Impact Factor ISSN: 1449-1907
- Issue 9; 2026
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Volume 23; 2026
- Past Issues
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Conclusion
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2015; 12(9):719-726. doi:10.7150/ijms.12612 This issue Cite
Research Paper
Genomic Copy Number Variations in the Myelodysplastic Syndrome and Acute Myeloid Leukemia Patients with del(5q) and/or -7/del(7q)
Rui Zhang1,2, Young-Mi Kim1, Xianfu Wang1, Yan Li2, Xianglan Lu1, Andrea R. Sternenberger1, Shibo Li1 ![]() , Ji-Yun Lee3
, Ji-Yun Lee3 ![]()
1. Department of Pediatrics, The University of Oklahoma Health Sciences Center, Oklahoma City, OK 73104, USA
2. Department of Hematology, The First Affiliated Hospital of China Medical University, Shenyang, Liaoning, P.R. China
3. Department of Pathology, College of Medicine, Korea University, Seoul, South Korea.
Received 2015-5-5; Accepted 2015-8-17; Published 2015-9-1
Abstract
The most common chromosomal abnormalities in myelodysplastic syndrome (MDS) and acute myeloid leukemia (AML) are -5/del(5q) and -7/del(7q). When -5/del(5q) and -7/del(7q) coexist in patients, a poor prognosis is typically associated. Given that -5/del(5q) and/or -7/del(7q) often are accompanied with additional recurrent chromosomal alterations, genetic change(s) on the accompanying chromosome(s) other than chromosomes 5 and 7 may be important factor(s) affecting leukemogenesis and disease prognosis. Using an integrated analysis of karyotype, FISH and array CGH results in this study, we evaluated the smallest region of overlap (SRO) of chromosomes 5 and 7 as well as copy number alterations (CNAs) on the other chromosomes. Moreover, the relationship between the CNAs and del(5q) and -7/del(7q) was investigated by categorizing the cases into three groups based on the abnormalities of chromosomes 5 and 7 [group I: cases only with del(5q), group II: cases only with -7/del(7q) and group III: concurrent del(5q) and del(7q) cases]. The overlapping SRO of chromosome 5 from groups I and III was 5q31.1-33.1 and of chromosome 7 from groups II and III was 7q31.31-q36.1. A total of 318 CNAs were observed; ~ 78.3% of them were identified on chromosomes other than chromosomes 5 and 7, which were defined as 'other CNAs'. Group III was a distinctive group carrying the most high number (HN) CNAs, cryptic CNAs and 'other CNAs'. The loss of TP53 was highly associated with del(5q). The loss of ETV6 was specifically associated with group III. These CNAs or genes may play a secondary role in disease progression and should be further evaluated for their clinical significance and influence on therapeutic approaches in patients with MDS/AML carrying del(5q) and/or -7/del(7q) in large-scale, patient population study.
Keywords: MDS, AML, del(5q), -7/del(7q), Copy number alterations
Introduction
Whole or partial deletion of chromosomes 5 and 7 [-5/del(5q) and -7/del(7q)] are considered to be the most recurrent genetic abnormalities consisting of 10-20% of myeloid malignancies including both myelodysplastic syndrome (MDS) and acute myeloid leukemia (AML) [1-3]. It has been proposed that haploinsufficiency of tumor suppressor genes on the common deleted regions (CDRs) of chromosomes 5 and 7 contributes to the pathogenesis of MDS and AML[4,5]. However, the exact mechanism and genes being affected are still under investigation [6]. The prognostic value of -5/del(5q) and -7/del(7q) as the sole abnormality is poor in most MDS and AML cases, except del(5q) syndrome in MDS that has a favorable prognosis [7-9]. Given that -5/del(5q) and/or -7/del(7q) often are accompanied with additional recurrent chromosomal alterations [10-12], genetic change(s) on the accompanying chromosome(s) other than chromosomes 5 and 7 may be important factor(s) affecting leukemogenesis and disease prognosis. Recently, it has been reported that alterations of the XPD and TP53 genes were associated specifically with the risk of developing AML with -5/del(5q) and/or -7/del(7q) [13,14]. Moreover, the significance of the co-existence of -5/del(5q) and -7/del(7q) in the progression of MDS and AML needs to be explored since ~40% of MDS and AML cases with-5/del(5q) also have -7/del(7q) [10,11].
Therefore, using an integrated analysis of karyotype, fluorescence in situ hybridization (FISH) and array comparative genomic hybridization (CGH) results in the present study, we evaluated interesting genes in the smallest region of overlap (SRO) of chromosomes 5 and 7 as well as copy number alterations (CNAs) on the other chromosomes. Moreover, the link between the genomic alterations and del(5q) and/or -7/del(7q) was investigated by categorizing the cases into three groups based on the abnormalities of chromosomes 5 and 7. Group I consisted of cases only with del(5q), group II were cases only with -7/del(7q) and group III included the cases with concurrent del(5q) and del(7q).
Materials and Methods
Patient samples
This research project was approved by both the Ethical Committee of the First Affiliated Hospital of China Medical University and the Institutional Review Board (IRB) at the University of Oklahoma Health Sciences Center (OUHSC) (IRB#13100).
Retrospectively, twenty six samples were collected from 2006 - 2010 at the Genetics Laboratory at OUHSC and found to be positive for del(5q) and/or -7/del(7q) by conventional cytogenetic, FISH and/or array CGH analyses. Of the 26 cases, twenty two were bone marrow and four were leukemic blood samples with initial diagnoses of MDS (n=6) or AML (n=20). The diagnosis was made according to the criteria of the French-American-British (FAB) Cooperative Group. The ratio of male to female patients was 17 to 9 and the median age of the patients was 59 years old, ranging from 2 to 73 years old (Table S1).
Conventional cytogenetics and FISH
Overnight cultures of 22 [unstimulated bone marrow (n=18) and leukemic blood (n=4)] out of a total of 26 samples were established and harvested according to our standard laboratory protocols. Chromosome studies were not performed on the remaining four bone marrow samples because it was not requested by physicians. Chromosome preparations were treated and stained by Trypsin-Leishman (GTL) banding. The chromosomal abnormalities were described according to the International System for Human Cytogenetic Nomenclature (ISCN). Subsequent FISH analyses were performed on four cases (cases 4, 11, 15 and 18) using a series of probes which included LSID5S23, D5S721(5p15.2)/EGR1(5q31) (cases 4 and 15), LSI ELN(7q11.23)/D7S486(7q31)(cases 11, 15 and 18), CEP7 (case 15) (Abbott Molecular Inc., Des Plaines, IL) and homebrewed probe RP11-836E15 (21q22.3) (case 15) (BAC clone from Invitrogen Corporation, Carlsbad, CA) according to our standard laboratory protocols [15].
Array CGH
Genomic DNA was isolated from all 26 samples using a commercially available DNA extraction kit (Puregene blood kit, QIAGEN Inc., Valencia, CA). The manufacturer's protocol was followed on a 720k oligonucleotides chip purchased from Roche/NimbleGen System Inc. (Madison, WI). Commercially available pooled normal control DNA was used for reference (Promega Corporation, Madison, WI). The patient DNA and the reference DNA were labeled with either Cyanine 3 (Cy-3) or Cyanine 5 (Cy-5) by random priming (Trilink Biotechnologies, San Diego, CA) and then hybridized to the chip via incubation in the MAUI hybridization system (BioMicro Systems, Salt Lake City, UT). After the 40-hour hybridization at 42°C, the slides were washed and scanned using a MS200 scanner (Roche NimbleGen System Inc., Madison, WI). NimbleScan version 2.4 and the SignalMap version 1.9 were applied for data analysis (NimbleGen System Inc, Madison, WI). The genomic locations were retrieved from the National Center for Biotechnology Information (NCBI) build 36 (hg 18). Frequently affected regions recently detected as copy number polymorphisms (CNPs) were excluded from data analysis according to the CNP database generated in our laboratory and the Database of Genomic Variants (http://projects.tcag.ca/variation).
Statistical analysis
Correlation of genomic CNAs and del(5q) and/or -7/del(7q) were assessed using Fisher's exact test for qualitative variables and the Chi-square test for quantitative factors. All statistical analyses were performed using SPSS version 17.0 and a p-value ≤0.05 was considered statistically significant.
Results
Grouping of del(5q) and/or -7/del(7q) by integrated analysis
The integrated analysis of 26 cases positive for del(5q) and/or -7/del(7q) was performed by combining karyotype, FISH and array CGH results (Table S1). Of the 26 cases, four cases (cases 4, 11, 15 and 18) were analyzed by GTL banding, FISH and array CGH, 18 cases were analyzed by GTL banding and array CGH and four cases (cases 6, 17, 25 and 26) were analyzed only by array CGH. There is a discrepancy in the results of GTL banding and array CGH analyses due to the different levels of resolution of each technique. The cases determined to be monosomy 5 (cases 4, 5, 21 and 23) or monosomy 7 (cases 19, 21 and 24) by GTL banding analysis were further classified as del(5q), del(7q) or rearrangements involving chromosomes 5 or 7 by array CGH and/or FISH. Three cases originally defined as derivative chromosomes 5 (case 24) and 7 (cases 20 and 23) by GTL banding were also refined as del(5q) and del(7q) after array CGH analysis. The karyotype analysis of case 17 only showed constitutional trisomy 21 (Down syndrome), whereas array CGH not only confirmed the gain of chromosome 21 but also uncovered additional genomic imbalances, including the loss of chromosome 7 which was confirmed by FISH. Additionally, one case (case 3) with a del(5)(q31q35) determined by GTL banding analysis was re-designated as a large gain of 5p combined with a cryptic loss of 5q by array CGH. Based on the results and to better understand the association between CNAs and del(5q) and/or -7/del(7q), the cases were categorized into three groups based on the abnormalities of chromosomes 5 and 7. The first group consisted of six cases of del(5q), the second group included 11 cases with -7/del(7q) and the third group contained nine cases that had both del(5q) and del(7q).
Total CNAs detected by array CGH
A total of 318 CNAs from 26 cases were observed with sizes ranging from 0.028Mb to 170.9 Mb which encompassing gene(s); 32.7% (104/318) of the CNAs were gains and 67.3% (214/318) of the CNAs were losses. Cryptic CNAs, sizes smaller than 5Mb, accounted for 50.9% (162/318), which were undetectable by GTL banding (Table S2, Figure S1). Of these cryptic CNAs, 63 were gains and 99 were losses. Since the average number of CNAs per case was 12 (ranging from 1 to 40), the cases with 12 or more CNAs were defined as high number (HN) CNAs. The ratio of the number of cases with HN CNAs to the number of total cases of each group was compared between the three groups (Table 1). Group III had the most cases with HN CNAs; group II did not include any cases with HN CNAs. These two groups (II and III) were the only groups statistically different (p ≤0.05) from each other based on the Chi-square test results.
Comparison of complex CNAs between the three groups
| Group I | Group II | Group III | p value | |||
|---|---|---|---|---|---|---|
| I vs III | II vs III | I vs II | ||||
| Number of cases with HN CNAs / number of total cases in each group | 2/6 | 0/11 | 8/9 | 0.0889 | ≤ 0.0001 | 0.1103 |
Delineation of SROs of del(5q) and -7/del(7q) in the three groups
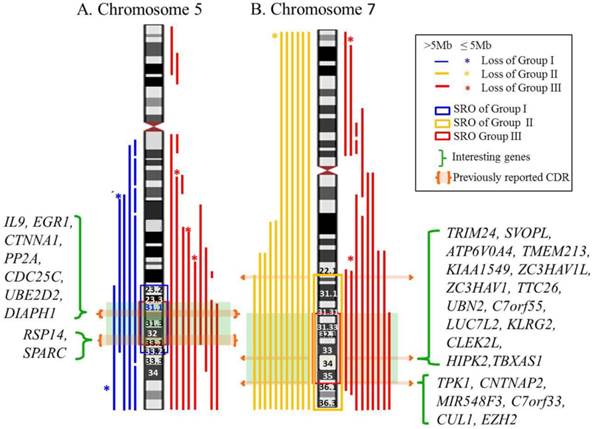
Genomic segmental losses of chromosome 5 were found in 15 cases consisting of six cases from group I and nine cases from group III (Table S3). Eight cases had a simple 5q deletion and seven cases had a complex deletion, defined as a deletion with more than two nonconsecutive deletions, of 5q. Monosomy (n=6) and genomic deletion (n=14) of chromosome 7 were observed in 20 cases, which included 11 cases from group II and nine cases from group III. Interestingly, all six cases of monosomy 7 belonged to group II. Of the other 14 cases, 12 cases were simple deletions of 7q and two cases were complex deletion of 7q. The SRO of chromosomes 5 and 7 in the three groups were mapped (Figure 1). The SRO of group I is 5q23.2-q33.2 (122,435,964-154,319,730bp: hg18) and ~ 32Mb in size. The SRO of group III is 5q31.1-33.1 (130,562,020-150,625,216bp: hg18) and ~ 20Mb in size, which is the common SRO of groups I and III of chromosome 5. The SRO of group II is 7q22.1-qter (102,913,354-158,816,034bp: hg18) and ~ 56Mb in size. The SRO of group III is 7q31.31-q36.1 (119,547,309-149,033,790bp: hg18) and ~ 29Mb in size, which is the common SRO of groups II and III of chromosome 7.
Map of SRO of chromosomes 5 and 7 in the three groups. A. The SRO of chromosome 5. The SRO of group I is 5q23.2-q33.2 (122,435,964-154,319,730bp: hg18) and ~ 32Mb in size. The SRO of group III is 5q31.1-33.1 (130,562,020-150,625,216bp: hg 18) and ~ 20Mb in size, which is the common SRO of groups I and III of chromosome 5. B. The SRO of chromosome 7. The SRO of group II is 7q22.1-qter (102,913,354-158,816,034bp: hg 18) and ~ 56Mb in size. The SRO of group III is 7q31.31-q36.1 (119,547,309-149,033,790bp: hg 18) and ~ 29Mb in size, which is the common SRO of groups II and III of chromosome 7. Interesting genes in each SRO are listed.
Delineation of 'other CNAs' in the three groups
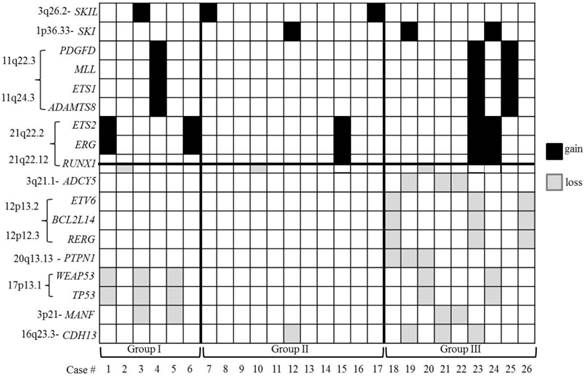
The CNAs on chromosomes other than chromosomes 5 and 7 were defined as 'other CNAs'. A total of 249 'other CNAs', which accounted for ~ 78.3% of all the CNAs, were observed with a striking distribution throughout the genome, except on chromosome Y. The average number of 'other CNAs' was 9.58 per case and thus, the cases with 10 or more 'other CNAs' were defined as cases with HN 'other CNAs' and 'other CNAs' which occurred in at least two or more cases was defined as recurrent 'other CNAs'. To characterize the features of the 'other CNAs', several parameters, including 1) the average number of 'other CNAs' per case, 2) the average number of recurrent 'other CNAs' per case and 3) the ratio of cases with HN 'other CNAs' to the number of total cases of each group, were compared between the three groups (Table 2). Comparing groups II and III, significant differences (p<0.05) were observed on all the parameters. Comparing groups I and III, no statistical difference (p≥0.05) was found on any parameter except the average number of recurrent 'other CNAs'. Group I showed a significantly higher average number of 'other CNAs' compared to group II. Of the 249 'other CNAs', 68 were recurrent 'other CNAs' (Table S4, Figure S2). Notably, most of the recurrent 'other CNAs' were found in group III (97.1%) and none were found in group II only. Moreover, 69.1% of the recurrent 'other CNAs' were also cryptic CNAs and all of them found in group III. Interesting tumor-associated genes based on the known gene function from UCSC genome browser (http://genome.ucsc.edu/) and NCBI (http://www.ncbi.nlm.nih.gov/gene/) were selected and separately listed from recurrent 'other CNAs' (Figure 2, Tables S5). From this additional analysis, genomic segmental losses of 3q21.1, 12p13.2, 12p13.1-p12.3 and 20q13.13 harboring ADCY5, ETV6 (Figure S3A), BCL2L14, RERG and PTPN1 were specially found in group III only. The most common segmental aberration in this study (5 cases) was the loss of various size of 17p harboring TP53 (17p13.1) (Figure S3B), which was observed in both groups I and III. In addition, both gain (3 cases) and loss (2 cases) of 21q22.12, encompassing RUNX1, with various breakpoints was recurrently observed in all three groups.
Comparison of 'other CNAs' between the three groups using several parameters
| Group I | Group II | Group III | p value | |||
|---|---|---|---|---|---|---|
| I vs III | II vs III | I vs II | ||||
| Average number of “other CNAs”/case (mean±SEM) | 12.00±5.398 | 1.818±0.7841 | 17.44±1.952 | 0.2916 | ≤ 0.0001 | 0.0230 |
| Average number of recurrent “other CNAs”/case (mean±SEM) | 1.263±0.08163 | 1.053±0.05263 | 1.621±0.08011 | 0.0043 | 0.0004 | 0.0892 |
| Cases with HN “other CNAs”/cases in each group (number /number) | 3/6 | 0/11 | 8/9 | 0.2352 | ≤ 0.0001 | 0.0294 |
SEM: Standard Error of the Mean
The distribution of the selected interesting tumor-associated genes located in recurrent 'other CNAs'. A black shadow indicates a gain of genes and a grey shadow indicates a loss of genes. Note that the losses of the 12p13 region encompassing the ETV6 gene and the 17p13.1 region encompassing the TP53 gene do not occur simultaneously in group III.
Discussion
It is important to understand precise and comprehensive genomic information for the diagnosis and management of MDS/AML. Array CGH and single nucleotide polymorphism array (SNP-A) are a powerful new tool to assess the chromosomal imbalances at the level of the whole genome. Recently, it has been widely used to overcome the limited capacity of conventional cytogenetic analysis and improve the abnormality detection rate for MDS/AML [16,17,18,19]. The cohort of patients with del(5q) and/or -7/del(7q) in this study was characterized by an integrated analysis of karyotype, FISH and array CGH results. Previous cytogenetic studies have shown -5 (monosomy 5) and del(5q) were the most common chromosomal abnormalities in MDS/AML. However, the present study, as well as a recent high-density single nucleotide polymorphism array (SNP-A) study of 1,155 cases with MDS/AML, demonstrated that the cases with putative -5 identified by karyotype analysis actually retained primarily the short arm of chromosome 5 [20]. This result confirmed findings in previous studies [21,22]. Also, the loss of heterozygosity (LOH) 7 has been detected by SNP-A and showed carrying similar prognosis to -7/del(7q) [23,24]. This novel characterization indicates that array CGH is a necessary and powerful complementary tool for the correct diagnosis of MDS/AML patients. Not only does it reveal cryptic CNAs beyond the resolution of G-banding, but it also refines the chromosomal abnormalities found by G-banding and can characterize genomic alterations in cases that cannot be cultured or cultures which fail to grow.
The CDRs on the long arms of chromosomes 5 and 7 underline the importance to pinpoint the candidate genes serving as haploinsufficient tumor suppressors [4,5,25]. Previously identified CDRs of 5q were classified into two CDRs consisting of a distal CDR (5q32-q33.1), which has been associated with 5q- syndrome and a proximal CDR (5q31.2-q31.3), which has been related to MDS/AML [26]. Interesting genes suggested in these CDRs were RPS14 located in the 5q distal CDR and EGR1, CTNNA1, DIAPH1 and IL9 found in the 5q proximal CDR, which play roles as a key tumor suppressor genes [25,26,27]. Although previous research described a CDR for 7q22 [4,6], a recent single nucleotide polymorphism array (SNP-A) analysis in 1,450 patients with myeloid malignancy proposed that the CDRs on 7q involve 7q34 and 7q35-q36 as well [28].The interesting genes suggested in these CDRS were TRIM24, SVOPL, ATP6V0A4, TMEM213, KIAA1549, ZC3HAV1L, ZC3HAV1, TTC26, UBN2, C7orf55, LUC7L2, KLRG2, CLEK2L, HIPK2, TBXAS1, TPK1, CNTNAP2, MIR548F3, C7orf33, CUL1, EZH2 [28]. In our cohort of patients, expanded SRO covering 5q23.2-q33.2 and 7q31.31-q36.1 were identified on chromosomes 5 and 7 encompassing previously identified CDRs of 5q and 7q, which is likely due to the small number of cases.
The high frequency of the losses of 5q and 7q combined with a loss of 17p has been reported in the complex karyotype background [12,29]. This suggests the possibility of other genomic aberrations accompanying del(5q) and/or -7/del(7q) that may also contribute to the development or prognosis of MDS/AML. As no difference of SRO on 5q and 7q are shown between the three groups in the present study, further investigation to differentiate the recurrent 'other CNAs' between the groups was initiated. Group III was a distinctive group that carried the most HN CNAs, cryptic CNAs (<5Mb) and 'other CNAs', including recurrent and HN 'other CNAs', of the three groups in this study. Although group III showed a significantly higher average number of recurrent 'other CNAs' compared to group I (Table 2), both groups shared recurrent 'other CNAs' and target genes (Table S4, Figure S2). Thus, it can be hypothesized that del(5q) may be the earlier event in the progression of MDS/AML cases with concurrent del(5q) and -7/del(7q), whereas 7q deletion as well as other genomic aberrations may play a secondary role in the late stage of leukemogenesis.
Several studies have already reported the significant genomic aberrations encompassing leukemogenic genes such as gains of 11q23.3 (MLL), 8q24.13-q24.21 (MYC), 21q22.2 (EGR and TMPRSS2) and 12p13.32 (CCND2) and losses of 9p21.3 (CDKN2A), 12p13.31p13.2 (ETV6), 16q22.1 (CBFB), 17p13 (TP53), 17q11.2 (NF1), 18p11.2 (PTPN2) and 21q22.12 (RUNX1), which were found in our study as well [30-37]. Our study not only identified previously reported tumor-related genes harbored in the recurrent CNAs but also assessed these genes based on three categorized groups: del(5q) (group I), -7/del(7q) (group II), and concurrent del(5q) and del(7q) (group III) (Figure 2), even if we have the limitation that LOH cannot be detected by our study. Gains of PDGFD, MLL, ETS1 and ADAMTS8 and losses of MANF, TP53 and WRAP53 were presumed to be highly associated with del(5q) because they were common in both groups I and III. A recent study related to pathogenesis of therapy related myeloid neoplasms (t-MN) with a complex karyotype and del(5q) and/or -7/del(7q) found that TP53 mutations were pre-existing in hematopoetic stem-cells, and that TP53 mutated cells survived chemotherapy and radiation therapy for primary cancer to give rise to t-MN. This indicates that aberration of TP53 may be happened in the early stage of disease and associated with prognosis [38]. However, the loss of ETV6, a frequent deletion in acute leukemia cases with translocations, was shown to be particularly associated with group III. Recently, the cryptic CNA del(12p), involving the ETV6 gene as well as point mutation of ETV6 gene, has been reported in AML and MDS with clinical significance [17,39-41]. It is thought to be a complementary step that, in cooperation with certain critical cytogenetic events such as t(12;21) or +8, drives leukemogenesis. However, in contrast to a study by Wall and colleagues who demonstrated a high association of ETV6 deletion and monosomy 7 in MDS/AML cases [42], all the deletions of ETV6 in our study were found in group III [concurrent del(5q) and del(7q)] and none of the cases were in group II [-7/del(7q) only)]. Also, aberrations of ADCY5, BCL2L14, RERG and PTPN1 that have been previously reported in MDS and leukemia cases were shown to be highly associated in the cases with concurrent del(5q) and del(7q) (group III) [43-45]. These CNAs or genes may play a secondary role in disease progression and should be further evaluated for their clinical significance and influence on therapeutic approaches in patients with MDS/AML carrying del(5q) and/or -7/del(7q) in large-scale, patient population study.
Conclusion
An integrated analysis combining karyotype, FISH and array CGH results performed on MDS/AML samples with del(5q) and/or -7/del(7q) allowed us to group patients by genomic imbalances and to suggest candidate genes located on chromosomes other than 5 and 7 that may affect the maintenance and progression of a patient's malignant phenotype. Group III, with concurrent del(5q) and -7/del(7q), was a distinctive group that carried the most HN CNAs, cryptic CNAs (<5Mb) and 'other CNAs', including recurrent and HN 'other CNAs'. Loss of TP53 was more highly associated with del(5q) than del(7q). However, the loss of ETV6 was shown to be particularly associated with concurrent del(5q) and del(7q) (group III). The CNAs or genes, such as TP53 and ETV6, identified in our study may play an important role in the disease progression and should be further evaluated for their clinical significance and influence on therapeutic approaches in patients with MDS/AML carrying del(5q) and/or -7/del(7q) in large scaled patient population study.
Supplementary Material
Table S1 Summary of 26 cases of MDS/AML with del(5q) and/or -7/del(7q). Table S2 Summary of genomic CNAs detected by array CGH. Table S3 Characterizations of the deletion of chromosomes 5 and 7 in the three groups. Table S4 Recurrent 'other CNAs' in three groups. Table S5 Selected interesting tumor-associated genes* from the recurrent 'other CNAs'. Figure S1 An overview of the genomic imbalances found in 26 MDS/AML cases with del(5q) and/or -7/del(7q). Figure S2 The group and size distribution of 68 recurrent 'other CNAs'. Figure S3 Losses of 12p13.2 including ETV6 gene in three cases (A) and 17p harboring TP53 gene in four cases (B) were detected by array CGH. Red vertical line indicates the location gene is located.
Competing Interests
The authors declare that they have no competing interests.
Acknowledgements
This work was supported by the Basic Science Research Program (2014R1A2A2A01003566) from the National Research Foundation of Korea (NRF) grant, which is funded by the Ministry of Education, Science and Technology (MEST), Republic of Korea and a grant K1507791 from Korea University.
References
1. Desangles F. -7/del(7q) in adults. Atlas Genet Cytogenet Oncol Haematol. 1999 http://AtlasGeneticsOncology.org/Anomalies/del7qID1093.html
2. Nolte F, Hofmann WK. Myelodysplastic syndromes: molecular pathogenesis and genomic changes. Ann Hematol. 2008;87:777-95
3. Kanehira K, Ketterling RP, Van Dyke DL. Del(5q) in myeloid neoplasms. Atlas Genet Cytogenet Oncol Haematol. 2009 http://AtlasGeneticsOncology.org/Anomalies/ del5qID1092.html
4. Curtissa NP, Bonifasa JM, Lauchle JO, Balkman JD, Kratz CP, Emerling BM. et al. Isolation and analysis of candidate myeloid tumor suppressor genes from a commonly deleted segment of 7q22. Genomics. 2005;85:600-7
5. Joslin JM, Fernald AA, Tennant TR, Davis EM, Kogan SC, Anastasi J. Haploinsufficiency of EGR1, a candidate gene in the del(5q), leads to the development of myeloid disorders. Blood. 2007;110:719-26
6. Wong JC, Zhang Y, Lieuw KH, Tran MT, Forgo E, Weinfurtner K. et al. Use of chromosome engineering to model a segmental deletion of chromosome band 7q22 found in myeloid malignancies. Blood. 2010;115:4524-32
7. The Association of Clinical Cytogeneticists (ACC) Professional Standards Committees. Professional guidelines for clinical cytogenetics; association for clinical cytognetics; acute myeloid leukemia & myelodysplastic syndromes (V1.00). 2012.
8. Greenberg PL, Tuechler H, Schanz J, Sanz G, Garcia-Manero G, Solé F. et al. Revised international prognostic scoring system for myelodysplastic syndromes. Blood. 2010;120:2454-65
9. NCCN Clinical Practice Guidelines in Oncology (NCCN Guidelines®); 2001. National Comprehensive Cancer Network Inc. http://www.nccn.org/professionals/physician_gls/f_guidelines.asp
10. Schoch C, Haferlach T, Bursch S, Gerstner D, Schnittger S, Dugas M. et al. Loss of genetic material is more common than gain in acute myeloid leukemia with complex aberrant karyotype: a detailed analysis of 125 cases using conventional chromosome analysis and fluorescence in situ hybridization including 24-color FISH. Genes Chromosomes Cancer. 2002;35:20-9
11. Van Limbergen H, Poppe B, Michaux L, Herens C, Brown J, Noens L. et al. Identification of cytogenetic subclasses and recurring chromosomal aberrations in AML and MDS with complex karyotypes using M-FISH. Genes Chromosomes Cancer. 2002;33:60-72
12. Alvarez S, Cigudosa JC. Gains. losses and complex karyotypes in myeloid disorders: a light at the end of the tunnel. Hematol Oncol. 2005;23:18-25
13. Smith AG, Worrillow LJ, Allan JM. A common genetic variant in XPD associates with risk of 5q- and 7q-deleted acute myeloid leukemia. Blood. 2007;109:1233-6
14. Rücker FG, Schlenk RF, Bullinger L, Kayser S, Teleanu V, Kett H. et al. TP53 alterations in acute myeloid leukemia with complex karyotype correlate with specific copy number alterations, monosomal karyotype, and dismal outcome. Blood. 2012;119:2114-21
15. Zhang R, Kim YM, Yang X, Li Y, Li S, Lee J. A possible 5'-NRIP1/UHRF1-3' fusion gene detected by array CGH analysis in a Ph+ ALL patient. Cancer Genet. 2011;204:687-91
16. Casas S, Aventı́n A, Fuentes F, Vallespí T, Granada I, Carrió A. et al. Genetic diagnosis by comparative genomic hybridization in adult de novo acute myelocytic leukemia. Cancer Genet Cytogenet. 2004;153:16-25
17. Paulsson K, Heidenblad M, Strömbeck B, Staaf J, Jönsson G, Borg A. et al. High-resolution genome-wide array-based comparative genome hybridization reveals cryptic chromosome changes in AML and MDS cases with trisomy 8 as the sole cytogenetic aberration. Leukemia. 2006;20:840-6
18. Arenillas L. et al. Single nucleotide polymorphism array karyotyping: a diagnostic and prognostic tool in myelodysplastic syndromes with unsuccessful conventional cytogenetic testing. Genes Chromosomes Cancer. 2013;52:1167-77
19. Thiel A. et al. Comprehensive array CGH of normal karyotype myelodysplastic syndromes reveals hidden recurrent and individual genomic copy number alterations with prognostic relevance. Leukemia. 2011;25:387-99
20. Jerez A, Gondek LP, Jankowska AM, Makishima H, Przychodzen B, Tiu RV. et al. Topography, clinical, and genomic correlates of 5q myeloid malignancies revisited. J Clin Oncol. 2012;30:1343-9
21. Bram S, Swolin B, Rödjer S, Stockelberg D, Ögärd I, Bäck H. Is monosomy 5 an uncommon aberration? Fluorescence in situ hybridization reveals translocations and deletions in myelodysplastic syndromes or acute myelocytic leukemia. Cancer Genet Cytogenet. 2003;42:107-14
22. Galván AB, Mallo M, Arenillas L, Salido M, Espinet B, Pedro C, Florensa L. et al. Does monosomy 5 really exists in myelodysplastic syndromes and acute myeloid leukemia? Leuk Res. 2010;34:1242-5
23. Xu X, Johnson EB, Leverton L, Arthur A, Watson Q, Chang FL. et al. The advantage of using SNP array in clinical testing for hematological malignancies--a comparative study of three genetic testing methods. Cancer Genet. 2013;206:317-26
24. O'Keefe C, McDevitt MA, Maciejewski JP. Copy neutral loss of heterozygosity: a novel chromosomal lesion in myeloid malignancies. Blood. 2010; 115: 2731-9. Shannon KM, Le Beau MM. Cancer: hay in a haystack. Nature. 2008;451:252-3
25. Eisenmann KM, Dykema KJ, Matheson SF, Kent NF, De Ward AD, West RA. et al. 5q- myelodysplastic syndromes: chromosome 5q genes direct a tumor-suppression network sensing act in dynamics. Oncogene. 2009;28:29-3441
26. Jerez A, Sugimoto Y, Makishima H, Verma A, Jankowska AM, Przychodzen B. et al. Loss of heterozygosity in 7q myeloid disorders: clinical associations and genomic pathogenesis. Blood. 2012;119:6109-17
27. Schoch C, Kern W, Kohlmann A, Hiddemann W, Schnittger S, Haferlach T. Acute myeloid leukemia with a complex aberrant karyotype is a distinct biological entity characterized by genomic imbalances and a specific gene expression profile. Genes Chromosomes Cancer. 2005;43:227-38
28. Ebert BL. Deletion 5q in myelodysplastic syndrome: a paradigm for the study of hemizygous deletions in cancer. Leukemia. 2009;23:1252-6
29. Akagi T, Ogawa S, Dugas M, Kawamata N, Yamamoto G, Nannya Y. et al. Frequent genomic abnormalities in acute myeloid leukemia/myelodysplastic syndrome with normal karyotype. Haematologica. 2009;94:213-23
30. Bajaj R, Xu F, Xiang B, Wilcox K, Diadamo AJ, Kumar R. et al. Evidence-based genomic diagnosis characterized chromosomal and cryptic imbalances in 30 elderly patients with myelodysplastic syndrome and acute myeloid leukemia. Mol Cytogenet. 2011;4:3-12
31. Radtke I, Mullighan CG, Ishii M, Su X, Cheng J, Ma J. et al. Genomic analysis reveals few genetic alterations in pediatric acute myeloid leukemia. Proc Natl Acad Sci USA. 2009;106:12944-9
32. Slovak ML, Smith DD, Bedell V, Hsu YH, O'Donnell M, Forman SJ. Assessing karyotype precision by microarray-based comparative genomic hybridization in the myelodysplastic/myeloproliferative syndromes. Mol Cytogenet. 2010;3:23-36
33. Starczynowski DT, Vercauteren S, Telenius A, Sung S, Tohyama K, Brooks-Wilson A. et al. High-resolution whole genome tiling path array CGH analysis of CD34+ cells from patients with low-risk myelodysplastic syndromes reveals cryptic copy number alterations and predicts overall and leukemia-free survival. Blood. 2008;112:3412-24
34. Suela J, Alvarez S, Cifuentes F, Largo C, Ferreira BI, Blesa D. et al. DNA profiling analysis of 100 consecutive de novo acute myeloid leukemia cases reveals patterns of genomic instability that affect all cytogenetic risk groups. Leukemia. 2007;21:1224-31
35. Tyybäkinoja A, Elonen E, Piippo K, Porkka K, Knuutila S. Oligonucleotide array-CGH reveals cryptic gene copy number alterations in karyotypically normal acute myeloid leukemia. Leukemia. 2007;21:571-4
36. Walter MJ, Payton JE, Ries R, Shannon WD, Deshmukh H, Zhao Y. Acquired copy number alterations in adult acute myeloid leukemia genomes. Proc Natl Acad Sci USA. 2009;106:12950-95
37. Wong TN, Ramsingh G, Young AL, Miller CA, Touma W, Welch JS, Et al. Role of TP53 mutations in the origin and evolution of therapy-realted acrute myeloid leukemia. Nature. 2015;26:552-5
38. Moorman AV, Schwab C, Ensor HM, Russell LJ, Morrison H, Jones L. et al. IGH@ translocations, CRLF2 deregulation, and microdeletions in adolescents and adults with acute lymphoblastic leukemia. J Clin Oncol. 2012;30:3100-8
39. Zakaria Z, MdAhid MF, Ismail A, Ten SK, Mohamad Nor N, Kamaluddin NR. et al. Chromosomal aberrations in ETV6/RUNX1-positive childhood acute lymphoblastic leukemia using 244 K oligonucleotide array comparative genomic hybridization. Mol Cytogenet. 2012;5:41-6
40. Bejar R, Stevenson K, Abdel-Wahab O, Galili N, Nilsson B, Garcia-Manero G. et al. Clinical effect of point mutations in myelodysplastic syndromes. N Engl J Med. 2011;364:2496-506
41. Wall M, Rayeroux KC, Mackinnon RN, Zordan A, Campbell LJ. ETV6 deletion is a common additional abnormality in patients with myelodysplastic syndromes or acute myeloid leukemia and monosomy 7. Haematologica. 2012;97:1933-6
42. Al-Shehhi H, Konn ZJ, Schwab CJ, Erhorn A, Barber KE, Wright SL. et al. Abnormalities of the der(12)t(12;21) in ETV6-RUNX1 acute lymphoblastic leukemia. Genes Chromosomes Cancer. 2013;52:202-13
43. Brobeil A, Bobrich M, Graf M, Kruchten A, Blau W, Rummel M. et al. PTPIP51 is phosphorylated by Lyn and c-Src kinases lacking dephosphorylation by PTP1B in acute myeloid leukemia. Leuk Res. 2011;35:1367-75
44. Kuang SQ, Tong WG, Yang H, Kruchten A, Blau W, Rummel M. et al. Genome-wide identification of aberrantly methylated promoter associated CpG islands in acute lymphocytic leukemia. Leukemia. 2008;22:1529-38
45. Lee S, Kwon HC, Kim SH, Oh SY, Lee JH, Lee YS. Identification of genes underlying different methylation profiles in refractory anemia with excess blast and refractory cytopenia with multilineage dysplasia in myelodysplastic syndrome. Korean J Hematol. 2012;47:186-93
Author contact
![]() Corresponding author: Shibo Li, MD. FACMG., 1122 NE 13th Street, Suite 1400, Oklahoma City, OK 73104. Tel: +1-405-271-3590; Fax: +1-405-271-7117; Email: Shibo-liedu or Ji-Yun Lee, Ph.D., 73 Inheon-Ro, Seoungbuk-Gu, Seoul, 136-705 South Korea. Tel: +82-2-290-6141; Fax: +82-2-953-3130; Email: jiyun-leeac.kr
Corresponding author: Shibo Li, MD. FACMG., 1122 NE 13th Street, Suite 1400, Oklahoma City, OK 73104. Tel: +1-405-271-3590; Fax: +1-405-271-7117; Email: Shibo-liedu or Ji-Yun Lee, Ph.D., 73 Inheon-Ro, Seoungbuk-Gu, Seoul, 136-705 South Korea. Tel: +82-2-290-6141; Fax: +82-2-953-3130; Email: jiyun-leeac.kr