Impact Factor ISSN: 1449-1907
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Issue 4; 2026
- Volume 23; 2026
- Past Issues
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Conclusions
Abbreviations
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2015; 12(1):83-91. doi:10.7150/ijms.10101 This issue Cite
Research Paper
Nerve Growth Factor Protects the Ischemic Heart via Attenuation of the Endoplasmic Reticulum Stress Induced Apoptosis by Activation of Phosphatidylinositol 3-Kinase
Ke Wei, Li Liu, Fei Xie, Xuechao Hao, Jie Luo, Su Min ![]()
Department of Anesthesiology, The First Affiliated Hospital of Chongqing Medical University, Chongqing, China.
Received 2014-7-14; Accepted 2014-11-3; Published 2015-1-1
Abstract
Background: Increased expression of nerve growth factor (NGF) has been found in the myocardium suffered from ischemia and reperfusion (I/R). The pro-survival activity of NGF on ischemic heart has been supposed to be mediated by phosphatidylinositol 3-kinase/protein kinase B (PI3K/Akt) signaling pathway. Endoplasmic reticulum (ER) stress, which is activated initially as a defensive response to eliminate the accumulated unfolded proteins, has shown a critical involvement in the ischemia induced myocardial apoptosis. This study was aimed to investigate whether NGF induced heart protection against I/R injury includes a mechanism of attenuation of ER stress-induced myocardial apoptosis by activation of PI3K/Akt pathway.
Methods: Isolated adult rat hearts were perfused with a Langendörff perfusion system. Hearts in the Sham group were subjected to 225 min of continuous Krebs-Henseleit buffer (KHB) perfusion without ischemia. Hearts in I/R group were perfused with KHB for a 75-min of equilibration period followed by 30 min of global ischemia and 120 min of KHB reperfusion. Hearts in the NGF group accepted 45 min of euilibration perfusion and 30 min of NGF pretreatment (with a final concentration of 100 ng/ml in the KHB) before 30 min of global ischemia and 120 min of reperfusion. Hearts in K252a and LY294002 groups were pretreated with either a TrkA inhibitor, K252a or a phosphatidyl inositol 3-kinase inhibitor, LY294002 for 30 min before NGF (100 ng/ml) administration. Cardiac hemodynamics were measured from the beginning of the perfusion. Cardiac enzymes and cardiac troponin I (cTnI) were assayed before ischemia and at the end of reperfusion. Myocardial apoptosis rate was measured by TUNEL staining, and expression of glucose-related protein 78 (GRP78), CCAAT/enhancer-binding protein homologous protein (CHOP), caspase-12, total- and phospho-(Ser473)-Akt were assessed by Western blot analyses.
Results: NGF pretreatment significantly improved the recovery of post-ischemia cardiac hemodynamics. Reduced creatine kinase-MB (CK-MB), lactate dehydrogenase (LDH) activity and cTnI levels, as well as decreased myocardial apoptosis ratio were observed in the NGF group. The improvement of NGF on recovery of cardiac function and alleviation of myocardial injury were completely abolished by K252a or LY294002. GRP78, caspase-12 and CHOP were highly expressed in ischemic myocardium, while NGF significantly inhibited the overexpression of these proteins which were involved in ER stress-induced myocardial apoptosis. NGF pretreatment also induced phosphorylation of Akt. When the activation of PI3K/Akt pathway is blocked by LY294002, the NGF induced suppression of the apoptosis-related proteins expression was reversed.
Conclusions: NGF pretreatment may protect the ischemic heart via inhibition of the ER stress-induced apoptosis; this pro-survival effect is mediated by PI3K/Akt pathway.
Keywords: ischemia/reperfusion injury, nerve growth factor, endoplasmic reticulum, apoptosis.
Introduction
Nerve growth factor (NGF) is one of the representative members of the neurotrophin family, which includes brain derived neurotrophic factor (BDNF), neurotrophin-3 (NT-3), and neurotrophin-4/5 (NT-4/5). It can be synthesized and secreted by both immature and mature cardiac myocytes, and its expression level changes following myocardial injury. Studies on cardiovascular diseases have proved the impact of neurotrophins on heart formation, angiogenesis and regeneration of cardiac sympathetic nerves [1-3]. Recent studies further demonstrated a pro-survival activity of NGF on the ischemic myocardium. Overexpressed NGF and its high-affinity receptor, tyrosine kinase (TrkA), were observed both in the ischemic rat and human hearts [4, 5]. In another study by Caporali et al., NGF was found to protect cardiomyocytes from hypoxia/reoxygenation or angiotensin induced apoptosis [6]. Although little is known about the mechanism of NGF induced pro-survival effect on ischemic myocardium, some studies have attributed it to the activation of the phosphatidylinositol 3-kinase (PI3K) signaling pathway [6, 7].
As a highly dynamic and multifunctional signaling organelle in eukaryotic cells, the endoplasmic reticulum (ER) is closely involved in the synthesis and folding of proteins, calcium homeostasis, and biosynthesis of lipids. Under certain pathological conditions, such as ischemia, hypoxemia, and ATP depletion, when unfolded proteins accumulate in the ER, the transmembrane sensors activate the unfolded protein response (UPR) to eliminate and degrade the unfolded and misfolded proteins. However, when these adaptation responses fail to deal with the unfolded proteins, cell apoptosis is triggered [8]. Several ER stress-related signaling pathways have been proposed to be associated with this programmed cell death, including the activation of CHOP and caspase-12 [9-11].
PI3K/Akt signaling pathway has been thought to mediate the anti-apoptotic process in a series of studies [12]. In a rabbit autoimmune cardiomyopathy model, Mao et al. reported a cardio-protective effect of darbepoetin alfa on attenuating ER stress-induced apoptosis by activation of phosphatidylinositol 3-kinase/protein kinase B (PI3K/Akt) signaling pathway [13]. Studies on PC12 cells also observed a PI3K/Akt mediated protection on ER stress-induced apoptosis after the treatment of exogenous NGF [14, 15]. However, in the ischemic heart, the relationship between NGF and the ER stress-induced apoptosis is still unknown, neither is the role of PI3K/Akt pathway in the NGF induced pro-survival process.
In present study, the impact of NGF on ER stress-induced myocardial apoptosis was investigated in isolated rat hearts undergoing total ischemia and reperfusion (I/R). In addition, role of PI3K/Akt pathway on this NGF triggered protection was assessed with PI3K inhibitor LY294002.
Materials and Methods
Animals
All experiments were approved by the Institutional Animal Care and Use Committee of Chongqing Medical University. All animals received humane care in compliance with the Guide for the Care and Use of Laboratory Animals of the U.S. National Institutes of Health (NIH Publication No.85-23, revised 1996). Adult male Wistar rats with body weight between 200-220 g were used.
Isolated I/R heart model
Rats were anesthetized with pentobarbital sodium (40 mg/kg, intraperitoneally) and administered heparin (150U/kg, intraperitoneally). Then, hearts were rapidly isolated and connected to the Langendörff perfusion system. Krebs-Henseleit buffer (KHB) retrogradely perfused the heart via aorta. The perfusion pressure was maintained at 70 cmH2O. The perfusate was bubbled with a 95% O2-5% CO2 gas mixture, and the bubbling rate was adjusted to maintain a physiological pH (7.35-7.45). The perfusate temperature was maintained at 38°C. The basilar part of the pulmonary artery was cut to allow coronary perfusate flow. A water-filled latex balloon, connected via a catheter to a pressure transducer (Powerlab), was inserted in the left ventricle. The pressure transducer was connected to a computerized chart recorder system (Macintosh Quardra610, Maclab charts 3.6v/s) to record the left ventricular developed pressure (LVDP), left ventricular end-diastolic pressure (LVEDP) and maximum increase rate and decrease rate of left ventricular pressure (±dp/dtmax).
Chemicals
NGF from rat, K252a and LY294002 were obtained from Sigma-Aldrich (St. Louis, Missouri, USA) and were dissolved in dimethyl sulfoxide (DMSO) before being added to the buffer. The final concentration of DMSO was <0.1%. KHB was composed as follows: NaCl 118.5 mM, NaHCO 325 mM, KCl 4.8 mM, MgSO4 1.2 mM, KH2PO4 1.2 mM, CaCl2 2.5 mM, Glucose 11 mM.
Experimental protocol
The experimental protocol is showed in Figure 1. The hearts (n = 30) were randomly assigned to one of the five groups (n = 6 for each group):
Schematic diagram of the experimental protocol.
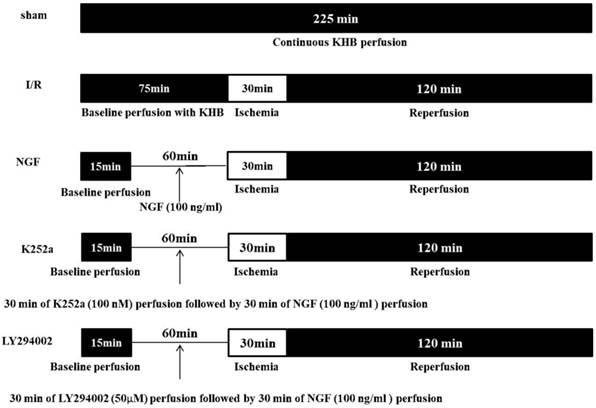
Sham group: hearts were subjected to 225 min of continuous KHB perfusion without I/R.
I/R Group: the hearts were subjected to a stabilization period of KHB perfusion for 75 min followed by 30 min of global ischemia and 120 min of reperfusion.
NGF group: after 45 min of stabilization period, the hearts were perfused with KHB contained with 100 ng/ml of NGF for 30 min followed by I/R.
LY294002 group and K252a group: 50 μM of PI3K inhibitor LY294002 or 100 nM of TrkA receptor inhibitor K252a was perfused for 30 min before NGF administration, then the hearts underwent I/R.
Hemodynamics examination during I/R
LVDP, LVEDP,±dp/dtmax and coronary flow rate (CFR) were continuously measured during the whole perfusion process.
Biochemical assay
Coronary effluent samples were collected for determination of the activity of creatine kinase (CK-MB) and lactate dehydrogenase (LDH) by auto-analyzer (AU5400; Olympus Diagnostics, NY, USA) at the end of stabilization period (as the baseline level, BL) and the end of reperfusion. Concentration of cardiac troponin I (cTnI) was also measured with the same sample by automated chemiluminescence system (ACS 180; Bayer Corp., NY, USA).
TUNEL Staining
At the end of reperfusion, hearts were quickly removed from the perfusion system. Myocardium tissues in the left ventricle were cut into 2-mm thick specimens. The samples were then fixed in 4% precooled paraformaldehyde for 72 h and embedded in paraffin. Paraffin-embedded tissues were sectioned into 5-μm thick slices. Apoptosis was measured by using the terminal dUTP nick-labeling (TUNEL) assay according to the manufacturer's instructions (Roche Diagnostics, Basel, Switzerland). Five fields from each heart were randomly selected with an optical microscope and analyzed in a blinded manner. The apoptotic rate was calculated as the ratio of TUNEL-positive nuclei to the total number of cardiac myocyte nuclei.
Western blot analysis
Expression of GRP78, CHOP, caspase-12, total and phospho-(Ser473)-Akt (p-Akt) were measured by western blot. Left ventricular tissues were homogenized using lysis buffer (Beyotime, China), and the supernatants were collected after centrifugation at 12,000 × g for 15 min at 4°C. After quantitative analysis of protein concentration, total proteins were separated by sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE), transferred to polyvinylidene fluoride membranes (Millipore, Billerica, MA, USA), blocked with 5% non-fat milk in Tris buffered saline for 1 h at 37°C, and then incubated overnight at 4°C with anti-Akt (Cell signaling, dilution: 1:1000), anti-phospho-Akt (Ser473, Santa Cruz, dilution: 1:200), anti-caspase-12 (Santa Cruz, dilution: 1:200), anti-GRP 78 (Signalway Antibody Co., Ltd, dilution: 1:3000), and anti-CHOP (Beyotime, dilution: 1:1000) as primary antibodies. After incubation for 1 h at 37°C with secondary antibody, bands were seen using the enhanced chemiluminescence kit (Beyotime) according to the manufacturer's protocol. All the results were normalized to glyceraldehyde-3-phosphate dehydrogenase (GAPDH) levels and expressed as fold intensity compared with the Sham group.
Statistical analysis
All values were expressed as mean ± standard deviation (SD). Statistics were performed using SPSS 20.0. Two-way repeated-measures analysis of variance (ANOVA) followed by post-hoc t-test with Bonferroni correction were used for multiple comparisons of LVDP, LVEDP,±dp/dt and CFR. One-way ANOVA and Student-Neuman-Keuls post-test were used to compare the concentration of cardiac enzyme and cTnI, apoptotic rate, expression levels of GRP78, CHOP, caspase-12, Akt, and p-Akt. Liner correlation analysis was used to evaluate the correlation between apoptotic rate and GRP78, CHOP expression. P value <0.05 was considered statistically significant.
Results
Changes of cardiac function
The baseline levels of LVDP, LVEDP ,±dp/dt and CFR were similar among the groups (Fig.2). After 30 min of global ischemia, LVDP, ±dp/dt and CFR values in the I/R, NGF and LY294002 group were dramatically decreased compared with BL and with those in the Sham group (P < 0.05 vs. Sham group).
NGF pretreatment promoted the recovery of cardiac function and coronary flow after ischemia and reperfusion (n=6). (A) The left ventricular developed pressure (LVDP) at the end of stabilization period of perfusion (BL) and different time during reperfusion; (B) The left ventricular end-diastolic pressure (LVEDP) at BL and during reperfusion; (C,D) Maximum increase rate and decrease rate of LVDP (±dp/dtmax ) at BL and during reperfusion; (E) Coronary flow rate at BL and different time after reperfusion. * P<0.05; # P<0.01; ‡ P<0.001; § P<0.0001.
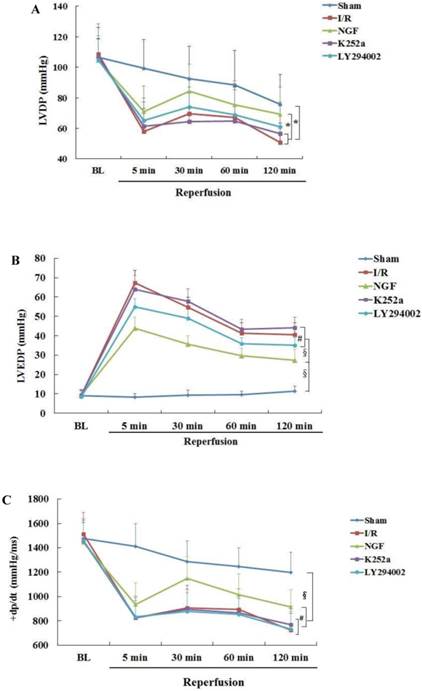
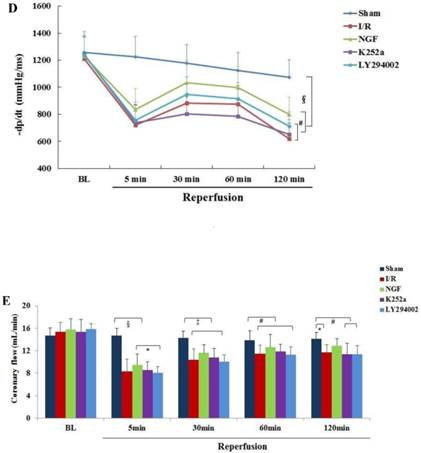
Compared with I/R group, a better recovery of LVDP, LVEDP and ±dp/dt in the NGF group was observed during reperfusion. NGF pretreatment also improved the post-ischemic coronary perfusion, although no statistical significance was found between the NGF group and I/R group. The benefit of NGF on post-ischemic heart function can be completely abolished by K252a or LY294002.
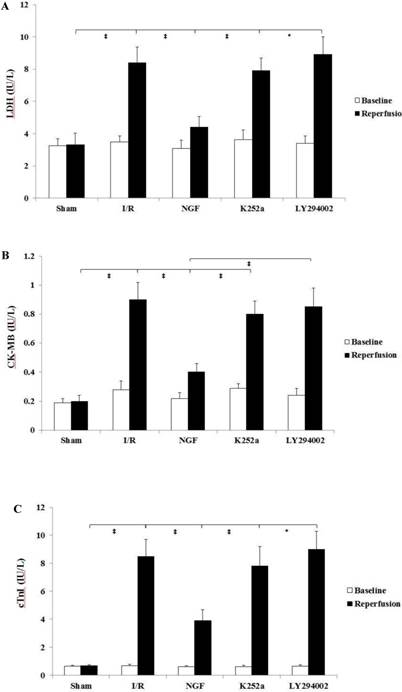
Release of cardiac enzymes
The pre-ischemic level of cardiac enzymes and cTnI were with no statistical differences among the groups (Fig. 3). At the end of reperfusion, NGF pretreatment significantly decreased the cardiac enzymes and cTnI release compared with group I/R. This cardio-protective effect by NGF against I/R injury was reversed when K252a or LY294002 was used.
NGF pretreatment limited the cardiac enzyme and cTnI release (n=6). (A and B) Concentration of CK-MB and LDH in coronary effluent at the end of stabilization period (baseline) and the end of reperfusion. (C) Concentration of cardiac troponin I in the coronary effluent at the end of stabilization period (baseline) and the end of reperfusion. The values are expressed as means±SD, * P<0.05; ‡ P<0.001.
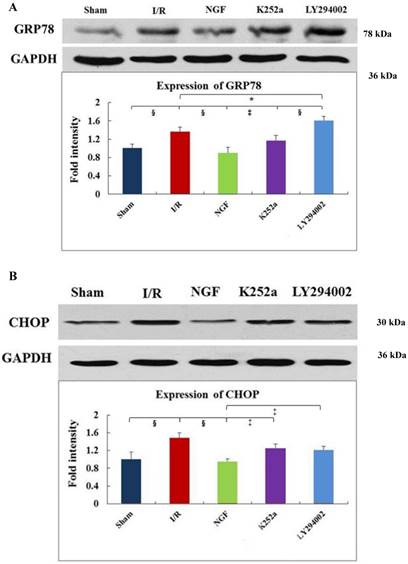
GRP78, CHOP, and caspase-12 expression
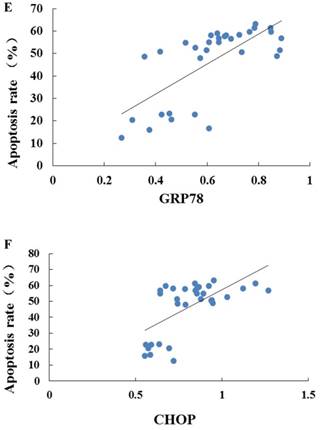
As indicated by Fig 4 A~C, levels of GRP78, CHOP, and caspase-12 expression were markedly up-regulated in the hearts undergoing I/R. NGF pretreatment significantly attenuated the post-ischemic high expression of GRP78, caspase-12 and CHOP. This attenuation was completely blocked when K252a or LY294002 was used. The inhibition on the expression of caspase-12, however, was completely blocked by K252a and partly blocked by LY294002. Furthermore, the level of GRP78 (r= 0.6343, P<0.05; Fig.4E) and CHOP (r= 0.4495, P<0.05; Fig.4F) was positively correlated with the cell apoptosis rate.
Expression of GRP78, caspase-12, CHOP, and total Akt and phosphorylated Akt protein in the myocardium in the Sham, I/R, NGF, K252a and LY294002 group at the end of reperfusion was analyzed by western blot (A~D). Representative immunoblots are shown on the top of the bar graphs. P-Akt is expressed as the ratio of the total Akt. Expression levels of the proteins are shown as fold intensity compared with that in the Sham group. * P < 0.05, # P < 0.01, ‡ P < 0.001, § P < 0.0001. The correlation between apoptosis rate and GRP78, CHOP is presented in (E) and (F).
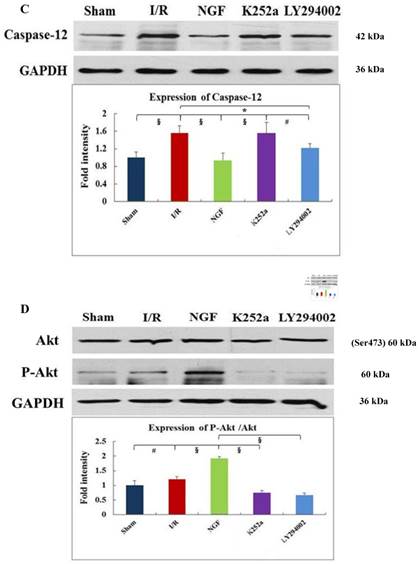
Akt and Phosphorylation of Akt
NGF pretreatment may cause a high expression of p-Akt during I/R. compared with I/R group, ratio of p-Akt/Akt was significantly increased at the end of reperfusion (Fig.4D). However, this NGF induced activation of Akt can be completely inhibited by LY294002 or K252a.
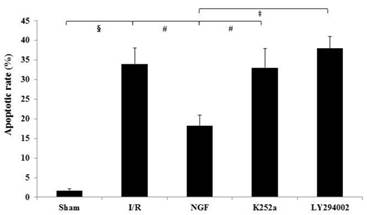
Apoptotic rate
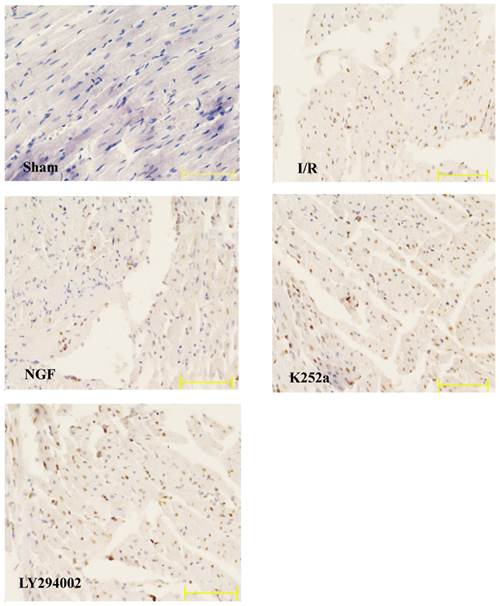
The apoptotic rate of the I/R group was significantly increased after I/R (Fig.5). NGF pretreatment dramatically reduced the apoptotic rate, while K252a and LY294002 abolished this pro-survival effect.
The apoptotic rate of cardiac myocytes in the Sham, I/R, NGF, K252a and LY294002 group. The representative micrographs of TUNEL-stained left ventricular tissue sections are shown above the bar graphs. TUNEL-positive nuclei stained by alkaline phosphatase appear red. Magnification 400×. The apoptotic rate was calculated as the ratio of TUNEL-positive nuclei to the total number of cardiac myocytes nuclei. # P < 0.01; ‡ P < 0.001; § P < 0.0001.
Discussion
The present study demonstrates that exogenous NGF pretreatment may protect the heart from I/R injury and reduce the myocardial apoptosis. The expression of several apoptosis-related proteins involved in ER stress is suppressed during this process. The NGF induced pro-survival activity, along with the down-regulation of the apoptosis-related proteins, can be abolished by PI3K inhibitor LY294002. All these results indicate that NGF may protect the ischemic heart by attenuating the ER stress-induced apoptosis via the mediation of PI3K/Akt signaling pathway.
As a member of neurotrophins with pro-survival effect on cardiovascular system, NGF was found to be crucial in maintaining normal myocardial performance [16]. The cardiovascular activity was initially attributed to promotion of the neuroregeneration process. In an in vivo dog model with left anterior descending coronary occlusion, Abe and his colleagues observed that both exogenous and endogenous NGF may protect the heart from neural stunning [17]. However, later studies found the role of endogenous NGF in several other pathological processes, including angiogenesis, endothelial cells and cardiomyocyte survival, cardiac repair following myocardial infarction [4, 18, 19]. To investigate a non-neurogenic cardioprotective effect of exogenous NGF, we used a denervated heart model to avoid influence of preload and afterload, neural and humoral regulation on heart function. Similar with the result from another in vivo study on rat hearts [20], we observed a protective effect of exogenous NGF on myocardium from I/R injury, as well as an improvement of post-ischemic cardiac function in isolated perfused rat hearts.
The present study demonstrated a potential association between NGF and ER stress-induced apoptosis during I/R. The ER stress, which is also known as the unfolded protein response (UPR), is characterized by the up-regulation of molecular chaperone GRP78 and activation of apoptosis. GRP78 is always combined with several critical transmembrane ER signaling proteins, i.e. activating transcription factor 6 (ATF6), PKR-like ER kinase (PERK), and inositol-requiring enzyme 1 (IRE1) under normal circumstance. These transmembrane proteins are released from GRP78 binding during ER stress and initiating specific apoptosis pathway, which includes the transcriptional induction of C/EBP homologous protein (CHOP) and the activation of caspase-12, -9, and -3. Recent studies reached contradictory results regarding the impact of NGF on the expression of GRP78. In PC12 cells, Mao et al. found that NGF may lead to down-regulation of GRP78 and CHOP, attenuates norepinephrine-induced ER stress and the consequent apoptosis [15]. Nevertheless, in another study with the same cells, NGF was found to enhance the expression of GRP78 and attenuate the 2-deoxy-d-glucose triggered ER stress [21]. In present study, NGF lead to reduced expression of GRP78 after I/R, as well as decreased level of CHOP and caspase-12, both of which were directly related to the extent of myocardial apoptosis. Consequently, our results suggested that the pro-survival activity of NGF on ischemic myocardium was mediated by down-regulation of the ER stress-triggered overexpression of GRP78.
The signaling pathway involved in attenuation of ER stress-induced myocardial apoptosis by NGF remains obscure. A relationship between PI3K/Akt pathway and the pro-survival activity of NGF on ER stress-induced apoptosis in PC12 cells has been proposed [21]. A series of studies revealed that the anti-apoptotic activity of neurotrophins on neurocytes is mediated by activation of the PI3K/Akt signaling pathway [22-25]. Similarly in the myocardium, the pro-survival activity of NGF was also found to be linked with the activation of PI3K/Akt pathway [6]. In present study, Akt was activated following NGF pretreatment, and decreased myocardial injury and apoptosis rate were observed. This anti-apoptotic effect of NGF can be completely abolished by PI3K inhibitor LY294002, thus a role of PI3K/Akt pathway in mediating the cardioprotection of NGF on ischemia induced myocardial apoptosis is confirmed. Furthermore, NGF induced down-regulation of the ER stress-related proteins, i.e. GRP78, CHOP, and caspase-12 were also completely or partially diminished by the PI3K inhibitor, indicating a potential relationship between PI3K/Akt pathway and ER stress-induced apoptosis.
Previous researches have studied the effect of exogenous NGF on cultured cardiac myocytes or rat hearts, the doses of NGF, LY294002 and K252a in present study are chosen according to these studies[6, 20], and also the results of our preliminary tests. Furthermore, to investigate the impact of NGF on early recovery of ischemic heart after reperfusion, NGF was administered before total ischemia with a pretreatment protocol in this study. Pharmacological posttreatment, as well as pretreatment, has shown significant cardioprotection in a series of studies. A number of pharmacological agents have been reported in experimental studies to reduce myocardial infarct size when administered at the onset of myocardial reperfusion, e.g., anesthetics, adenosine, atrial natriuretic peptides, etc. [26-28]. It remains unclear if cardioprotection from pretreatment and posttreatment use the same mechanism [29]. To sudden unexpected ischemia-reperfusion, posttreatment is more practical compared with pretreatment. However, when ischemia/reperfusion injury is predicable, such as cardiac arrest during cardiopulmonary bypass, whether pretreatment is with advantage over posttreatment on cardioprotection needs to be further studied.
It is of concern that the influence of NGF on the expression of caspase12 is only partially abolished by LY294002, thus another pathway may be existed in mediating the anti-apoptosis besides PI3K/Akt. Moreover, although the expression of apoptosis-related proteins included in ER stress is down-regulated by NGF, it is still to be further elucidated whether the ER stress response itself or the apoptosis-related proteins involved in ER stress (e.g. GRP78) is the target of NGF.
Conclusions
In summary, this study demonstrated with an isolated heart model that NGF may protect ischemic heart from I/R injury via attenuating the ER stress-induced apoptosis, this process is mediated by activating of PI3K/Akt pathway.
Abbreviations
Akt: protein kinase B; ATF6: activating transcription factor 6; BDNF: brain derived neurotrophic factor; CHOP: CCAAT/enhancer-binding protein homologous protein; CK-MB: creatine kinase-MB; CFR: coronary flow rate; cTnI: cardiac enzymes and cardiac troponin I; ±dp/dtmax: maximum increase rate and decrease rate of left ventricular pressure; DMSO: dimethyl sulfoxide; ER: endoplasmic reticulum; GAPDH: glyceraldehyde-3-phosphate dehydrogenase;GRP78: glucose-related protein 78; I/R: ischemia and reperfusion; IRE1: inositol-requiring enzyme 1; KHB: Krebs-Henseleit buffer; LDH: lactate dehydrogenase; LVDP: left ventricular developed pressure; LVEDP: left ventricular end-diastolic pressure; NGF: nerve growth factor; NT-3: neurotrophin-3; NT-4/5: neurotrophin-4/5; PERK: PKR-like ER kinase; PI3K: phosphatidylinositol 3-kinase; TrkA: Tropomyosin kinase A; TUNEL: terminal deoxynucleotidyl transferase-mediated dUTP nick-endlabeling; UPR: unfolded protein response.
Acknowledgements
This study was supported by grants from the National Natural Science Foundation of China (Project number: 81000066) and the grants for National Key Clinical Specialty construction. We thank Lixue Chen, PhD. (from the experiment center of the First Affiliated Hospital of Chongqing Medical University) for his respectable help in this study.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Caporali A, Emanueli C. Cardiovascular actions of neurotrophins. Physiol Rev. 2009;89:279-308
2. Lee R, Kermani P, Teng KK, Hempstead BL. Regulation of cell survival by secreted proneurotrophins. Science. 2001;294:1945-1948
3. Snider WD. Functions of the neurotrophins during nervous system development: what the knockouts are teaching us. Cell. 1994;77:627-638
4. Meloni M, Caporali A, Graiani G. et al. Nerve growth factor promotes cardiac repair following myocardial infarction. Circ Res. 2010;106:1275-1284
5. Hiltunen JO, Laurikainen A, Vakeva A, Meri S, Saarma M. Nerve growth factor and brain-derived neurotrophic factor mRNAs are regulated in distinct cell populations of rat heart after ischaemia and reperfusion. J Pathol. 2001;194:247-253
6. Caporali A, Sala-Newby GB, Meloni M. et al. Identification of the prosurvival activity of nerve growth factor on cardiac myocytes. Cell Death Differ. 2008;15:299-311
7. Shimoke K, Kishi S, Utsumi T. et al. NGF-induced phosphatidylinositol 3-kinase signaling pathway prevents thapsigargin-triggered ER stress-mediated apoptosis in PC12 cells. Neurosci Lett. 2005;389:124-128
8. Glembotski CC. The role of the unfolded protein response in the heart. J Mol Cell Cardiol. 2008;44:453-459
9. Oyadomari S, Mori M. Roles of CHOP/GADD153 in endoplasmic reticulum stress. Cell Death Differ. 2004;11:381-389
10. Hitomi J, Katayama T, Taniguchi M, Honda A, Imaizumi K, Tohyama M. Apoptosis induced by endoplasmic reticulum stress depends on activation of caspase-3 via caspase-12. Neurosci Lett. 2004;357:127-130
11. Nakagawa T, Zhu H, Morishima N. et al. Caspase-12 mediates endoplasmic-reticulum-specific apoptosis and cytotoxicity by amyloid-beta. Nature. 2000;403:98-103
12. Hyoda K, Hosoi T, Horie N, Okuma Y, Ozawa K, Nomura Y. PI3K-Akt inactivation induced CHOP expression in endoplasmic reticulum-stressed cells. Biochem Biophys Res Commun. 2006;340:286-290
13. Mao W, Iwai C, Liu J, Sheu SS, Fu M, Liang CS. Darbepoetin alfa exerts a cardioprotective effect in autoimmune cardiomyopathy via reduction of ER stress and activation of the PI3K/Akt and STAT3 pathways. J Mol Cell Cardiol. 2008;45:250-260
14. Shimoke K, Amano H, Kishi S, Uchida H, Kudo M, Ikeuchi T. Nerve growth factor attenuates endoplasmic reticulum stress-mediated apoptosis via suppression of caspase-12 activity. J Biochem. 2004;135:439-446
15. Mao W, Iwai C, Keng PC, Vulapalli R, Liang CS. Norepinephrine-induced oxidative stress causes PC-12 cell apoptosis by both endoplasmic reticulum stress and mitochondrial intrinsic pathway: inhibition of phosphatidylinositol 3-kinase survival pathway. Am J Physiol Cell Physiol. 2006;290:C1373-1384
16. Govoni S, Pascale A, Amadio M. et al. NGF and heart: Is there a role in heart disease? Pharmacol Res. 2011;63:266-277
17. Abe T, Morgan DA, Gutterman DD. Protective role of nerve growth factor against postischemic dysfunction of sympathetic coronary innervation. Circulation. 1997;95:213-220
18. Emanueli C, Salis MB, Pinna A, Graiani G, Manni L, Madeddu P. Nerve growth factor promotes angiogenesis and arteriogenesis in ischemic hindlimbs. Circulation. 2002;106:2257-2262
19. Cantarella G, Lempereur L, Presta M. et al. Nerve growth factor-endothelial cell interaction leads to angiogenesis in vitro and in vivo. FASEB J. 2002;16:1307-1309
20. Strande JL, Routhu KV, Lecht S, Lazarovici P. Nerve growth factor reduces myocardial ischemia/reperfusion injury in rat hearts. J Basic Clin Physiol Pharmacol. 2013;24:81-84
21. Kishi S, Shimoke K, Nakatani Y. et al. Nerve growth factor attenuates 2-deoxy-d-glucose-triggered endoplasmic reticulum stress-mediated apoptosis via enhanced expression of GRP78. Neurosci Res. 2010;66:14-21
22. Dudek H, Datta SR, Franke TF. et al. Regulation of neuronal survival by the serine-threonine protein kinase Akt. Science. 1997;275:661-665
23. Shimoke K, Yamada M, Ikeuchi T, Hatanaka H. Synthetic lipid products of PI3-kinase which are added to culture medium prevent low K+-induced apoptosis of cerebellar granule neurons via Akt kinase activation. FEBS Lett. 1998;437:221-224
24. Brunet A, Datta SR, Greenberg ME. Transcription-dependent and -independent control of neuronal survival by the PI3K-Akt signaling pathway. Curr Opin Neurobiol. 2001;11:297-305
25. Shimoke K, Chiba H. Nerve growth factor prevents 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced cell death via the Akt pathway by suppressing caspase-3-like activity using PC12 cells: relevance to therapeutical application for Parkinson's disease. J Neurosci Res. 2001;63:402-409
26. Sanada S, Komuro I, Kitakaze M. Pathophysiology of myocardial reperfusion injury: preconditioning, postconditioning, and translational aspects of protective measures. Am J Physiol Heart Circ Physiol. 2011;301:H1723-1741
27. Mahaffey KW, Puma JA, Barbagelata NA. et al. Adenosine as an adjunct to thrombolytic therapy for acute myocardial infarction: results of a multicenter, randomized, placebo-controlled trial: the Acute Myocardial Infarction STudy of ADenosine (AMISTAD) trial. J Am Coll Cardiol. 1999;34:1711-1720
28. Kitakaze M, Asakura M, Kim J. et al. Human atrial natriuretic peptide and nicorandil as adjuncts to reperfusion treatment for acute myocardial infarction (J-WIND): two randomised trials. Lancet. 2007;370:1483-1493
29. Ovize M, Baxter GF, Di Lisa F. et al. Postconditioning and protection from reperfusion injury: where do we stand? Position paper from the Working Group of Cellular Biology of the Heart of the European Society of Cardiology. Cardiovasc Res. 2010;87:406-423
Author contact
![]() Corresponding author: Su Min, MD., Professor and Chairman, Department of Anesthesiology, The First Affiliated Hospital of Chongqing Medical University, Chongqing 400016, China. Phone and Fax: 86-23-89011068; E-mail: wk202448com.
Corresponding author: Su Min, MD., Professor and Chairman, Department of Anesthesiology, The First Affiliated Hospital of Chongqing Medical University, Chongqing 400016, China. Phone and Fax: 86-23-89011068; E-mail: wk202448com.