Impact Factor ISSN: 1449-1907

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2015; 12(6):487-493. doi:10.7150/ijms.10982 This issue Cite
Research Paper
Pim-2 Modulates Aerobic Glycolysis and Energy Production during the Development of Colorectal Tumors
Xue-hui Zhang1, Hong-liang Yu1,2, Fu-jing Wang2, Yong-long Han3, Wei-liang Yang2 ![]()
1. Daqing Oilfield General Hospital, Zhongkang Street 9, Daqing, 163001, China
2. The Second Affiliated Hospital of Harbin Medical University, Road Xuefu 246, Harbin, 150086, China
3. The Sixth People's Affiliated Hospital of Shanghai Jiao Tong University, Road Yishan 600, Shanghai, 200233, China
Received 2014-11-3; Accepted 2015-4-10; Published 2015-6-8
Abstract
Tumor cells have higher rates of glucose uptake and aerobic glycolysis to meet energy demands for proliferation and metastasis. The characteristics of increased glucose uptake, accompanied with aerobic glycolysis, has been exploited for the diagnosis of cancers. Although much progress has been made, the mechanisms regulating tumor aerobic glycolysis and energy production are still not fully understood. Here, we demonstrate that Pim-2 is required for glycolysis and energy production in colorectal tumor cells. Our results show that Pim-2 is highly expressed in colorectal tumor cells, and may be induced by nutrient stimulation. Activation of Pim-2 in colorectal cells led to increase glucose utilization and aerobic glycolysis, as well as energy production. While knockdown of Pim-2 decreased energy production in colorectal tumor cells and increased their susceptibility to apoptosis. Moreover, the effects of Pim-2 kinase on aerobic glycolysis seem to be partly dependent on mTORC1 signaling, because inhibition of mTORC1 activity reversed the aerobic glycolysis mediated by Pim-2. Our findings suggest that Pim-2-mediated aerobic glycolysis is critical for monitoring Warburg effect in colorectal tumor cells, highlighting Pim-2 as a potential metabolic target for colorectal tumor therapy.
Keywords: Pim-2, Aerobic glycolysis, Apoptosis, Warburg effect
Introduction
Cancer cell energy metabolism deviates significantly from that of normal tissues. In mammalian cells, glycolysis is down-regulated by oxygen, which allows mitochondria to oxidize pyruvate and generate large amounts of ATP [1]. However, cancer cells perform higher rates of aerobic glycolysis with products of pyruvate and lactate, known as Warburg effect [2]. Although aerobic glycolysis was initially thought as supplement of disrupted mitochondrial respiration, recent studies declare that it may act as a driving force for tumor transformation and proliferation [3,4]. It is thought that cancer cells take this metabolic transformation not only to meet energy demand but also to maintain the redox homeostasis [3]. Due to the preference of aerobic glycolysis, cancer cells can be selectively targeted by disruption of their glucose metabolism [5-7]. Despite considerable progress, how aerobic glycolysis is precisely regulated needs further elucidation. Targeted killing of cancer cells without toxicity to normal cells, is one of the most significant considerations in cancer chemotherapy. Thus, understanding the regulatory mechanism of tumor glucose metabolism is necessary for the design and development of anticancer drugs.
Tumorigenic reliance on glycolysis is highly correlated with many intracellular signaling factors, such as hexokinase [8], phosphofructokinase [9], and pyruvate kinase [10]. These glycolytic factors are consistently and significantly expressed in cancer cells. Meanwhile, oncogenes such as Ras, Src, and Myc have also been found to promote glycolysis by increasing the expression of glucose transporters and glycolytic enzymes [11]. Mammalian target of rapamycin complex I (mTORC1) signaling is known as a master regulator of aerobic glycolysis [12,13], which is also consistently activated in many cancers [14]. mTORC1 signaling controls glycolysis not only by regulating glycolytic gene transcription via HIF1-α (hypoxia-inducible factor 1-α) [15], but also by modulating glycolytic enzyme expression, such as PKM2 (the M2 splice isoform of pyruvate kinase) [16]. Thus, factors that involve mTORC1 signaling activation may have potential to modulate aerobic glycolysis in cancer cells. To further identify factors involved in tumor aerobic glycolysis, we focused on Pim-2, a member of the proviral integration of Moloney virus family of oncogenic serine/threonine kinases, which have been reported to activate mTORC1 signaling under special conditions [17].
Pim-2, together with Pim-1 and 3, is attributed to a serine/threonine kinase family encoded by proto-oncogenes [17]. Pim-2 gene expression is modulated at both transcriptional and translational levels by numerous cytokines (especially IL-3) [18]. Pim-2 plays an important role in tumor cell growth, differentiation, and survival [19,20]. For example, Pim-2 phosphorylates oncogene Myc and leads to an increase in Myc protein stability and thereby an increase in transcriptional activity [21]. Also, Pim-2 can phosphorylate Bad or activate NF-κB to promote cancer survival [22,23]. Again, Pim-2 has been found to compensate for mTORC1 signaling activation and is involved in tumor cell growth [24]. Nevertheless, it is still largely unclear through which pathways Pim-2 promotes tumor cell growth and survival, and how Pim-2 is involved in tumor cell metabolism.
To identify the role of Pim-2 in tumor development, we investigated the expression pattern and functions of Pim-2 in colorectal tumor cells. We found that Pim-2 is highly expressed in colorectal tumor cells and its expression was induced by nutrient status. Overexpression of Pim-2 in colorectal cells led to increased glycolysis and energy production. While Pim-2 knockdown decreased aerobic glycolysis and increases cell susceptibility to apoptosis. Moreover, inhibition of mTORC1 signaling activity via rapamycin reduced Pim-2 mediated glycolysis, suggesting that the effect of Pim-2 on glycolysis may be partly dependent on mTORC1 activation. All these findings establish Pim-2 as a key regulator of aerobic glycolysis in colorectal tumor cells, and will help us to understand the tumor regulatory mechanism of aerobic glycolysis and offer a novel target for improving cancer therapy.
Material and methods
Chemicals and materials
The inhibitor of mTORC1 signaling rapamycin was purchased from Sigma-Aldrich (St. Louis, MO, USA). Cell medium, trypsin and fetal bovine serum (FBS) were obtained from Hyclone (Hyclone, Logan, Utah). The anti-Pim-2 antibody was from Santa Cruz (Santa Cruz, California, USA). The actin and HA-tagged antibodies were from Millipore (Billerica, MA, USA). Anti-cleaved caspase 3, anti-Bax, anti-Bcl-2, anti-p-p70S6K1 and anti-p-p4EBP-1 antibodies were purchased from Cell Signaling Technology (Beverly, MA, USA). Other chemicals were of the highest purity available.
Cell culture and transfections
In present study, human colorectal carcinoma cells HCT116, HT29 and SK/S were obtained from the American Type Culture Collection (Manassas, VA, USA), and NCM460 non-transfected human colonic epithelial cells were purchased from INCELL Corporation (San Antonio, TX, USA) [25]. HCT116 cells were cultured in DMEM and NCM460 in M3 media with 10% FBS plus 1% antibiotics at 37°C with constant humidity. As for cell starvation, cultured HCT116 cells were 0.5% FBS for 16 h and incubated with dPBS for 2 h. The final re-feeding was performed by adding DMDM full media to starved cells for 1 h.
For Pim-2 overexpression, a HA-tagged Pim-2 construct was generated in NCM460 cells by subcloning the PCR-amplified human Pim-2 coding sequence into pRK5-HA vectors. To reduce the endogenous Pim-2 protein level in HCT116 cells, small interfering RNAs against Pim-2 were obtained from Shanghai GenePharma (China), with the sequence of CUCGAAGUCGCACUGCUAU. When the cells were 80-90% confluent, they were transfected using Lipofectamine™ 2000, and the cells were harvested 24 h after transfection. For inhibition of mTORC1 activity in HCT116 cells, 100 μM rapamycin was applied to cells for 24 h to block mTORC1 activity.
RNA extraction and real-time PCR
Whole cell RNA for reverse transcription was extracted from cells using Trizol reagent (Invitrogen, Carlsbad, CA, USA). Quantitative real-time PCR was performed using the Bio-Rad iQ5 system using Bio-Rad proprietary iQ5 software (Hercules, CA, USA), and the relative gene expression was normalized to actin as the internal control. Primer sequences for SYBR Green probes of target genes were as following Table 1.
Primer sequences of target genes in this study.
| Name | Primer sequence (5′→3′) |
|---|---|
| Pim-2-F | ACTCCAGGTGGCCATCAAAG |
| Pim-2-R | TCCATAGCAGTGCGACTTCG |
| Actin-F | GAGACCTTCAACACCCCAGC |
| Actin-R | ATGTCACGCACGATTTCCC |
Cell lysates preparation and western blots
For western blots, prepared cells were trypsinized and harvested, washed with PBS once and resuspended in PBS buffer containg 1% Triton X-100 and protease inhibitors. After sonication, lysates were centrifuged at 13 000 rpm for 5 min. The protein concentration was determined so that equivalent amounts of lysate were added to an equal volume of 2X Laemmli buffer and boiled for 10 min. For western blot analysis, proteins were separated by SDS-PAGE and transferred to a PVDF membrane. All the processes of western blots were according to standard method. After exposure to Kodak films, protein quantification was carried out using ImageJ.
Metabolic examination
All the metabolic examinations, including glucose consumption, pyruvate and lactate production and ATP production, were performed according to the manufacturer's instructions (Biovision). Briefly, a total of 1 × 106 cells per well were seeded in 6-well plates for 24 h, with or without pharmacological manipulations. Then, the cells were washed, harvested, and homogenized in assay buffer, and the medium was collected to assess glucose consumption. Samples were mixed with respective reaction buffers and read by fluorescence at Ex/Em = 535/590 nm in a microplate reader to measure the product concentration. All the final results were normalized to cell numbers for quantification.
Statistical analysis
Quantitative data are shown as mean ± SEM using ANOVA with post-hoc tests for comparisons. The p-values of 0.05 (*), 0.01 (**) and 0.001 (***) were considered as the levels of significance for the statistical tests.
Results
Pim-2 is highly expressed in colorectal tumor cells
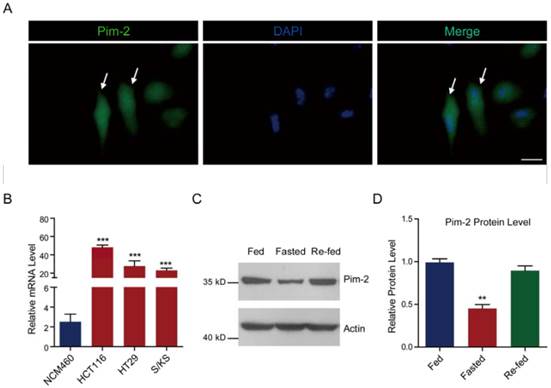
To determine whether colorectal-derived Pim-2 retains high expression, we assessed Pim-2 expression in several human colorectal tumor cells. We carried out Pim-2 immunostaining to directly visualize Pim-2 localization in HCT116 colorectal tumor cells. Green fluorescence indicated that Pim-2 was widely expressed in both the cytosol and nucleus of HCT116 cells, which is consistent with previous reports of other types of tumor cells (Fig. 1A) [26]. To further validate the expression pattern of Pim-2 in colorectal tumor cells, we assessed Pim-2 expression in colorectal tumor cells compared to NCM460 colorectal epithelial cells. The results of real-time PCR assays showed that Pim-2 mRNA levels were significantly high in colorectal tumor cells, such as HCT116, HT29, and S/KS cells (Fig. 1B). Moreover, we found that when colorectal tumor cells were starved, Pim-2 protein levels reduced by 54.9 % compared to normal-fed cells, while cell re-feeding activated Pim-2 protein levels (Fig. 1C and D). The altered Pim-2 levels according to nutrient status indicate that Pim-2 may be critical in tumor cell metabolism. Taken together, these results suggest that Pim-2 is highly expressed in colorectal tumor cells, which may play an important role in tumor development.
Pim-2 is highly expressed in colorectal tumor cells. (A) Images showing the Pim-2 expression pattern in cultured HCT116 human colorectal tumor cells. Green fluorescence indicates Pim-2, and blue indicates DAPI. Bar 25 μm. (B) Real-time PCR results showing that Pim-2 mRNA levels were significantly high in colorectal tumor cells. Results are the average of four independent experiments. Data represent mean ± SEM. ***p<0.001. (C-D) Western blots and histograms showing that the Pim-2 protein level was reduced by starvation and restored by re-feeding in HCT116 cells. Results are the average of four independent experiments. Data represent mean ± SEM **p<0.01.
Pim-2 promotes glycolysis and energy production in colorectal epithelial cells
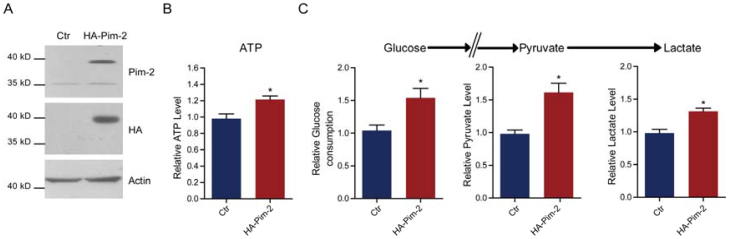
To examine how Pim-2 participates in colorectal tumor development, we investigate whether ectopic overexpression of Pim-2 in colorectal epithelial cells would alter cell metabolism. For the first, we constructed an HA-tagged Pim-2 vector to overexpress Pim-2 in NCM460 colorectal epithelial cells. Both Pim-2 and HA blots indicated Pim-2 overexpression in NCM460 cells (Fig. 2A). Notably, the endogenous Pim-2 level was much lower than the exogenous level. Next, we assayed energy production with Pim-2 overexpression. The results show that Pim-2 overexpression increased ATP production by 21.4%, indicating that energy production was indeed induced by Pim-2 (Fig. 2B). As for aerobic glycolysis promoting energy production by glucose conversion to pyruvate and lactate, we examined glucose consumption as well as pyruvate and lactate production. Results showed that after Pim-2 overexpression, glucose consumption increased by 53.9%, pyruvate by 61.4% and lactate by 31.4% compared to control (Fig. 2C). The upregulated axis of glucose/pyruvate/lactate indicates that Pim-2 overexpression may promote aerobic glycolysis, which may generate higher amounts of ATP to meet the energy demand of tumor development.
Pim-2 promotes glycolysis and energy production in colorectal epithelial cells. (A) Western blots showing Pim-2 overexpression in NCM460 human colorectal epithelial cells. (B-C) Biochemical results showing that Pim-2 overexpression increased ATP (B), glucose consumption, pyruvate and lactate production (C) in NCM460 cells. Results are the average of four independent experiments. Data represent mean ± SEM *p<0.05.
Pim-2 knockdown reduces glycolysis and energy production
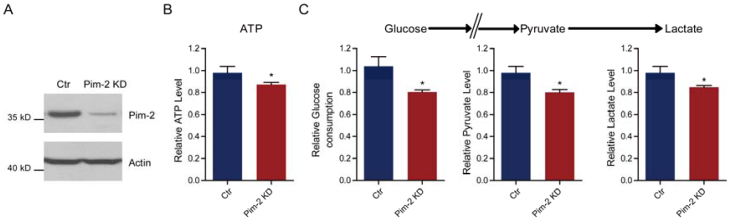
Since Pim-2 overexpression in NCM460 colorectal epithelial cells could activate aerobic glycolysis, we proposed that Pim-2 may be responsible for the development of colorectal tumors by providing an energy source. To test this hypothesis, we knocked down endogenous Pim-2 expression in HCT116 colorectal tumor cells and examine whether aerobic glycolysis was reduced. Similarly, the biochemical results confirmed Pim-2 knockdown in HCT116 cells (Fig. 3A). We found that, with Pim-2 knockdown, ATP production was reduced by 12.7% in colorectal tumor cells (Fig. 3B), along with reduced glucose consumption (19.7%), pyruvate (19.9%) and lactate (15.2%) productions (Fig. 3C). Thus, reduced Pim-2 protein levels may decrease aerobic glycolysis, suggesting that Pim-2 might be critical for glucose metabolism in colorectal tumor cells.
Pim-2 knockdown reduces glycolysis and energy production. (A) Western blots showing Pim-2 knockdown in HCT116 colorectal tumor cells. (B-C) Biochemical results showing that Pim-2 knockdown decreased ATP (B), glucose consumption, pyruvate and lactate production (C) in HCT116 cells. Results are the average of four independent experiments. Data represent mean ± SEM *p<0.05.
Pim-2 knockdown increases cell susceptibility to apoptosis
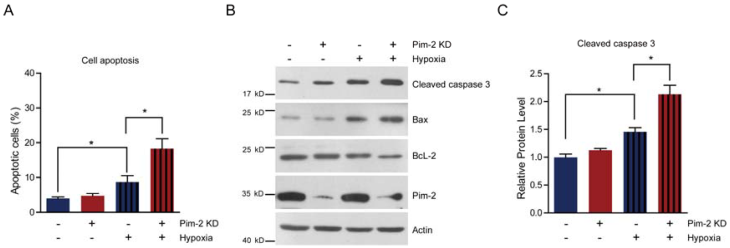
Next, we examined cell survival under basal and stress conditions by Pim-2 knockdown. The quantitative results show that there is no signficant difference between control and Pim-2 knockdown cells in terms of cell viability under normoxic conditions. However, Pim-2 knockdown led to increase cell apoptosis under hypoxic conditions, suggesting that Pim-2 is important for the survival of colorectal tumor cells (Fig. 4A). To assess apoptosis, we examined apoptotic markers in both control and Pim-2 knockdown cells. Results show that loss of Pim-2 indeed activated the apoptotic marker cleaved caspase-3 under hypoxic conditions, and increased expression of the apoptotic protein Bax, with decreased Bcl-2 expression (Fig. 4B and C). The increased ratio of Bax/Bcl-2 together with caspase 3 cleavage is hallmarks of cell apoptosis. Therefore, our findings suggest that Pim-2 knockdown may enhance susceptibility to hypoxia-mediated apoptosis.
Pim-2 knockdown increases cell susceptibility to apoptosis. (A) Histograms showing that Pim-2 knockdown increased apoptosis of HCT116 cells under hypoxic conditions. Results are the average of four independent experiments. Data represent mean ± SEM. *p<0.05. (B-C) Western blots and histograms showing that Pim-2 knockdown increased cleaved caspase 3 and Bax protein levels and decreased Bcl-2 protein levels under hypoxic conditions. Results are the average of four independent experiments. Data represent mean ± SEM *p<0.05.
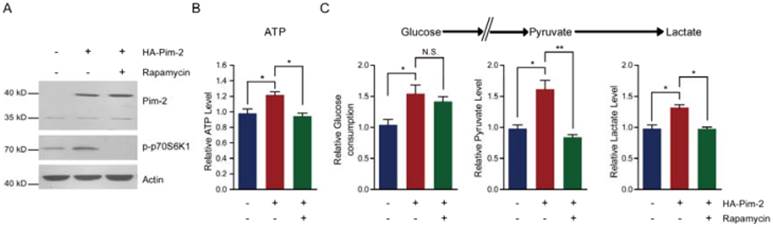
Inhibition of mTORC1 signaling by rapamycin reduces Pim-2 mediated glycolysis
To elucidate the molecular mechanism of how Pim-2 regulates aerobic glycolysis, we assessed energy production with Pim-2 overexpression and mTORC1 inhibition. The results show that overexpression of Pim-2 activated mTORC1 signaling (indicated by p-p70S6K1 and p4EBP-1), while rapamycin inhibited mTORC1 signaling in the presence of HA-Pim-2 (Fig. 5A). These data indicate that rapamycin can block HA-Pim-2 mediated mTORC1 activation. Moreover, HA-Pim-2 mediated glycolysis was restored to normal levels by rapamycin treatment (Fig. 5B and C). Based on these results, Pim-2 regulates aerobic glycolysis through a mechanism that might be partly dependent on mTORC1 signaling.
Inhibition of mTORC1 signaling by rapamycin reduces Pim-2 mediated glycolysis. (A) Western blots showing that rapamycin treatment inhibited mTORC1 activity under both basal and HA-Pim-2 overexpression conditions in NCM460 cells. (B-C) Biochemical results showing that rapamycin treatment restored Pim-2 induced ATP (B), glucose consumption, pyruvate and lactate production (C) in NCM460 cells. Results are the average of four independent experiments. Data represent mean ± SEM *p<0.05 and **p<0.01. N.S., not statistically significant.
Discussion
Upregulation of glycolytic metabolic pathways in majority of invasive cancers is the result of adaptation to environmental pressures [27]. Because cancer cells prefer aerobic glycolysis as their energy source, it provided a rationale in many previous studies by targeting certain glycolytic enzymes for cancer therapy [7,28]. Thus, elucidating the molecular mechanisms of tumor glycolysis may render the glycolytic regulators as targets for cancer therapy.
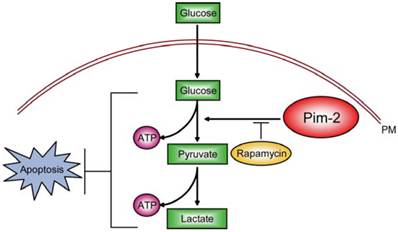
Among these potential targets, we propose that Pim-2 may be a novel and ideal target. In current study, we demonstrated that Pim-2 is highly expressed in colorectal tumor cells and promotes aerobic glycolysis for tumor development. Knockdown of Pim-2 in colorectal tumor cells led to reduced glycolysis and energy production, increasing cell susceptibility to apoptosis. Moreover, the effect of Pim-2 on aerobic glycolysis may be partly dependent on mTORC1 signaling, because inhibition of mTORC1 signaling by rapamycin reversed Pim-2 mediated aerobic glycolysis (Fig. 6). Our work uncovers novel relationships between Pim-2 and tumor cell metabolism, and offers new targets to colorectal cancer therapy.
Model. Schematic representation to highlight the molecular link between Pim-2, aerobic glycolysis and cell survival in colorectal tumor cells. Pim-2 promotes aerobic glycolysis and energy production to maintain tumor survival. Rapamycin treatment inhibits mTORC1 signaling and at least partly reverses Pim-2 mediated aerobic glycolysis.
Cancer cells commonly exhibit up-regulated aerobic glycolysis. This biological adaptation to metabolic changes occurs due to mitochondrial dysfunction, hypoxia, and oncogenic signals [7]. These alterations in energy metabolism provide a survival advantage to cancer cells [29]. However, the biological dependency of cancer cells on glycolysis for energy generation also provides a biochemical basis to preferentially kill cancer cells by inhibiting glycolysis [30]. Here, our studies afford compelling evidence showing that colorectal Pim-2 plays an important role in aerobic glycolysis and tumor development. When endogenous Pim-2 expression was knocked down or Pim-2 mediated glycolysis was blocked by rapamycin, cell susceptibility to apoptosis was dramatically increased due to a lack of energy production. Thus, Pim-2 may be a potential target for clinical cancer therapy by disrupting tumor energy source.
In previous studies, Pim-2 was found to function as an inhibitor of apoptosis that is transcriptionally regulated by a variety of proliferative signals [31]. For example, Pim-2 expression maintains high levels of NF-κB activity with its anti-apoptotic function [31]. Pim-2 can act as a binding partner of PKM2 to directly phosphorylate PKM2 and regulate glycolysis [32]. Moreover, Pim-2 may interact with HIF-1α as a co-factor, and enhance the protective responses to hypoxia [33]. All these studies strongly implicate that Pim-2 participates in tumor development through metabolic pathways. Here, we further identify that Pim-2 is a critical regulator of aerobic glycolysis in colorectal tumor cells. Pim-2 is required for tumor energy production and survival. Notably, the effect of Pim-2 on glycolysis could be partly restored by mTORC1 inhibitor rapamycin, suggesting that Pim-2 may regulate glycolysis via mTORC1 signaling. Although Pim-2 could be involved in mTORC1 activation under certain conditions, blocking mTORC1 activity by rapamycin had a similar effect as Pim-2 knockdown. According to these facts, we assume that Pim-2 may regulate aerobic glycolysis via mTORC1 signaling, by either promoting HIF-1α/glycolytic gene expression [15] or targeting at PKM2 to increase pyruvate production [34].
Conclusion
The present findings demonstrate that Pim-2 might be critical for aerobic glycolysis and energy production in colorectal tumor cells. The effect of Pim-2 on aerobic glycolysis seems to be partly through mTORC1 signaling. Our findings suggest that Pim-2 mediated aerobic glycolysis is critical for controlling Warburg effect in colorectal tumor cells, highlighting Pim-2 as a potential metabolic target for colorectal tumor therapy.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Gatenby RA, Gillies RJ. Why do cancers have high aerobic glycolysis? Nat Rev Cancer. 2004;4:891-9
2. Cairns RA, Harris IS, Mak TW. Regulation of cancer cell metabolism. Nat Rev Cancer. 2011;11:85-95
3. Chen W, Wang Q, Bai L. et al. RIP1 maintains DNA integrity and cell proliferation by regulating PGC-1alpha-mediated mitochondrial oxidative phosphorylation and glycolysis. Cell Death Differ. 2014;21:1061-70
4. Sebastian C, Zwaans BM, Silberman DM. et al. The histone deacetylase SIRT6 is a tumor suppressor that controls cancer metabolism. Cell. 2012;151:1185-99
5. Mathupala SP, Rempel A, Pedersen PL. Aberrant glycolytic metabolism of cancer cells: a remarkable coordination of genetic, transcriptional, post-translational, and mutational events that lead to a critical role for type II hexokinase. J Bioenerg Biomembr. 1997;29:339-43
6. Levine AJ, Puzio-Kuter AM. The control of the metabolic switch in cancers by oncogenes and tumor suppressor genes. Science. 2010;330:1340-4
7. Pelicano H, Martin DS, Xu RH. et al. Glycolysis inhibition for anticancer treatment. Oncogene. 2006;25:4633-46
8. Patra KC, Wang Q, Bhaskar PT. et al. Hexokinase 2 is required for tumor initiation and maintenance and its systemic deletion is therapeutic in mouse models of cancer. Cancer Cell. 2013;24:213-28
9. Park YY, Kim SB, Han HD. et al. Tat-activating regulatory DNA-binding protein regulates glycolysis in hepatocellular carcinoma by regulating the platelet isoform of phosphofructokinase through microRNA 520. Hepatology. 2013;58:182-91
10. Christofk HR, Vander Heiden MG, Harris MH. et al. The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature. 2008;452:230-3
11. Fritz V, Fajas L. Metabolism and proliferation share common regulatory pathways in cancer cells. Oncogene. 2010;29:4369-77
12. Düvel K, Yecies JL, Menon S. et al. Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Mol Cell. 2010;39:171-83
13. Zha X, Wang F, Wang Y. et al. Lactate dehydrogenase B is critical for hyperactive mTOR-mediated tumorigenesis. Cancer Res. 2011;71:13-8
14. Guertin DA, Sabatini DM. Defining the role of mTOR in cancer. Cancer Cell. 2007;12:9-22
15. Cheng SC, Quintin J, Cramer RA. et al. mTOR- and HIF-1α-mediated aerobic glycolysis as metabolic basis for trained immunity. Science. 2014;345:1250684
16. Nemazanyy I, Espeillac C, Pende M. et al. Role of PI3K, mTOR and Akt2 signalling in hepatic tumorigenesis via the control of PKM2 expression. Biochem Soc Trans. 2013;41(4):917-22
17. Narlik-Grassow M, Blanco-Aparicio C, Carnero A. The PIM family of serine/threonine kinases in cancer. Med Res Rev. 2014;34:136-59
18. Kapelko-Słowik K, Urbaniak-Kujda D, Wołowiec D. et al. Expression of PIM-2 and NF-κB genes is increased in patients with acute myeloid leukemia (AML) and acute lymphoblastic leukemia (ALL) and is associated with complete remission rate and overall survival. Postepy Hig Med Dosw (Online). 2013;67:553-9
19. Fox CJ, Hammerman PS, Cinalli RM. et al. The serine/threonine kinase Pim-2 is a transcriptionally regulated apoptotic inhibitor. Genes Dev. 2003;17:1841-54
20. Nawijn MC, Alendar A, Berns A. For better or for worse: the role of Pim oncogenes in tumorigenesis. Nature Rev Cancer. 2011;11:23-34
21. Zhang Y, Wang Z, Li X. et al. Pim kinase-dependent inhibition of c-Myc degradation. Oncogene. 2008;27:4809-19
22. Yan B, Zemskova M, Holder S. et al. The PIM-2 kinase phosphorylates BAD on serine 112 and reverses BAD-induced cell death. J Biol Chem. 2003;278:45358-67
23. Ren K, Zhang W, Shi Y. et al. Pim-2 activates API-5 to inhibit the apoptosis of hepatocellular carcinoma cells through NF-kappaB pathway. Pathol Oncol Res. 2010;16:229-37
24. Lin YW, Beharry ZM, Hill EG. et al. A small molecule inhibitor of Pim protein kinases blocks the growth of precursor T-cell lymphoblastic leukemia/lymphoma. Blood. 2010;115:824-33
25. Song HT, Qin Y, Yao GD. et al. Astrocyte elevated gene-1 mediates glycolysis and tumorigenesis in colorectal carcinoma cells via AMPK signaling. Mediators Inflamm. 2014;2014:287381
26. Brault L, Menter T, Obermann EC. et al. PIM kinases are progression markers and emerging therapeutic targets in diffuse large B-cell lymphoma. Br J Cancer. 2012;107:491-500
27. Smallbone K, Gatenby RA, Gillies RJ. et al. Metabolic changes during carcinogenesis: potential impact on invasiveness. J Theor Biol. 2007;244:703-13
28. Gatenby RA, Gillies RJ.Glycolysis in cancer. a potential target for therapy. Int J Biochem Cell Biol. 2007;39:1358-66
29. Ganapathy-Kanniappan S, Geschwind JF. Tumor glycolysis as a target for cancer therapy: progress and prospects. Mol Cancer. 2013;12:152
30. Jang M, Kim SS, Lee J. Cancer cell metabolism: implications for therapeutic targets. Exp Mol Med. 2013;45:e45
31. Hammerman PS, Fox CJ, Cinalli RM. et al. Lymphocyte transformation by Pim-2 is dependent on nuclear factor-kappaB activation. Cancer Res. 2004;64:8341-8
32. Yu Z, Zhao X, Huang L. et al. Proviral insertion in murine lymphomas 2 (PIM2) oncogene phosphorylates pyruvate kinase M2 (PKM2) and promotes glycolysis in cancer cells. J Biol Chem. 2013;288:35406-16
33. Yu Z, Zhao X, Ge Y. et al. A regulatory feedback loop between HIF-1α and PIM2 in HepG2 cells. PLoS One. 2014;9:e88301
34. Sun Q, Chen X, Ma J. et al. Mammalian target of rapamycin up-regulation of pyruvate kinase isoenzyme type M2 is critical for aerobic glycolysis and tumor growth. Proc Natl Acad Sci U S A. 2011;108:4129-34
Author contact
![]() Corresponding author: Prof. Wei-liang Yang, The Second Affiliated Hospital of Harbin Medical University, Road Xuefu 246, Harbin, 150086, China. Tel. and Fax: 86-451-8660475; E-mail: yangweiliang163.com or yangweiliang08com
Corresponding author: Prof. Wei-liang Yang, The Second Affiliated Hospital of Harbin Medical University, Road Xuefu 246, Harbin, 150086, China. Tel. and Fax: 86-451-8660475; E-mail: yangweiliang163.com or yangweiliang08com