2015; 12(9):748-758. doi:10.7150/ijms.12177 This issue Cite
Research Paper
Effects of Selenium Yeast on Oxidative Stress, Growth Inhibition, and Apoptosis in Human Breast Cancer Cells
Chih-Hung Guo1,2,3,4 ![]() , Simon Hsia1,4, Min-Yi Shih1, Fang-Chin Hsieh1, Pei-Chung Chen4,5,6
, Simon Hsia1,4, Min-Yi Shih1, Fang-Chin Hsieh1, Pei-Chung Chen4,5,6 ![]()
1. Institute of Biomedical Nutrition, Hung-Kuang University, Taichung 433, Taiwan
2. Department of Medical Research, China Medical University Hospital, Taichung 404, Taiwan
3. Department of Health and Nutrition Biotechnology, Asia University, Taichung 413, Taiwan
4. Taiwan Nutraceutical Association, Taipei 115, Taiwan
5. College of Life Sciences, National Chung Hsing University, Taichung 402, Taiwan
6. College of Engineering, National Chiao Tung University, Hsinchu 300, Taiwan
Received 2015-3-19; Accepted 2015-8-17; Published 2015-9-5
Abstract
Recent evidence suggests that selenium (Se) yeast may exhibit potential anti-cancer properties; whereas the precise mechanisms remain unknown. The present study was aimed at evaluating the effects of Se yeast on oxidative stress, growth inhibition, and apoptosis in human breast cancer cells. Treatments of ER-positive MCF-7 and triple-negative MDA-MB-231 cells with Se yeast (100, 750, and 1500 ng Se/mL), methylseleninic acid (MSA, 1500 ng Se/mL), or methylselenocysteine (MSC, 1500 ng Se/mL) at a time course experiment (at 24, 48, 72, and 96 h) were analyzed. Se yeast inhibited the growth of these cancer cells in a dose- and time-dependent manner. Compared with the same level of MSA, cancer cells exposure to Se yeast exhibited a lower growth-inhibitory response. The latter has also lower superoxide production and reduced antioxidant enzyme activities. Furthermore, MSA (1500 ng Se/mL)-exposed non-tumorigenic human mammary epithelial cells (HMEC) have a significant growth inhibitory effect, but not Se yeast and MSC. Compared with MSA, Se yeast resulted in a greater increase in the early apoptosis in MCF-7 cells as well as a lower proportion of early and late apoptosis in MDA-MB-231 cells. In addition, nuclear morphological changes and loss of mitochondrial membrane potential were observed. In conclusion, a dose of 100 to 1500 ng Se/mL of Se yeast can increase oxidative stress, and stimulate growth inhibitory effects and apoptosis induction in breast cancer cell lines, but does not affect non-tumorigenic cells.
Keywords: Selenium Yeast, Oxidative Stress, Apoptosis, Breast Cancer Cells
1. Introduction
Breast cancer is a complex disease, and its prognosis depends on tumor heterogeneity and host characteristics. Several prognostic factors such as estrogen receptor (ER)-alpha, progesterone receptor (PR), and human epidermal growth factor receptor-2 (HER-2) expression have been well identified as predictive and therapeutic criteria. Three major subtypes of breast cancers include positive expression of ER and PR, HER-2-positive expression, and triple-negative tumors (negative expression of ER, PR, and HER-2)[1]. Current treatment strategies, which combine treatments with surgery, radiotherapy, and anti-cancer drugs, have certainly improved survival rate, but many patients still develop metastatic diseases [2]. Different from the conventional treatment strategies, nutritional supplement interventions have hold promises for cancer patients and have become the alternative treatments.
Selenium (Se) is well-known as an essential trace mineral and an important component of antioxidant enzyme glutathione peroxidase (GPx), which is critical for scavenging reactive oxygen species (ROS) and maintaining the oxidation-reduction (redox) balance [3]. Recent studies have shown that oxidative stress is related to unrepaired DNA damage, mutating tumor suppressor genes, carcinogenesis, and tumor's angiogenesis [4], whereas oncogene-induced oxidative stress activates the tumor suppressor pathway, and in turn contributes to elimination of tumor cells [5]. There was new evidence indicating the protective effects of Se on oxidative damage and its anti-cancer properties [6]. However, the possible mechanisms of Se involved in anti-cancer activity remain unknown.
Zhao et al [7] have indicated that nutritional levels of Se supplementation can maintain the balance of anti-oxidants and pro-oxidants system; whereas the anti-cancer action of Se may require supra-nutritional levels. Recently, the facts that oral Se supplementation (400 μg elemental Se per day) in healthy adults and cancer patients received supplements consisting of 1600-3200 μg Se have been documented, and their plasma Se concentrations reached over 1000 ng/mL [8]. Furthermore, several forms of Se compounds have been proved to stimulate apoptosis in some types of tumor cells. For example, inorganic selenite induces pro-oxidative apoptotic death of tumor cells [9]; whereas selenite is also toxic to normal cells at high dose levels. Organic Se compounds such as methylseleninic acid (MSA) and methyl-selenocysteine (MSC) induced apoptosis of cultured tumor cells; whereas the oxidative capacity of metabolites was found [10,11]. As shown from the studies, it appears that the potential pro-oxidant effect of these Se compounds may be the mechanism that induces apoptotic death of tumor cells. Additionally, Se yeast is often employed in nutritional supplements and there is no direct evidence of toxicity [12]. As shown in an earlier study, supplementation with Se yeast alone or in combination with chemotherapeutic drugs could induce apoptosis in certain tumor cell lines [13]. However, there is limited information regarding the redox potentials of Se yeast in breast tumor cells.
The purpose of the present research was to evaluate the dose-response effects of supra-supplementation of Se yeast on the redox regulation and incidence of apoptosis in human breast tumor cells. Furthermore, the impact of Se compounds (in the forms of Se yeast, MSC, and MSA) on the survival rate and redox potential of primary human mammary epithelial cells (HMEC) was also determined.
2. Materials and Methods
2.1. Cells and Reagents
ER-positive MCF-7 and triple-negative MDA-MB-231 human breast cancer cell lines, and primary HMEC (derived from normal adult mammary glands) were obtained from American Type Culture Collection (Rockville, MD) and Lonza Inc (Walkersville, MD, USA). The MSA (95%) and MSC (95%) were purchased from Sigma-Aldrich (Saint Louis, MO, USA). Se yeast was supplied by Do Well Laboratories Inc (Irvine, CA 92614, USA). All other chemicals were of the highest grade commercially available.
Both MCF-7 and MDA-MB-231 cells were cultured in Dulbecco's modified eagle medium containing 10% fetal bovine serum, and the medium were supplemented with L-glutamine (2 mM), streptomycin (100 μg/mL), penicillin (100 units/mL), and amphotericin B (0.25 μg/mL). In addition, HMEC was cultured in mammary epithelial basal medium containing growth factor. Cells were grown in 25 or 75 cm2 flasks at an atmosphere of 5% CO2 and in humified 95% air, and the medium were changed every other day. Before each experiment, cells were seeded in 12-well or 96-well plates and 10 cm dishes in a humid incubator with 5% CO2. Further, MCF-7 cells were cultured with 17β-estradiol (E2; 10 nM) alone, or a combination of E2 (10 nM) and 4-hydroxy- tamoxifen (Tam; 100 nM). Supplemental Se includes Se yeast (100, 750, and 1500 ng Se/mL), MSA (1500 ng Se/mL), and MSC (1500 ng Se/mL) were chosen. Cells without Se treatment served as controls (Ctrl). Finally, these cells were collected at different time points. All experiments were repeated at least three times.
2.2. Preparation of Selenium Yeast Extraction
0.1 g of Se yeast powder was added to 1x phosphate buffered saline (PBS). Then, we disrupted the yeast cell wall using a bead milling and ultra-sonication methods. After three minutes, the pancreatic enzymes (trypsin and α-chymotrypsin) were added to help with further digestion for 12h in a 37 °C water bath. Finally, the supernatant solution was collected after eliminating the enzyme activity and centrifugation.
2.3. Determination of Se Contents
Based on the protocols from our previous work [3], atomic absorption spectrophotometry (932AA, GBC, Melbourne, Australia) with the accessory hydride generation system (HG 3000, GBC, Melbourne, Australia) was used for determining Se concentrations of Se yeast extraction and medium during the following experiments. In brief, samples were digested for a total of 10.5 h with an initial temperature of 60 °C for 1-1.5 h, followed by increasing temperatures by 20 °C increments and finally heated up to 225 °C for 2 h in a mixture of 3.2 mL nitric acid (16N), and 0.8 mL concentrated perchloric acid. Accuracy of the methods was confirmed by comparing with standard reference materials (SeronormTM level 2, NO0371, Nycomed, Oslo, Norway).
2.4. Nuclear Morphological Observation
Both MDA-MB-231 and MCF-7 cells were incubated in a 3-cm dish at a density of 1 × 105 cells/well. After treatment with various forms of Se for 48 or 72 h, the cells were washed with 1x PBS and fixed in 10% paraformaldehyde for 30 min at room temperature and then washed again with 1x PBS. Additionally, 0.2 % of Triton X-100 was added to increase the permeability of cells, and we used 1% bovine serum albumin to block the reaction. Nuclei staining with 4',6'-diamidino-2-phenylindole (DAPI) was then visualized with a fluorescence microscope (Zeizz Axio Observer Z1; Carl Zeiss Micro Imaging GmbH, Jena, Germany) for determination of nuclear morphological changes.
2.5. Analysis of Cell Viability
5 x 103 cells were seeded in 96-well plates and the cells were treated with different concentrations of Se yeast (100, 750, and 1500 ng Se/mL), MSC (1500 ng Se/mL), or MSA (1500 ng Se/mL) for various periods (24, 48, 72 and 96 h). According to XTT Cell Viability Assay Protocol, XTT labeling mixture was added to each well at a final concentration of 0.5 mg/mL, and then the plate was further incubated for 4 h. XTT-containing media were removed, and a solution containing 10% sodium dodecyl sulfate plus 0.01 M HCl and 5% isobutyl alcohol was added to each well and mixed thoroughly to dissolve the formed crystal formazan. Following that the cells were incubated at 37 °C for another 4 h, the light absorption of samples was measured at 590 nm. Viability was expressed as a percentage of absorbance values in treated cells relative to that in control cells.
2.6. Early/Late Apoptosis Assay
Cells were seeded in 12-well plates for 24 h, followed by treating with different types of Se for 24, 48, 72, and 96 h, respectively. After the exposure period, the harvested cells were washed with 1x PBS and then suspended in the binding buffer. An aliquot (100 μl) of the solution was then incubated with proper amounts of annexin V-fluorescein isothiocyanate (FITC) and propidium iodide (PI) in dark for 5 min at room temperature, and 400 μl of the binding buffer was added to each sample. Finally, the stained cells in each field were analyzed on the flow cytometer (Partec CyFlow ML, Partec GmbH, Münster, Germany) within 0.5 h. The data revealed that intact cells were primarily negative for both annexin V-FITC and PI, and that early apoptotic cells were positive for annexin V-FITC and late apoptotic cells or necrotic cells were positive to both annexin V-FITC and PI.
2.7. Assessment of Mitochondrial Membrane Potential
MDA-MB-231 cells (8 x 104) were seeded in 12-well plates. After 24 h of incubation, cells were treated with different concentrations of Se yeast (100, 750, and 1500 ng Se/mL), MSC (1500 ng Se/mL), and MSA (1500 ng Se/mL) for various periods (24, 48, 72, and 96 h), respectively. The harvested cells were incubated with rhodamine 123 (Rh-123) for 30 min in dark at 37°C, and re-suspended in 1x PBS. Finally, the mitochondrial membrane potential was measured by determining Rh-123 fluorescence in the flow cytometry, and the fluorescence emission at 515 nm was quantified.
2.8. Measurement of intracellular ROS
Lucigenin (bis-N-methylacridinium nitrate)-amplified chemiluminescence is widely used to measure intracellular ROS production. Further, the light emission can be produced when lucigenin is excited by superoxide anion (O2-)[14]. Finally, the addition of antioxidant enzyme SOD to cells should prove that the chemiluminescent signal was derived from O2-. The cultured cells at a density of 1 × 106 cells were initially plated in 10-cm dish for 24 h. The cells were then pre-incubated with Se yeast, MSA or MSC (1500 ng Se), after 6, 12, 24, 48, 72, and 96 h, the cells were washed twice with 1x PBS. The treated cells were further incubated with 1 mL of Lucigenin solution for the quantification of the O2- status. Negative controls were also prepared by replacing cell suspension with 1x PBS. The chemiluminescence measurements were subsequently monitored for 200 sec at 37 °C using CLA-FSI Chemiluminescence Analyzing system (Tohoku Electronic Industrial Co., Ltd., Sendal, Japan). Each assay was performed six times and data are expressed as chemilu- minescence counts per 1x104 cells.
2.9. Measurement of Cytosolic SOD and GPx Activities
The activity of superoxide dismutase (SOD) was determined using a commercial kit (Randox, San Diego). In briefly, MCF-7 (1 x 106) or MDA-MB-231 (5 x 105) cells were initially seeded in 10-cm dish for 24 h. The cells were then incubated with Se yeast, MSA or MSC (1500 ng Se). The harvested cells were washed with 1x PBS and sonicated on ice in a bath-type ultrasonicator. After centrifugation, the obtained supernatants were then collected to determine the enzyme activity at 540 nm using ELISA microplate spectrophotometer Multiskan Ascent, Thermo Labsystems).
In addition, glutathione peroxidase (GPx) activity was performed with GPx assay kits (Cayman Chemical, Ann Arbor, Michigan, USA) as previously described [3], according to the manufacturer's instructions. Oxidized glutathione, produced upon reduction of an organic hydroperoxide by GPx, was recycled to its reduced state by glutathione reductase and NADPH. The oxidation of NADPH to NADP+ is accompanied by a decrease in absorbance at 340 nm; it was directly proportional to the GPx activity. Protein content was also determined using the Coomassie protein assay (Pierce, Rockford, IL, USA) with bovine serum albumin as the standard.
2.10. Statistical Analysis
Data were expressed as mean ± SEM. One-way ANOVA (analysis of variance) followed by post-hoc analysis with Duncan's multiple-range tests was used to determine statistically significant difference (p < 0.05).
3. Results
3.1. Effect of Growth Inhibition by Se
Compared with the un-treated group, the SeY groups treated with different concentrations of 100, 750, or 1500 ng Se/ml (in the form of Se yeast) significantly inhibited the growth of E2-incubated MCF-7 cells treated with and without tamoxifen in a dose- and time-dependent manner (Figure 1, p < 0.05). The growth inhibitory effect of these MCF-7 cells was also observed in both MSA and MSC groups (1500 ng Se/mL) when cells were treated with Se for 24, 48, 72, and 96 h, respectively. Furthermore, the inhibitory effects of Se-treated groups were observed as follow: MSA 1500 > MSC 1500 > SeY 1500 > SeY 750 > SeY 100. And, the E2+Tam group treated with tamoxifen exhibited growth-inhibitory effects in the MCF-7 cells under E2 stimulated condition; the combination of Se with tamoxifen would further increase the inhibitory effects of tamoxifen alone.
Growth inhibition in (A)MCF-7 cells cultured with E2, (B)MCF-7 cells cultured with E2 and tamoxifen, (C)MDA-MB-231 cells, and (D)HEMC after 24, 48, 72, and 96 h incubation by Se yeast, MSC, and MSA. Results are expressed as relative reading (mean ± SD) at least from three independent experiments. Ctrl=controls; SeY 100=selenium yeast (100 ng Se/ml); SeY 750=selenium yeast (750 ng Se/ml); SeY 1500=selenium yeast (1500 ng Se/ml); MSC 1500=methylselenocysteine (1500 ng Se/ml); MSA 1500=methylseleninic acid (1500 ng Se/ml); E2=17β-estradiol; E2 + TAM=17β-estradiol and tamoxifen.
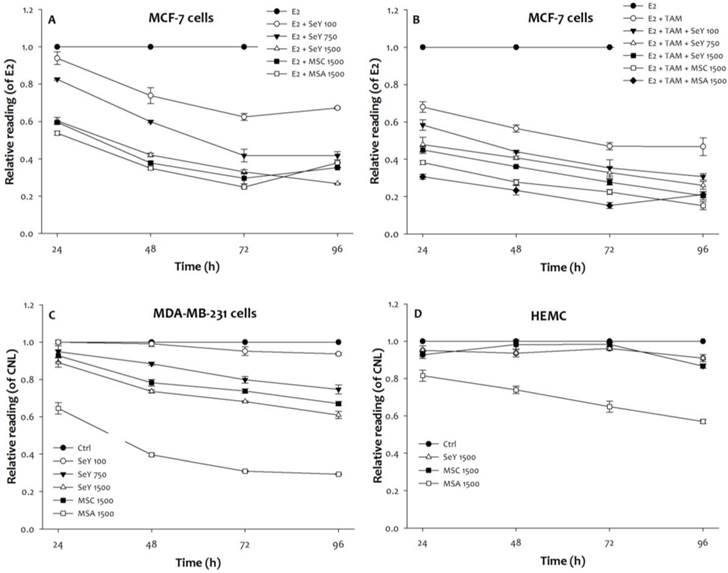
The growth-inhibitory properties of MDA-MB-231 cells after 24 h of incubation were observed as follow: MSA 1500 > MSC 1500, SeY 1500 > CNL, SeY 100, and SeY 750. After 48 and 72 h of incubation, SeY 750 group had increased inhibitory effects as compared with the SeY 100 and Ctrl groups. After 96 h of incubation, there was higher growth inhibitory in all groups treated with Se as compared with Ctrl group.
Additionally, we observe the incubated HMEC cells of MSA 1500 group at above incubation periods have a significantly growth inhibition as compared with that value of MSC 1500 and SeY 1500 groups. Non-significant influence on the growth-inhibitory effects of cells on both MSC 1500 and SeY 1500 groups was observed.
3.2. Changes of Se Levels in Medium
Compared with initial values, MCF-7, MDA-MB-231, or HMEC cells treated with MSA (1500 ng Se/mL) exhibits an accelerated decline in medium Se concentrations (Figure 2). By contrast, there was non-significant sharp decrease in medium Se concentrations from 0 to 96 h at Se yeast or MSC (1500 ng Se/mL) treatments.
The Se levels of media was quantified in (A)MCF-7 cells cultured with E2, (B)MCF-7 cells cultured with E2 and tamoxifen, (C)MDA-MB-231 cells, and (D)HEMC after 0, 6, 12, 24, 48, 72, or 96 h incubation. Results are expressed as relative reading (mean ± SD) at least from three independent experiments. SeY 1500=selenium yeast (1500 ng Se/ml); MSC 1500=methylselenocysteine (1500 ng Se/ml); MSA 1500=methylseleninic acid (1500 ng Se/ml); E2=17β-estradiol; E2 + TAM=17β-estradiol and tamoxifen.
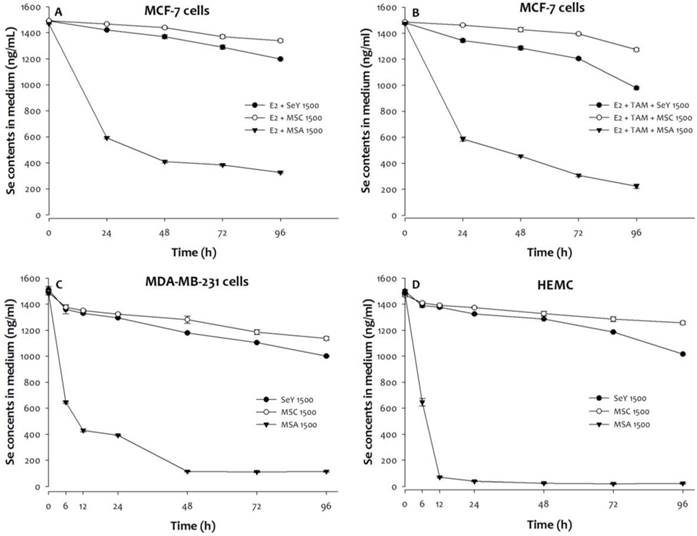
A significant decrease in media concentrations of Se was found in MSA 1500 group and followed by SeY 1500 and MSC 1500 groups when MCF-7 cells incubation was under a combination with E2 and tamoxifen condition. In addition, the tamoxifen treatment further reduced the Se concentrations in media of three different forms of Se groups.
3.3. ROS production induced by Se
Compared with the untreated MCF-7 cells, more superoxide product generated in all Se-treated groups was observed. Moreover, the SeY 1500 and the MSC 1500 groups have lower concentrations of superoxide than the MSA 1500 group at any time point (Figure 3). The MDA-MB-231 cells in the MSC 1500 and the SeY 1500 groups showed lower superoxide levels than those in the MSA 1500 group at the 6th, 12th, 24th, 48th, and 72th h. After 96 h of incubation, there were no significant differences in superoxide product among the different groups. For HMEC cells, Se yeast or MSC (1500 ng Se/mL) did not cause a significant increase in superoxide production; whereas MSA treatment induced cells to produce more superoxide radicals.
Superoxide generation in (A)MCF-7 cells cultured with E2, (B)MCF-7 cells cultured with E2 and tamoxifen, (C)MDA-MB-231 cells, and (D)HEMC after 6, 12, 24, 48, 72, and 96 h incubation, as well as (E)MCF-7 cells and (F)MDA-MB-231 cells after incubation of 48 h by Se yeast, MSC, and MSA. Results are expressed as relative reading (mean ± SD) at least from three independent experiments. SeY 1500=selenium yeast (1500 ng Se/ml); MSC 1500=methylselenocysteine (1500 ng Se/ml); MSA 1500=methylseleninic acid (1500 ng Se/ml).
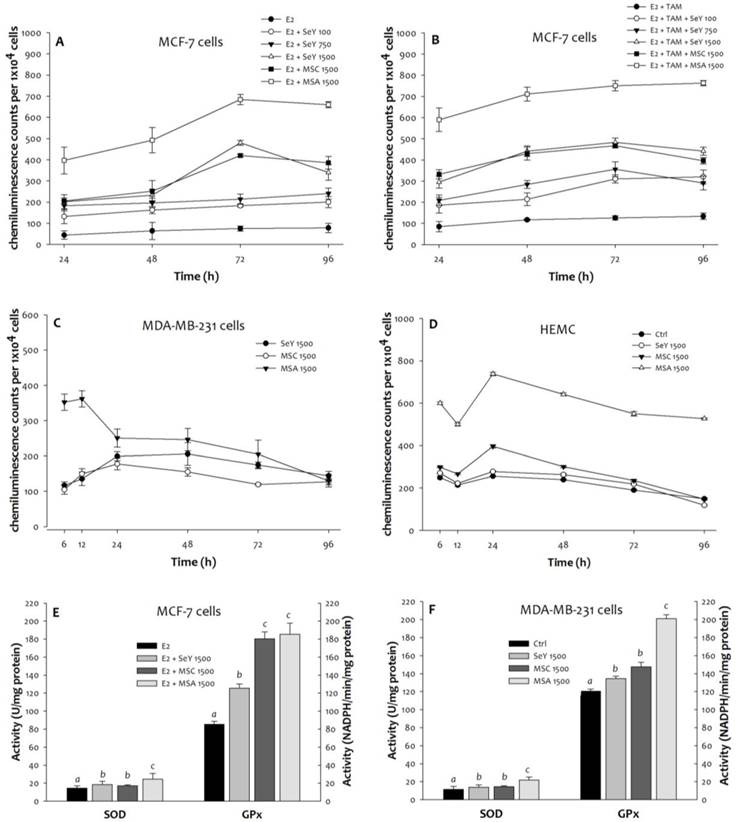
3.4. Antioxidant Enzyme Activities of GPx and SOD
Throughout the time course experiments, we further examined the activities of antioxidant enzymes. After 48 h of incubation, the MCF-7 cells in the MSA 1500 group showed a significantly higher SOD activity, as well as markedly higher GPx activity in both MSA 1500 and MSC 1500 groups than the SeY 1500 group (Figure 3). For MDA-MB-231 cells, both SeY 1500 and MSC 1500 groups had lower activity of SOD and GPx as compared with MSA 1500 group.
3.5. Early/Late Apoptosis Induction by Se
For early apoptosis, three SeY groups showed a raised degree of the MCF-7 cell under E2 with/without tamoxifen condition in a time- and dose-dependent manner. The SeY 1500 group has also significantly higher early apoptosis when compared with the MSA 1500 and MSC 1500 groups (Figure 4). In contrast, higher late apoptosis of MCF-7 cells in both MSA and MSC groups than those by SeY groups were observed.
Se compounds induced early and late apoptosis in (A)(B)MCF-7 cells cultured with E2, (C)(D)MCF-7 cells cultured with E2 and tamoxifen, and (E)(F) MDA-MB-231 cancer cells after 24, 48, 72, and 96 h incubation. Results are expressed as relative reading (mean ± SD) at least from three independent experiments. Ctrl=controls; SeY 100=selenium yeast (100 ng Se/ml); SeY 750=selenium yeast (750 ng Se/ml); SeY 1500=selenium yeast (1500 ng Se/ml); MSC 1500=methylselenocysteine (1500 ng Se/ml); MSA 1500=methylseleninic acid (1500 ng Se/ml); E2=17β-estradiol; E2 + TAM=17β-estradiol and tamoxifen.
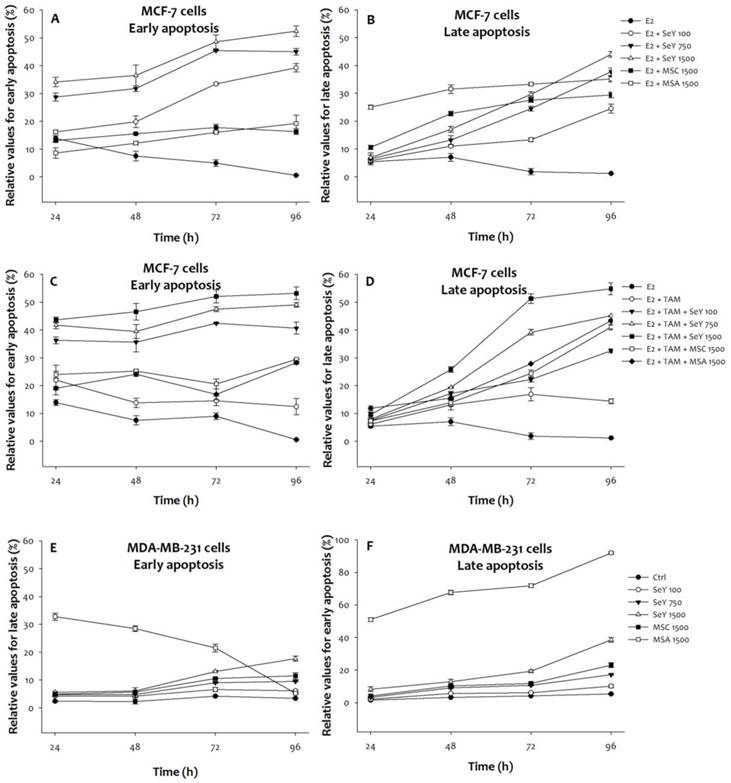
On the other hand, MSA 1500 group induced higher early apoptosis of MDA-MB-231 cells at the incubation of 24, 48 h, and 72 h, respectively. There were no significant differences of early apoptosis between SeY 1500 and MSC 1500 groups. Treatment with SeY induced early apoptosis of MDA-MB-231 cells in a dose-dependent manner at 72 h. After 96 h of incubation, SeY 1500 and MSC 1500 groups have the higher percentage of early apoptosis than MSA 1500 group. For late apoptosis, MSA 1500 group induced higher values at all different time points. Furthermore, the late apoptosis were observed as follow: MSA 1500 > SeY 1500 > MSC 1500 > SeY 750 > SeY 100.
3.6. Morphology Changes by Se
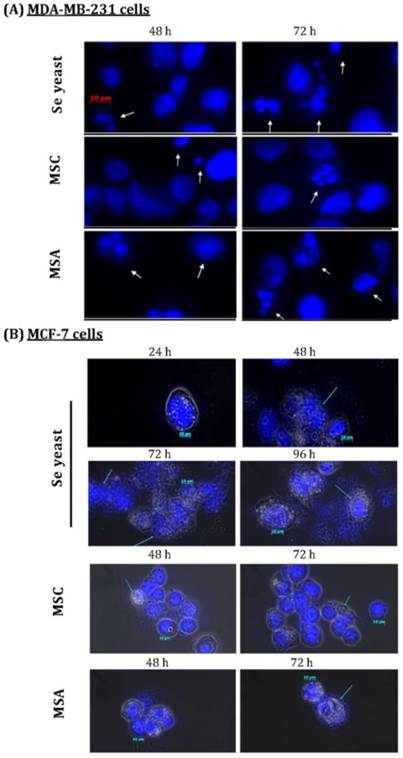
Exposure to Se showed morphology changes in the MCF-7 cells. Se yeast-treated cells (1500 ng Se/ml) had karyorrhexis, nucleus condensed, and DNA fragments at the 48th h, as well as apoptotic bodies at the 72th and 96th h (Figure 5). After MSC treatment, only the increased condensed was found. The E2-incubated cells in MSA 1500 group showed condensed nucleus at the 72th h, and apoptotic bodies at the 96th h; whereas apoptotic bodies occurred at the 72th h when incubated cells incubated with E2 and tamoxifen. On the other hand, three forms of Se treatment can show apoptotic bodies formation of MDA-MB-231 cells at the 48th h. Additionally, Se yeast and MSC-exposed cells have lower apoptotic bodies as compared with MSA.
Nuclear morphological changes induced by different Se compounds in (A)MDA-MB-231 cancer cells, as well as (B)MCF-7 cells cultured with E2 for different time periods. DAPI staining shows apoptotic nuclei; human breast cancer cells were exposure to different Se compounds (1500 ng Se/ml); MSC=methyl- selenocysteine; MSA=methylseleninic acid.
3.7. Mitochondrial Membrane Potential Changes by Se
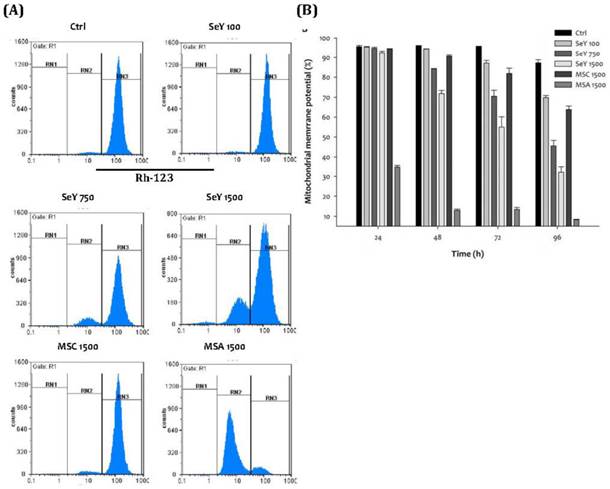
After incubation of 48 h, Se yeast treatment showed a decreased degree of normal mitochondrial membrane potential of the MDA-MB-231 cells in a dose-dependent manner. Furthermore, the SeY 1500 group has lower percentage of normal mitochondrial membrane potential as compared with the MSC 1500 group. The lowest percentages of normal mitochondrial membrane potential at the MSA 1500 group throughout the experimental period were observed (Figure 6).
(A)Depolarization of mitochondrial membrane potential in MDA-MB-231 cells treated with Se compounds after 48 h incubation; (B)Mitochondrial membrane potential decrease caused by treated with Se compounds after 24, 48, 72 and 96 h incubation. Results are expressed as relative reading (mean ± SD) at least from three independent experiments. Ctrl=controls; SeY 100=selenium yeast (100 ng Se/ml); SeY 750=selenium yeast (750 ng Se/ml); SeY 1500=selenium yeast (1500 ng Se/ml); MSC 1500=methylselenocysteine (1500 ng Se/ml); MSA 1500=methylseleninic acid (1500 ng Se/ml).
4. Discussion
In the present preliminary study, Se yeast (100, 750, and 1500 ng Se/ml; equivalent to 1.3, 9.5, and 19 μM Se) can inhibit cell growth and induce apoptosis in a time- and dose-dependent manner in ER-positive MCF-7 and triple-negative MDA-MB-231 human breast cancer cells. Se yeast (19 μM Se) induce lower amounts of ROS that may be the important cause of triggering early apoptosis as compared with the same level of MSA. Se yeast had the minimal effects on the growth inhibition and the production of ROS of non-tumorigenic HMEC cells. In contrast, MSA exposure led to excessive generation of ROS; the phenomenon would help explain why MSA accelerate tumor cell growth-inhibitory effect and induction of late apoptosis. Additionally, MSA treatment to HMEC cells resulted in a dramatic increase in growth-inhibitory effect and ROS production.
Growth inhibition by Se
A number of studies have demonstrated that the anti-cancer potential of Se focused primarily on several chemical forms include selenite, MSA and MSC, as well as their supra-nutritional levels [15,16]. Qi et al [17] pointed out that MSA (above 3.2 μM levels) could suppress the viability and induce the cell-cycle arrest of MDA-MB-231 cells. Moreover, the fact that growth inhibition of MCF-7 cells treatment with selenite (above 20 μM) or MSA (above 2 μM) was observed [18]. MSA (2.5 μM) exhibited a greater response on the proliferation inhibition effect and apoptosis in mouse mammary hyperplastic epithelial cell lines than that of MSC (25-50 μM)[19]. Obviously, MSC has lower anti-cancer effects as compared with MSA; the levels of the latter required for anti-cancer effects are above 2 μM.
Methylselenol, a major mono-methylated metabolite of MSA and MSC, may contribute to the anti-cancer activity of Se. Compared with MSA, an additional enzyme cysteine conjugate β-lyase is required for the conversion of MSC to methylselenol. Recently, Se yeast is often employed in nutritional supplements, which provides highly levels of bioactive organic forms of Se including selenomethionine, selenocysteine, MSC, and some peptides [20]. Likewise, the existence of methioninase and γ-lyase is necessary for selenomethionine to transform into methylselenol [11]. Thus, whether Se yeast can inhibit the growth of cancer cells similar to MSA or MSC, the matter will be further investigated.
In the present investigation we had found lower level of Se yeast (100 ng Se/ml; equivalent to 1.3 μM Se) can inhibit the growth of breast cancer cells and suggested that inhibitory effects by Se yeast through other mechanisms when compared with MSA. Furthermore, the comparison of the tumor growth-inhibitory effects by Se yeast and MSA (equal to 1500 ng Se/ml) were also determined. MSA exposure exhibited higher growth inhibition in the MCF-7 and MDA-MB-231 cells than Se yeast. The explanation for higher growth-inhibitory capacity may be partially related to higher Se uptake by these breast cancer cells. There was little information regarding the comparison of Se absorption among different Se compounds in vitro. The dose-response experiments were not available, which we consider to be a limitation. On the other hand, the intracellular metabolites production from Se compounds may be helpful in explaining our present results that both MSC and Se yeast have lower growth-inhibitory action in the treated cancer cells than those with MSA.
Oxidative stress by Se
The mechanism of Se, that is responsible for anti-cancer effects still remain largely unclear. It has been known for a long time that the diversely roles at Se-induced potential redox reaction are significantly linked to the risks and chemotherapy of cancers. MSA significantly decreased E2-induced ROS accumulation of T47D breast cancer cells [21]. Selenite also inhibited electromagnetic radiation-induced oxidative stress and reduced the mitochondrial dysfunction in MDA-MB-231 cells [22]. By contrast, some studies showed that selenite exposure induce the apoptosis of cancer cells by producing superoxide radicals [23,24]. The metabolites of selenite, Se nanoparticles, could induce oxidative stress that contributed to the cytotoxicity in cancer cells [25,26]. The metabolites of Se compounds may directly affect mitochondrial membranes and interfere with the redox status, which subsequently result in cell cycle arrest of cancer cells [23,26].
Our present results also indicated that the MSA-exposed MCF-7, MDA-MB-231, and HMEC cells have showed a greatest superoxide production as compared to the same level of Se yeast (1500 ng Se/ml). MSA treatment also led to increases in the activities of antioxidant enzymes SOD and GPx in these breast cancer cells. It appeared that the significant rise in ROS production may be attributed to Se metabolite formed quickly after MSA treatment. On the other hand, Trachootham et al [27] have previously demonstrated the existence of differential redox potential between normal and cancer cells. Relative to MCF-7 or MDA-MB-231 cells, the presence of high level of Se yeast or MSC did not increase superoxide production and reduce cellular growth that may be associated with the selenoprotein antioxidant protection in non-tumor HMEC cells. It will be necessary to further study the expression of selenoproteins in these cell lines, although the superoxide generation may be altered by culture conditions and the types of media.
Early/Late Apoptosis Induction
An excessive amount of ROS may disturb NAD+/NADH redox balance and mitochondrial function, as well as affect the cell growth [28]. The fact that increasing NAD+/NADH ratio would attenuate metastatic activity and facilitate apoptosis in cancer cells [29]. Apoptosis is a stage-dependent process such as early and late apoptotic events, as well as apoptotic cells undergo changes in morphology and biochemical events. A number of studies have showed that the anti-cancer properties of Se may be attributable to ROS-induced cancer cells apoptosis [25]. Swede et al [30] have pointed out that MSA (2.5 and 10 μM) inhibited lung cancer cell growth and induced late apoptosis after 24 h of incubation. In addition, exposure of 2.5 μM of MSA for 72 h induced the early apoptosis in other lung cancer lines [31].
The present results showed that Se yeast (at 1.3, 9.5, and 19 μM Se) or MSC exhibits higher early apoptosis, and that MSA (at 19 μM Se) induces higher late apoptosis of MCF-7 cells. It appeared that Se yeast caused moderate superoxide generation, which may initiate early apoptosis when compared with MSA; whereas MSA-induced more severe oxidative stress may significantly disrupt NAD+/NADH balance, which in turn led to early apoptotic cells become late apoptotic cells quickly. It is interesting to note that MCF-7 cells exposed to Se yeast, but not MSA, resulted in a dose-dependent induction of early apoptosis.
On the other hand, Se yeast caused a gradual increase in the time- and dose- dependent early/late apoptosis in the MDA-MB-231 cells. MSA (at 19 μM Se) treatment also induced markedly higher onset of both early and late apoptosis at a time course experiment; whereas MSA had less early apoptotic events at the 96th h. That is, after exposure to MSA resulted in cytotoxicity may cause large numbers of cells directly convert into late-stage apoptotic cells. For future research, the dose-response of MSA- induced apoptosis in differential breast cancer cells will be further confirmed.
Mitochondrial Membrane Potential
Proper mitochondrial membrane potential is critical for living cells. Se compounds such as selenite and selenomethionine can increase superoxide production and induce mitochondrial membrane depolarization [32]. Selenite induced a rapid superoxide burst and p53 activation, leading to loss of mitochondrial membrane potential [33]. MSA could aggressive mitochondrial membrane dysfunction in MDA-MB-231 cells [17]. Heikal [34] indicated that increased mitochondrial ROS production along with reduced mitochondrial membrane potential and intracellular NADH levels were observed during apoptosis process.
For further analysis of apoptosis, we evaluated the effects of different Se compounds on mitochondrial potential. MDA-MB-231 cells exposed to Se yeast or MSC cause loss of mitochondrial membrane potential that are less severe than that by same dose of MSA. All three forms of Se also altered mitochondrial membrane potential in a time-dependent manner. Additionally, cells exposed to Se yeast (9.5 μM Se) for 48 h have higher mitochondrial potential damage compared with those exposed to MSC (19 μM Se). The other mechanisms may involve the loss of mitochondrial potential by Se yeast. Furthermore, the morphological features that relate to apoptotic response in these breast cancer cells exposed to these Se compounds were also observed.
5. Conclusion
The present findings contribute to the field's understandings of Se yeast acting on human breast cancer cells in vitro. Se yeast (100, 750, and 1500 ng Se/mL) can induce apoptosis in human breast cancer cells. Exposure to Se yeast may cause Se metabolites in ER-positive MCF-7 and triple-negative MDA-MB-231 cancer cells to increase, which induce the generation of moderate amounts of ROS. Furthermore, these ROS production would lead to loss of mitochondrial membrane potential and speed up the large amounts of apoptotic cells in an early phase of apoptosis. And, less ROS production in HEMC cells by Se yeast may be eliminated by the synthesis of antioxidant selenoproteins. Therefore, exposure of Se yeast at the supra-nutritional levels may be more safe and effective anti-cancer agent as compared with MSA. Future studies are still needed to determine the dose-response effects of Se on the toxicity and early/late apoptosis in the breast cancer cells and HEMCs cells. More importantly, future studies are required to apply one design on animal model experiments.
Acknowledgements
The research is funded by Do Well Laboratories Inc., Irvine CA, U.S.A. (Irvine, CA 92614, USA). Great appreciation is extended to everyone who has contributed to the project.
Author Contributions
Conceived and designed the experiments: CHG, SH, PCC. Performed the experiments: CHG, SH, PCC. Analyzed the data: CHG, MYS, FCH. Contributed reagents/materials/ analysis tools: CHG, PCC. Wrote the paper: CHG
Competing Interests
The authors have declared that no competing interest exists.
References
1. Herrera AC, Panis C, Victorino VJ, Campos FC, Colado-Simão AN, Cecchini AL, Cecchini R. Molecular subtype is determinant on inflammatory status and immunological profile from invasive breast cancer patients. Cancer Immunol Immunother. 2012;61:2193-201
2. Schnitt SJ. Classification and prognosis of invasive breast cancer: from morphology to molecular taxonomy. Mod Pathol. 2010;23(Suppl 2):S60-4
3. Guo CH, Hsia S, Chen PC. Distribution of selenium and oxidative stress in breast tumor-bearing mice. Nutrients. 2013;5:594-607
4. Pani G, Galeotti T, Chiarugi P. Metastasis: cancer cell's escape from oxidative stress. Cancer Metastasis Rev. 2010;29:351-78
5. Inokuma T, Haraguchi M, Fujita F, Tajima Y, Kanematsu T. Oxidative stress and tumor progression in colorectal cancer. Hepatogastroenterology. 2009;56:343-7
6. Yazdi MH, Mahdavi M, Kheradmand E, Shahverdi AR. The preventive oral supplementation of a selenium nanoparticle-enriched probiotic increases the immune response and lifespan of 4T1 breast cancer bearing mice. Arzneimittel- forschung. 2012;62:525-31
7. Zhao R, Domann FE, Zhong W. Apoptosis induced by selenomethionine and methioninase is superoxide mediated and p53 dependent in human prostate cancer cells. Mol Cancer Ther. 2006;5:3275-84
8. Reid ME, Stratton MS, Lillico AJ, Fakih M, Natarajan R, Clark LC, Marshall JR. A report of high-dose selenium supplementation: response and toxicities. J Trace Elem Med Biol. 2004;18:69-74
9. Zhou N, Xiao H, Li TK, Nur-E-Kamal A, Liu LF. DNA damage-mediated apoptosis induced by selenium compounds. J Biol Chem. 2003;278:29532-7
10. Jung U, Zheng X, Yoon SO, Chung AS. Se-methylselenocysteine induces apoptosis mediated by reactive oxygen species in HL-60 cells. Free Radic Biol Med. 2001;31:479-89
11. Brozmanová J, Mániková D, Vlčková V, Chovanec M. Selenium: a double-edged sword for defense and offence in cancer. Arch Toxicol. 2010;84:919-938
12. Jacobson GK. Dietary selenium and selenized yeast. Agro Food Industry Hi Tech. 2010;21(n4):14-6
13. Vadgama JV, Wu Y, Shen D, Hsia S, Block J. Effect of selenium in combination with Adriamycin or Taxol on several different cancer cells. Anticancer Res. 2000;20:1391-414
14. Dikalov S, arrison DG. Methods for detection of mitochondrial and cellular oxygen species. Antioxid Redox Signal. 2014;20:372-82
15. Li GX, Lee HJ, Wang Z, Hu H, Liao JD, Watts JC, Combs GF Jr, Lü J. Superior in vivo inhibitory efficacy of methylseleninic acid against human prostate cancer over selenomethionine or selenite. Carcinogenesis. 2008;29:1005-12
16. Chen YC, Prabhu KS, Das A, Mastro AM. Dietary selenium supplementation modifies breast tumor growth and metastasis. Int J Cancer. 2013;133:2054-64
17. Qi Y, Fu X, Xiong Z, Zhang H, Hill SM, Rowan BG, Dong Y. Methylseleninic acid enhances paclitaxel efficacy for the treatment of triple-negative breast cancer. PLoS One. 2012;7:e31539
18. de Miranda JX, Andrade Fde O, Conti Ad, Dagli ML, Moreno FS, Ong TP. Effects of selenium compounds on proliferation and epigenetic marks of breast cancer cells. J Trace Elem Med Biol. 2014;28:486-91
19. Ip C, Thompson HJ, Zhu Z, Ganther HE. In vitro and in vivo studies of methylseleninic acid: Evidence that a monomethylated selenium metabolite is critical for cancer chemoprevention. Cancer Res. 2000;60:2882-6
20. Gammelgaard B, Gabel-Jensen C, Stürup S, Hansen HR. Complementary use of molecular and element-specific mass spectrometry for identification of selenium compounds related to human selenium metabolism. Anal Bioanal Chem. 2008;390:1691-706
21. Okuno T, Miura K, Sakazaki F, Nakamuro K, Ueno H. Methylseleninic acid (MSA) inhibits 17β-estradiol-induced cell growth in breast cancer T47D cells via enhance- ment of the antioxidative thioredoxin/thioredoxin reductase system. Biomed Res. 2012;33:201-10
22. Kahya MC, Nazıroğlu M, Çiğ B. Selenium reduces mobile phone (900 MHz)-induced oxidative stress, mitochondrial function, and apoptosis in breast cancer cells. Biol Trace Elem Res. 2014;160:285-93
23. Xiang N, Zhao R, Zhong W. Sodium selenite induces apoptosis by generation of superoxide via the mitochondrial-dependent pathway in human prostate cancer cells. Cancer Chemother Pharmacol. 2009;63:351-62
24. Wang Y, Fang W, Huang Y, Hu F, Ying Q, Yang W, Xiong B. Reduction of selenium- binding protein 1 sensitizes cancer cells to selenite via elevating extracellular glutathione: A novel mechanism of cancer-specific cytotoxicity of selenite. Free Radic Biol Med. 2015;79:186-96
25. Zou Y, Niu P, Yang J, Yuan J, Wu T, Chen X. The JNK signaling pathway is involved in sodium-selenite-induced apoptosis mediated by reactive oxygen in HepG2 cells. Cancer Biol Ther. 2008;7:689-96
26. Bao P, Chen Z, Tai RZ, Shen HM, Martin FL, Zhu YG. Selenite-induced toxicity in cancer cells is mediated by metabolic generation of endogenous selenium nanoparticles. J Proteome Res. 2015;14:1127-36
27. Trachootham D, Alexandre J, Huang P. Targeting cancer cells by ROS-mediated mechanisms: a radical therapeutic approach? Nat Rev Drug Discov. 2009;8:579-91
28. Ying W. NAD+/NADH and NADP+/NADPH in cellular functions and cell death: regulation and biological consequences. Antioxid Redox Signal. 2008;10:179-206
29. Santidrian AF, Matsuno-Yagi A, Ritland M, Seo BB, LeBoeuf SE, Gay LJ, Yagi T, Felding-Habermann B. Mitochondrial complex I activity and NAD+/NADH balance regulate breast cancer progression. J Clin Invest. 2013;123:1068-81
30. Swede H, Dong Y, Reid M, Marshall J, Ip C. Cell cycle arrest biomarkers in human lung cancer cells after treatment with selenium in culture. Cancer Epidemiol Biomarkers Prev. 2003;12(11 Pt 1):1248-52
31. Zhong W, Oberley TD. Redox-mediated effects of selenium on apoptosis and cell cycle in the LNCaP human prostate cancer cell line. Cancer Res. 2001;61:7071-8
32. Gonçalves AC, Barbosa-Ribeiro A, Alves V, Silva T, Sarmento-Ribeiro AB. Selenium compounds induced ROS-dependent apoptosis in myelodysplasia cells. Biol Trace Elem Res. 2013;154:440-7
33. Hu H, Jiang C, Schuster T, Li GX, Daniel PT, Lü J. Inorganic selenium sensitizes prostate cancer cells to TRAIL-induced apoptosis through superoxide/p53/Bax- mediated activation of mitochondrial pathway. Mol Cancer Ther. 2006;5:1873-82
34. Heikal AA. Intracellular coenzymes as natural biomarkers for metabolic activities and mitochondrial anomalies. Biomark Med. 2010;4:241-63
Author contact
![]() Corresponding authors: eillyguohk.edu.tw (C-H. Guo); chenpc.hkcom (P-C. Chen), Tel: 886-4-2631-8652 ext. 5807, 5808, Fax: 886-4-2633-8212
Corresponding authors: eillyguohk.edu.tw (C-H. Guo); chenpc.hkcom (P-C. Chen), Tel: 886-4-2631-8652 ext. 5807, 5808, Fax: 886-4-2633-8212