Impact Factor ISSN: 1449-1907
- Issue 9; 2026
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Volume 23; 2026
- Past Issues
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
General explanatory notes to the...
Introduction
Chemical procedures
Major achievements
Outlook
References
Footnotes

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2014; 11(7):697-706. doi:10.7150/ijms.8168 This issue Cite
Research Paper
A Peptide & Peptide Nucleic Acid Synthesis Technology for Transporter Molecules and Theranostics - The SPPS
Ruediger Pipkorn1 ![]() , Klaus Braun2, Manfred Wiessler2, Waldemar Waldeck3, Hans-Hermann Schrenk2, Mario Koch1, Wolfhard Semmler2, Dorde Komljenovic2
, Klaus Braun2, Manfred Wiessler2, Waldemar Waldeck3, Hans-Hermann Schrenk2, Mario Koch1, Wolfhard Semmler2, Dorde Komljenovic2
1. German Cancer Research Center, Dept. of Translational Immunology, INF 410, D-69120 Heidelberg, Germany
2. German Cancer Research Center, Dept. of Medical Physics in Radiology, INF 280, D-69120 Heidelberg, Germany
3. German Cancer Research Center, Division of Biophysics of Macromolecules, INF 580, D-69120 Heidelberg, Germany.
Received 2013-11-19; Accepted 2014-3-25; Published 2014-5-7
Abstract
Advances in imaging diagnostics using magnetic resonance tomography (MRT), positron emission tomography (PET) and fluorescence imaging including near infrared (NIR) imaging methods are facilitated by constant improvement of the concepts of peptide synthesis. Feasible patient-specific theranostic platforms in the personalized medicine are particularly dependent on efficient and clinically applicable peptide constructs. The role of peptides in the interrelations between the structure and function of proteins is widely investigated, especially by using computer-assisted methods.
Nowadays the solid phase synthesis (SPPS) chemistry emerges as a key technology and is considered as a promising methodology to design peptides for the investigation of molecular pharmacological processes at the transcriptional level. SPPS syntheses could be carried out in core facilities producing peptides for large-scale scientific implementations as presented here.
Keywords: Functional peptides, Ligation chemistry, Linker systems, Solid Phase Peptide Synthesis, Therapy, Diagnostics, Theranostics.
General explanatory notes to the peptide synthesis chemistry
Proteins play a pivotal role in various biological processes in living cells. They serve as structural construction materials, transmit messages inside and outside of cells, and are responsible for regulating the cellular metabolism. Proteins are composed of long chains of amino acids, shorter chains are called peptides. Peptides or proteins can be used to identify structures recognized by antibodies or dynamic interactions between nucleic acids (DNA/RNA) and proteins. The transport of substances through physiological membranes can be studied with the aid of such functional peptides. These peptides are indispensable to develop treatment approaches at the molecular level by triggering the up-or-down regulation of specific gene functions.
The demand for peptide-based pharmaceutical products like the human insulin and test materials for the pharmaceutical research is exponentially growing.
Introduction
One approach to meet the demand for peptide-based pharmaceutical products like the human insulin and test pieces for the pharmaceutical research is the combinatorial chemistry. It was introduced in the 1980s, based on the pioneering works of Furka [1-4], Geysen [5-8], Houghten [9-12], Lam [13-17] and Moos [18-21]. The combinatorial chemistry was considered to be a promising technology and traces back to health-economic questions raised in the 1930's [22-25].
Until 1922 the amount of insulin isolated from cattle pancreas satisfied the needs of the diabetic patient-centered care, but in the 1930s the difficulties in obtaining sufficient amounts of pancreas organs increased evidently [26-28]. The first chemical synthesis of insulin, composed of 51 amino acids was published by Meierhofer and Zahn in 1963 [29]. Earlier, the total chemical synthesis of insulin was not assumed to be a successful technical method and the interest for it remained solely academical. Semi synthesis strategies also failed to secure reproducible yield rates [30-33] and a lack of available insulin occurred. The dilemma was documented in the “National Diabetes Advisory Board“ (1976), revealing the predicted demand of insulin until the year 2000, a task which natural sources fulfill [34].
The solid phase peptide synthesis (SPPS) with the amino-protecting group strategy introduced in 1963 and 1972 by Merrifield [35, 36] and Carpino [37, 38], yielded encouraging results and was considered to be a solution to meet the foreseen insulin shortage. Starting in 1977 with genetic engineering and recombinant chemistry methods the production of sufficient amounts of human insulin was possible by gene transfer to microorganisms [39].
SPPS proved to be a valuable option for the insulin synthesis, yet failed to secure the production of sufficient amounts of insulin. In the SPPS technology the educts are covalently linked to carrier resins which are insoluble in the reaction medium conducting the complete synthesis steps. The advantages of the SPPS over the synthesis in solution are evident: simple reuse of reacting agents by filtration, and the feasibility of automation. Simultaneous syntheses of manifold different chemical compounds, either single or mixed substances are possible. The SPPS is not only qualified by the accurately defined synthesis steps and in the realization of pure and homogeneous reaction products with high yields, but also in the multifaceted compounds inconceivable in the combinatory chemistry. These relevant attributes predestined this technology as a key tool in the systematic development of active peptide-based pharmaceutical compounds like hormones, neurotransmitters, and as references in the protein research, like modified antibody fragments and conformation analysis [40, 41].
This technology can be carried out in core facilities and can be considered as the key technology for multiple functionalization procedures, like the ligation of complex modifications which allow
- the access to preparation of homogeneous proteins.
- the access to proteins suitable for biophysical, binding, targeting and structural studies
- the unlimited variability in the synthesis of the peptide chain with entirely new properties
- introduction of active site modification
- development of peptide-based pharmacologically active molecules
- the connection of proteins with (bio)-polymers, like nucleic acid derivatives (chimers)
Chemical procedures
The chemical procedures of the SPPS were optimized according to the chemical and physical properties of the reaction partners and their resulting reaction products. Here the multi-faceted peptide-based molecules and conjugates underline the diversity of the exemplified SPPS methodologies:
Basic principles of the SPPS
During the chemical synthesis the amino acids are connected with each other by peptide-bond formation of acid amides via a successive coupling reaction. For a directional course of the condensation reaction both, the N-terminal amino group of the first reactant and the C-terminal carboxyl-group of the other reactant must be protected. This is achieved by cleavable protection groups. To avoid undesired secondary reactions, the reactive side chains of the so-called trifunctional amino acids (Lys, Glu, Asp, Ser, Thr, Tyr, Cys) must also be protected in a reversible form.
In this method of the SPPS, the peptide is built up sequentially from the C- to the N-terminus on a polymeric resin (Figure 1). In the first step of the peptide synthesis the C-terminal amino acid is connected with its carboxyl group to the polymeric resin via a linker. This linker allows the cleavage of the peptide from the resin after the end of synthesis under specific conditions. The next following protected amino acid (in the sequence) is then activated by coupling reagents (e.g. an ester) and reacts with the free amino end of the peptide chain at the carboxyl-terminus.
illustrates the principle steps of the SPPS (as described above). In a final step, the peptide is released from the support and the side-chain protecting groups removed. Generally, side-chain protecting groups and resin linkage are chosen in a way that protecting groups can be removed and the assembled peptides released under the same conditions.
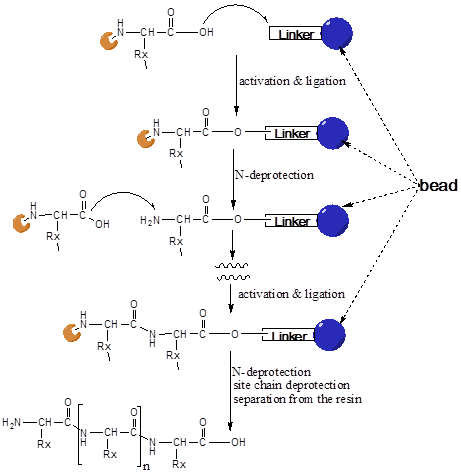
After deprotection by cleavage of the amino-protecting group, the next N-terminal protected amino acid ligation step follows. These coupling and cleavage cycles are repeated, until the peptide has reached the desired length. After finishing the couplings, the peptide is removed from the resin, i.e. the covalent bond between C-terminal amino acid and the anchor of the polymeric resin is separated. Dependent on the linker a peptide as free acid or a peptide as amide is released. The side chain protecting groups are in most cases split off at the same time.
Major achievements
Synthesis of pharmacologically active peptides
The insulin synthesis
The synthesis of the A- and B-chains in E. coli systems is documented but the oxidation of the insulin molecule was extremely inefficient. In 1979, Birr and Pipkorn described the first solid phase synthesis of the insulin A-chain harbouring selectively cleavable protecting groups. Under a stepwise oxidation of this A-chain and formation of the disulfide bridges with a natural B-chain the reaction product was combined to a peptide conjugate with biological hormone activity [42]. The reaction steps of the synthesis are illustrated in Figure 2 and described here in detail1.
illustrates the chemical route of the reaction steps started with the fully protected insulin A-chain upper line. Protecting groups used in the synthesis of the insulin A-chain. The detailed chemical procedure of the synthesis was described previously1. DDZ- = dimethyl-3,5-dimethoxybenzyloxycarbonyl residue; SBu- = t-butylthio; OBU-= t-butoxy; ACM-= acetamidomethyl; Bu-= t-butyl; MBZL- =4-methylbenzyl (modified from Pipkorn [42])
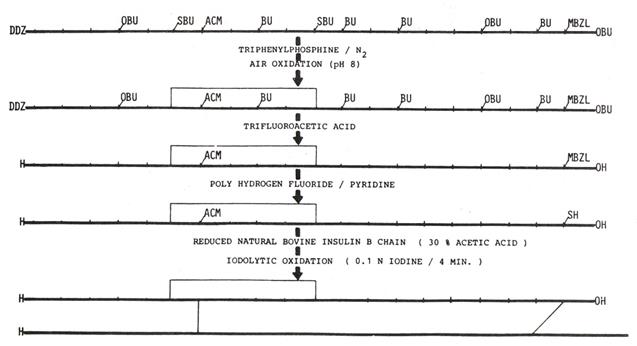
This first successful access to the A-chain of the pharmacologically active insulin by use of the SPPS was considered as a step to the multi-faceted field of peptide syntheses for functional and for structural determination studies of viral and non-viral proteins as described here:
Proalbumin functionalized with an arginine hexapeptide extension
Based on previous studies of the regulation of serum albumin [43] the investigations were engrossed by studying the regulatory function, the biosynthesis and the intracellular transport. The studies were directed to the pre-processed intracellular proalbumin molecule whose C-terminus consists of the hexapeptide Arg-Gly-Val-Phe-Arg-Arg. For this purpose we used two SPPS produced peptides on cross-linked solid phase polystyrene gels functionalized with 2-oxoethyl bromide acting as an anchor. The coupling procedures of carbonyldiimidazole to 1-hydroxybenzotriazole facilitated the preparations of the Arg-rich sequences as documented in 1982 [44].
A further object of the SPPS was the transforming protein pp60SRC as a peptide for functional analysis.
The synthesis of phosphoprotein pp60SRC
Transforming protein, phosphoprotein pp60SRC, encoded by the Rous sarcoma virus (SRC) acts as a tyrosine-specific phosphokinase [45, 46]. The synthesis was performed with short synthetic peptides (6-9 amino acids in length) corresponding to the sequences of different regions of the primary structure of pp60SRC [47].
We also synthesized long peptides for regional studies of the HIV-1 genome.
HIV-1 proteins p24 and gp41 for immunosuppressive studies
The variability of the SPPS allows the production of long peptides in high yields and purity sufficient for structural studies on human immunodeficiency virus type 1 (HIV-1) in p24 gag reactive sera. The tested sera reacted with (61-77 amino acids) p24-derived peptides in enzyme immunoassays (EIAs) [48]. Specificity and singularity of the reaction were demonstrated by further enhanced competition studies with synthetic peptides [49].
The effects of the peptides on the immunosuppressive properties caused by HIV and the influence on the number of T-helper lymphocytes and the impairment of their function were investigated by use of long peptides with overlapping epitopes within a narrow part of gp41 as shown in Table 1 [50].
lists the synthesized and the tested peptides and the amino acid sequences.*
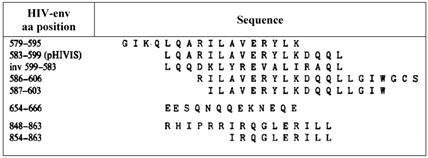
*) modified from Klasse [50].
Furthermore, for studies on HIV-1 infected patients' continuative anti-V3 IgG response, such designed peptides proved to accomplish needed serological analyses.
Antigenic peptides against V3 IgG
Serological studies aim to illuminate the differences between African and non-African HIV-1 strains. The antibody reactivity of 34 Swedish, 30 Tanzanian and 42 Zimbabwean HIV-1-positive sera to 67 synthetic peptides, synthesized by SPPS, were investigated and compared with sequences from North American and African HIV-1 isolates, derived from regions of gag and env known to be antigenic [49, 51, 52].
Peptides and peptide nucleic acids (PNAs) for delivery and targeting studies against the capsid assembly inhibitor (CAI) of HIV-1
The number of drug-resistant variants of HIV increased and the exploration of new alternative targets is necessary for the next generation of antiviral drug development. Here the synthesis and application of functional peptides [53] facilitating the passage across the cell membrane of CAI-directed PNAs (Figure 3) to the gag gene, expressed from the unspliced viral mRNA, are documented [53].
illustrates the schematized modular compositions of the investigated CAI-BioShuttle conjugates. The inhibitor peptide, control peptides, and the transmembrane transport module are connected with a sulfur bridge between the two cysteines (Single letter symbol C [bold]). Horizontally: ❶ represents the modules of the CAI-BioShuttle, ❷ the CAICTRL-BioShuttle, and ❸ and ❹ the BioShuttle connected to the reverse form of the CAI-inhibitor and the control, respectively. Vertically: ❶ CAI-Inhibitor, ❶ scrambled control and their corresponding peptides in reverse orientation ❸ and ❹ respectively. Middle column shows the transmembrane transport module. 1) Biotin; 2) scrambled sequence; 3) sulfur bridge (modified from Braun [53]).
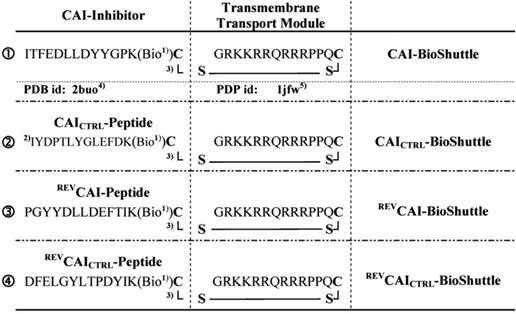
Peptides for neurotoxicity studies
Special synthesis of peptides for functional studies of Alzheimer's disease
Alzheimer's disease is characterized by progressive neuronal dysfunction, reactive gliosis, and the formation of amyloid plaques in the brain. The amyloid precursor protein (APP) copper-binding domain (CuBD) has a pivotal role in the Cu metabolism reducing Cu2+ to Cu+ and mediating copper-induced oxidation processes [54]. The APP CuBD is involved in the modulating Cu homeostasis and amyloid-β peptide production. The chemical synthesis of the tested APP CuBD peptides the Fmoc strategy was used as described [55].
Functionalization of catalytically active enzyme-fragments for structural studies
Myristoylation, phosphorylation and deamidation
Several isoenzymes of the catalytic subunit of cAMP-dependent protein kinase arise through posttranslational modifications of the enzyme outside the catalytic domain. A clustering of sites for such a modification exists at the N-terminus of the protein, where myristoylation (of Gly1), phosphorylation (at Ser10), and deamidation of Asn2 have been observed [56-58]. Biological significance of these is not yet fully understood. To learn more about the mutual influence of various modifications, myristoylation, deamidation, and phosphorylation, a set of hexadecapeptides based on the Cα gene product was synthesized by SPPS (peptide synthesis and purification are here described in detail2) with different combinations of these modifications including variations of the chain length of the fatty acid. As deamidation in vivo could lead to the formation of iso(β)Asp, peptides with this residue were added to the set. The structural behaviour of these peptides under the influence of solvent polarity was studied by CD-spectroscopy [59].
Chemical diagnostics and therapeutical approaches
Synthesis of an intracellular Gd-based contrast agent in magnetic resonance imaging (MRI)
The commonly used gadolinium (Gd3+)-based MRI-contrast agents are able to display the intercellular space very well, but they are not suitable for intracellular imaging. The goal of our study was to determine whether this contrast agent could be accumulated in tumor cells in vitro (HeLa cells) and in vivo (Dunning R3327 AT1 rat prostate adenocarcinoma) and whether the specificity of the PNA for the up-regulated c-myc mRNA in the cell cytoplasm would have an effect on contrast agent retention in the tumor cells [60]. To perform the synthesis of the peptide modules, we used the synthesis protocol3.
Synthesis of a MRI contrast agent for imaging of cell nuclei
Molecular imaging necessitates a sufficient amount of contrast agent within the cell. We realized the intracellular uptake and cell compartment specificity of the commonly used interstitial contrast agent gadolinium (Gd3+) ligated to a cell-nucleus addressed peptide module (NLS) which in turn was connected with a module composed of a cell penetrating peptide (CPP) facilitating the transfer across the cell membrane. By use of MRI, (Gd3+) was detected within DU-145 prostate cancer cells [61]4.
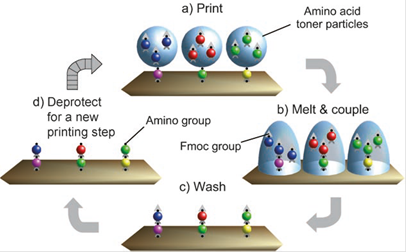
Synthesis of Peptide arrays
The aim of this further method is to synthesize and analyze as many peptides as possible, in order to, e.g. identify individual peptides that bind to a target protein. As documented, this one-bead-one-compound method identifies many different peptides readily (Figure 4) [62].
illustrates the particle-based Merrifield synthesis. a) A laser printer delivers Fmoc amino acid-OPfp esters embedded within toner particles to specific locations on a solid support, on which b) the particles are melted after transfer. Melting enables the amino acid derivatives to diffuse and undergo coupling to the support. A synthetic cycle is completed when c) excess monomers are washed away, and d) the Fmoc protecting group is removed. Repeated coupling cycles generate a peptide array (modified from Stadler [62]).
Reformulation of drugs
The pharmacologic potency as well as the adverse reactions of the highly efficient chemotherapeutic temozolomide (TMZ) is documented [63-65]. The re-formulation “old fashioned” drugs, like TMZ to the TMZ-BioShuttle conjugate, achieved an enhancement of the original potential of the TMZ. The TMZ-BioShuttle resulted in a higher pharmacological effect in glioblastoma cell lines dramatically with reduced doses. This permits the conclusion that a suitable chemistry could realize the ligation of pharmacologically active, but sensitive and highly unstable pharmaceutical ingredients without functional deprivation as first described by Waldeck in 2008 [66]. The synthesis steps were published by Pipkorn in 2009. The procedure steps of the intermediates of the solid phase are summarized here5 [67].
Syntheses of complex peptides and PNA modules consisting of cell-specific cleavage sides for delivery and targeting studies
BioShuttle peptides-based conjugates
Recent progress in genome research and in biochemistry techniques resulted in a better understanding of disease mechanisms, which led to the design and synthesis of novel diagnostic molecules and drugs with high specificity. In this context the central pharmacological question is: how to deliver sufficient amounts of substances to their target? Since successful drug delivery and targeting is a very complex problem, directed drug delivery is one of the most important goals of pharmaceutical research and development. This is illustrated and discussed in a review of the BioShuttle technology and outlines the use of combinatorial chemistry and SPPS in drug discovery [68, 69].
Clamp-BioShuttle-mediated oligonucleotid active transfer into nuclei of prostate cancer cells
Efficient and safe substance delivery is required for genetic interventions which are hampered by the risk of inflammatory reactions, inefficient delivery strategies, and unexplained specific local gene activation and its expression rate in the target tissue. A non-viral gene peptide-based carrier system for inactivated genes was developed with an efficiency of almost 100% cellular uptake of the bioconjugate called “Clamp-BioShuttle-phNIS-EGFP”. It shows a consistent gene expression which qualified the BioShuttle plasmid as a carrier tool in the technology for gene transfer, gene therapeutic approaches and for molecular diagnostics [70-73]6.
Modular peptides for studies of Cathepsin B addressed cleavable carrier and targeting conjugates
The treatment by effective doses of drugs can lead to adverse reactions and marked toxicity in patients. The main goal of every delivery system is, therefore a delivery of a precise amount of a drug to the desired location in order to achieve the necessary drug concentration in the target organ for effective treatment. The key problem still remains the achievement of curative doses in a pharmacologically active state in the desired target while avoiding side effects. We aimed to overcome this hurdle by the use of a “BioShuttle” carrier system which is able to deliver inactivated genetic material or functional peptides into cells and target tissues followed by a local activation. This BioShuttle carrier consists of the following modules: cell penetrating peptide (CPP), Cathepsin B cleavable spacer (CTBCS), cell nucleus address signal sequence (NLS) and a covalently linked functional peptide- or genetic-unit [74-76]. The synthesis procedure of the solid phase is described here7.
Development of theranostic molecules
Development of Fmoc-building blocks for synthesis of theranostic agents
Targeted therapeutic and molecular imaging strategies require suitable materials for facilitating disease diagnosis and therapy in a single step. Cell- and tissue-specific targeting is, therefore, carried out via theranostic agents, which have diagnostic and therapeutic properties in a single molecular formulation. The Wiessler group [77] developed a prototype of a such theranostic agent by use the Diels-Alder methodology with inverse electron demand (DARinv) [78], an efficient click chemistry technology to synthesize cyclic RGD-BioShuttle [79] constructs. These constructs carry both the near-infrared (NIR) imaging agent Cy7 [80] and the chemotherapeutic agent temozolomide (TMZ) [81]. They were synthesized by SPPS and could be considered as an example for modern family of drugs and diagnostics enabling personalized medicine [82, 83]
Outlook
The operational excellence of the SPPS fulfils the need of the chemical peptide synthesis of different functional peptides to study the interaction of structure and functional activity. Specific tasks about identification of anti-epitopes within protein sequences can also be addressed using individually synthesized peptides. Additionally dynamic cellular processes concerning DNA-, RNA- and protein levels can be measured and simulated with specific functional peptides. The process can also be understood in conjunction with inter-and intra-cellular transport of protein units (drug delivery & drug targeting [drug discovery]). A further potential of the SPPS is the development and design of tools helpful for monitoring of modern diagnostic processes. Molecular imaging (MRT, PET, SPECT) and fluorescence imaging respectively, are feasible by appropriate special synthetic peptides. Molecular modelling- and biocomputing-methods of synthetic peptides help to understand cellular structure and function. Developments of therapeutical approaches at the molecular level (antisense- and anti-Gene as well as si-RNA and opener/closer methodologies) are evident. Molecular imaging strategies for therapy monitoring make theranostic agents a promising tools not only in the expanding field of the personalized medicine but also in the nanotechnology field. This multi-faceted spectrum of applications was underpinned by a Scott Kahn's [84] assumption that “the solid phase chemistry can be the next generation of organic science”.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Furka A. Study on the possibilities of systematic searching for pharmaceutically useful peptides. Notarized Report (File number 36237/1982, in Hungarian). 1982
2. Sebestyen F, Dibo G, Kovacs A, Furka A. Chemical Synthesis of Peptide Libraries. Bioorganic & Medicinal Chemistry Letters. 1993;3:413-8
3. Furka A. History of Combinatorial Chemistry. Drug Development Research. 1995;36:1-12
4. Furka A. Combinatorial chemistry: 20 years on. Drug Discovery Today. 2002;7:1-4
5. Geysen HM, Wagner CD, Bodnar WM, Markworth CJ, Parke GJ, Schoenen FJ, Wagner DS, Kinder DS. Isotope or mass encoding of combinatorial libraries. Chemistry & Biology. 1996;3:679-88
6. Geysen HM. Combinatorial chemistry - past, present and future. Annals of Oncology. 1998;9:2
7. Geysen HM, Schoenen F, Wagner D, Wagner R. Combinatorial compound libraries for drug discovery: An ongoing challenge. Nature Reviews Drug Discovery. 2003;2:222-30
8. Fitzgerald SH, Sabat M, Geysen M. Survey of the diversity space coverage of reported combinatorial libraries. Journal of Combinatorial Chemistry. 2007;9:724-34
9. Blondelle SE, PerezPaya E, Dooley CT, Pinilla C, Houghten RA. Soluble Combinatorial Libraries of Organic, Peptidomimetic and Peptide Diversities. Trac-Trends in Analytical Chemistry. 1995;14:83-92
10. Houghten RA. Parallel array and mixture-based synthetic combinatorial chemistry: Tools for the next millennium. Annual Review of Pharmacology and Toxicology. 2000;40:273-82
11. Nefzi A, Ostresh JM, Yu JP, Houghten RA. Combinatorial chemistry: Libraries from libraries, the art of the diversity-oriented transformation of resin-bound peptides and chiral polyamides to low molecular weight acyclic and heterocyclic compounds. Journal of Organic Chemistry. 2004;69:3603-9
12. Lopez-Vallejo F, Caulfield T, Martinez-Mayorga K, Giulianotti MA, Nefzi A, Houghten RA, Medina-Franco JL. Integrating Virtual Screening and Combinatorial Chemistry for Accelerated Drug Discovery. Combinatorial Chemistry & High Throughput Screening. 2011;14:475-87
13. Salmon SE, Lam KS, Felder S, Yeoman H, Schlessinger J, Ullrich A, Krchnak V, Lebl M. Use of Large Combinatorial Chemical Libraries for Anticancer Drug Discovery. International Journal of Pharmacognosy. 1995;33:67-74
14. Hruby VJ, Shenderovich M, Lam KS, Lebl M. Design considerations and computer modeling related to the development of molecular scaffolds and peptide mimetics for combinatorial chemistry. Molecular Diversity. 1996;2:46-56
15. Aina OH, Sroka TC, Chen ML, Lam KS. Therapeutic cancer targeting peptides. Biopolymers. 2002;66:184-99
16. Lam KS, Renil M. From combinatorial chemistry to chemical microarray. Current Opinion in Chemical Biology. 2002;6:353-8
17. Lam KS. New aspects of natural products in drug discovery. Trends in Microbiology. 2007;15:279-89
18. Moos WH, Hurt CR, Morales GA. Combinatorial chemistry: oh what a decade or two can do. Molecular Diversity. 2009;13:241-5
19. Desai MC, Zuckermann RN, Moos WH. Recent Advances in the Generation of Chemical Diversity Libraries. Drug Development Research. 1994;33:174-88
20. Moos WH. Combinatorial chemistry at cross roads. Abstracts of Papers of the American Chemical Society. 1997;213:386-ORGN
21. Ng S, Goodson B, Ehrhardt A, Moos WH, Siani M, Winter J. Combinatorial discovery process yields antimicrobial peptoids. Bioorganic & Medicinal Chemistry. 1999;7:1781-5
22. Messeguer A, Cortes N. Combinatorial chemistry in cancer research. Clin Transl Oncol. 2007;9:83-92
23. Diller DJ. The synergy between combinatorial chemistry and high-throughput screening. Curr Opin Drug Discov Devel. 2008;11:346-55
24. McGee JE, Roy A. Patent review. Comb Chem High Throughput Screen. 2011;14:926-8
25. Oprea TI, Davis AM, Teague SJ, Leeson PD. Is there a difference between leads and drugs? A historical perspective. J Chem Inf Comput Sci. 2001;41:1308-15
26. Kvetensky J. [Historical evolution of knowledge of diabetes mellitus before the discovery of insulin. Review]. Vnitr Lek. 1972;18:67-72
27. Martin FI. The nature and use of purified insulins. Aust Fam Physician. 1976;5:514-25
28. Bliss M. The history of insulin. Diabetes Care. 1993;16(Suppl 3):4-7
29. Meienhofer J, Schnabel E, Bremer H, Brinkhoff O, Zabel R, Sroka W, Klostermayer H, Brandenburg D, Okuda T, Zahn H. [Synthesis of insulin chains and their combination to insulin-active preparations]. Z Naturforsch B. 1963;18:1120-1
30. Ruttenberg MA. Human insulin: facile synthesis by modification of porcine insulin. Science. 1972;177:623-6
31. Geiger R, Obermeier R. Insulin synthesis from natural chains by means of reversible bridging compounds. Biochem Biophys Res Commun. 1973;55:60-6
32. Obermeier R, Geiger R. A new semisynthesis of human insulin. Hoppe Seylers Z Physiol Chem. 1976;357:759-67
33. Obermeier R, Seipke G. Enzyme-Catalyzed Semisyntheses with Porcine Insulin. Process Biochemistry. 1984;19:29-32
34. United States.Congress.Senate.Committee on Labor and Public Welfare.Subcommittee on Health. National Diabetes Advisory Board act 1976. Washington: University of Michigan Library. 1976:1-116
35. Merrifield RB. Solid Phase Peptide Synthesis. I The Synthesis of a Tetrapeptide. J Am Chem Soc. 1963;85:2149-54
36. Marshall GR. Solid-phase synthesis: a paradigm shift. J Pept Sci. 2003;9:534-44
37. Carpino LA, Han GY. The 9-Fluorenylmethoxycarbonyl Amino-Protecting Group. J Org Chem. 1972;37:3404-9
38. Atherton E, Sheppard RC. The fluorenylmethoxycarbonyl amino protecting group. In: (ed.) Undenfriend S, Meienhofer J. The Peptides. New York: Academic Press. 1987:1-38
39. The MJ. Human insulin: DNA technology's first drug. Am J Hosp Pharm. 1989;46:S9-11
40. Scherkenbeck J, Lindell S. Applications of combinatorial chemistry in the agrosciences. Comb Chem High Throughput Screen. 2005;8:563-76
41. Lindell SD, Scherkenbeck J. Prospects for combinatorial chemistry in the agrosciences. Comb Chem High Throughput Screen. 2005;8:555-62
42. Birr C, Pipkorn R. Fully active insulin by selective formation of the disulfide bridges between a synthetic A-chain and natural B-chain. Angew Chem Int Ed Engl. 1979;18:536-7
43. Weigand K, Schmid M, Villringer A, Birr C, Heinrich PC. Hexa- and pentapeptide extension of proalbumin: feedback inhibition of albumin synthesis by its propeptide in isolated hepatocytes and in the cell-free system. Biochemistry. 1982;21:6053-9
44. Pipkorn R, Schmid M, Weigand K, Birr C. Carbonyldiimidazole/1-hydroxybenzotriazole activation in polymer phase synthesis of the arginine rich proalbumin hexapeptide extension. Int J Pept Protein Res. 1983;21:100-6
45. Hanafusa H, Halpern CC, Buchhagen DL, Kawai S. Recovery of avian sarcoma virus from tumors induced by transformation-defective mutants. J Exp Med. 1977;146:1735-47
46. Brugge JS, Steinbaugh PJ, Erikson RL. Characterization of the avian sarcoma virus protein p60src. Virology. 1978;91:130-40
47. Tamura T, Bauer H, Birr C, Pipkorn R. Antibodies against synthetic peptides as a tool for functional analysis of the transforming protein pp60src. Cell. 1983;34:587-96
48. Blomberg J, Vincic E, Jonsson C, Medstrand P, Pipkorn R. Identification of Regions of Hiv-1 P24 Reactive with Sera Which Give Indeterminate Results in Electrophoretic Immunoblots with the Help of Long Synthetic Peptides. Aids Research and Human Retroviruses. 1990;6:1363-72
49. Blomberg J, Lawoko A, Pipkorn R, Moyo S, Malmvall BE, Shao J, Dash R, Tswana S. A survey of synthetic HIV-1 peptides with natural and chimeric sequences for differential reactivity with Zimbabwean, Tanzanian and Swedish HIV-1-positive sera. AIDS. 1993;7:759-67
50. Klasse PJ, Pipkorn R, Blomberg J. Presence of antibodies to a putatively immunosuppressive part of human immunodeficiency virus (HIV) envelope glycoprotein gp41 is strongly associated with health among HIV-positive subjects. Proc Natl Acad Sci U S A. 1988;85:5225-9
51. Lawoko AL, Johansson B, Hjalmarsson S, Christensson B, Ljungberg B, Al-Khalili L, Sjolund M, Pipkorn R, Fenyo EM, Blomberg J. Comparative studies on neutralisation of primary HIV-1 isolates by human sera and rabbit anti-V3 peptide sera. J Med Virol. 1999;59:169-79
52. Lawoko A, Johansson B, Dash R, Falck L, Dietrich U, Pipkorn R, Nilehn B, Blomberg J. Continuity and discontinuity in the anti-V3 IgG response of human immunodeficiency virus type 1-infected persons in a cross-sectional and longitudinal study using synthetic peptides. J Infect Dis. 1995;172:682-90
53. Braun K, Frank M, Pipkorn R, Reed J, Spring H, Debus J, Didinger B, von der Lieth CW, Wiessler M, Waldeck W. HIV-1 capsid assembly inhibitor (CAI) peptide: structural preferences and delivery into human embryonic lung cells and lymphocytes. Int J Med Sci. 2008;5:230-9
54. Simons A, Ruppert T, Schmidt C, Schlicksupp A, Pipkorn R, Reed J, Masters CL, White AR, Cappai R, Beyreuther K. et al. Evidence for a copper-binding superfamily of the amyloid precursor protein. Biochemistry. 2002;41:9310-20
55. White AR, Multhaup G, Galatis D, McKinstry WJ, Parker MW, Pipkorn R, Beyreuther K, Masters CL, Cappai R. Contrasting, species-dependent modulation of copper-mediated neurotoxicity by the Alzheimer's disease amyloid precursor protein. J Neurosci. 2002;22:365-76
56. Carr SA, Biemann K, Shoji S, Parmelee DC, Titani K. n-Tetradecanoyl is the NH2-terminal blocking group of the catalytic subunit of cyclic AMP-dependent protein kinase from bovine cardiac muscle. Proc Natl Acad Sci U S A. 1982;79:6128-31
57. Jedrzejewski PT, Girod A, Tholey A, Konig N, Thullner S, Kinzel V, Bossemeyer D. A conserved deamidation site at Asn 2 in the catalytic subunit of mammalian cAMP-dependent protein kinase detected by capillary LC-MS and tandem mass spectrometry. Protein Science. 1998;7:457-69
58. Toner-Webb J, van Patten SM, Walsh DA, Taylor SS. Autophosphorylation of the catalytic subunit of cAMP-dependent protein kinase. J Biol Chem. 1992;267:25174-80
59. Tholey A, Pipkorn R, Bossemeyer D, Kinzel V, Reed J. Influence of myristoylation, phosphorylation, and deamidation on the structural behavior of the N-terminus of the catalytic subunit of cAMP-dependent protein kinase. Biochemistry. 2001;40:225-31
60. Heckl S, Pipkorn R, Waldeck W, Spring H, Jenne J, Der Lieth CW, Corban-Wilhelm H, Debus J, Braun K. Intracellular Visualization of Prostate Cancer Using Magnetic Resonance Imaging. Cancer Res. 2003;63:4766-72
61. Heckl S, Debus J, Jenne J, Pipkorn R, Waldeck W, Spring H, Rastert R, Der Lieth CW, Braun K. CNN-Gd(3+) Enables Cell Nucleus Molecular Imaging of Prostate Cancer Cells: The Last 600 nm. Cancer Res. 2002;62:7018-24
62. Stadler V, Felgenhauer T, Beyer M, Fernandez S, Leibe K, Guttler S, Groning M, Konig K, Torralba G, Hausmann M. et al. Combinatorial synthesis of peptide arrays with a laser printer. Angew Chem Int Ed Engl. 2008;47:7132-5
63. Stevens MF, Hickman JA, Langdon SP, Chubb D, Vickers L, Stone R, Baig G, Goddard C, Gibson NW, Slack JA. et al. Antitumor activity and pharmacokinetics in mice of 8-carbamoyl-3-methyl-imidazo[5,1-d]-1,2,3,5-tetrazin-4(3H)-one (CCRG 81045; M & B 39831), a novel drug with potential as an alternative to dacarbazine. Cancer Res. 1987;47:5846-52
64. Pagani E, Pepponi R, Fuggetta MP, Prete SP, Turriziani M, Bonmassar L, Lacal PM, Falcinelli S, Passarelli F, Guadagni F. et al. DNA repair enzymes and cytotoxic effects of temozolomide: comparative studies between tumor cells and normal cells of the immune system. J Chemother. 2003;15:173-83
65. Stupp R, Hegi ME. Methylguanine methyltransferase testing in glioblastoma: when and how? J Clin Oncol. 2007;25:1459-60
66. Waldeck W, Wiessler M, Ehemann V, Pipkorn R, Spring H, Debus J, Didinger B, Mueller G, Langowski J, Braun K. TMZ-BioShuttle- a reformulated temozolomide. Int J Med Sci. 2008;5:273-84
67. Pipkorn R, Waldeck W, Didinger B, Koch M, Mueller G, Wiessler M, Braun K. Inverse-electron-demand Diels-Alder reaction as a highly efficient chemoselective ligation procedure: Synthesis and function of a BioShuttle for temozolomide transport into prostate cancer cells. J Pept Sci. 2009;15:235-41
68. Pipkorn R, Waldeck W, Braun K. Synthesis and application of functional peptides as cell nucleus-directed molecules in the treatment of malignant diseases. J Mol Recognit. 2003;16:240-7
69. Pipkorn R, Waldeck W, Braun K. BioShuttle mediated plasmid transfer - A way to new genetic intervention. Biopolymers. 2007;88:536
70. Howl J, Nicholl ID, Jones S. The many futures for cell-penetrating peptides: how soon is now? Biochem Soc Trans. 2007;35:767-9
71. Pipkorn R, Waldeck W, Jenne JW, Didinger B, Braun K. Generation, application and quantification of clamp-BioShuttle carriers for plasmid delivery into nuclei of prostate cancer cells. Biochem Soc Trans. 2007;35:829-32
72. Braun K, von Brasch L, Pipkorn R, Ehemann V, Jenne J, Spring H, Debus J, Didinger B, Rittgen W, Waldeck W. BioShuttle-mediated plasmid transfer. Int J Med Sci. 2007;4:267-77
73. Pipkorn R, Waldeck W, von Brasch L, Didinger B, Koch M, Wiessler M, Braun K. Genetic Interventions by Clamp-PNA-BioShuttle. Chemistry Today. 2008;26:17-8
74. Pipkorn R, Waldeck W, Spring H, Jenne JW, Braun K. Delivery of substances and their target-specific topical activation. Biochim Biophys Acta. 2006;1758:606-10
75. Pipkorn R, Waldeck W, Didinger B, Braun K. Delivery of substances and their Target Cell-specific Activation. Proc Annu EPS Meet Gdansk. 2006
76. Pipkorn R, Waldeck W, Spring H, Jenne J, Braun K. Peptide synthesis of trojan horses for plasmid transfer and topical activation. Journal of Peptide Science. 2006;12:136
77. Wiessler M, Hennrich U, Pipkorn R, Waldeck W, Cao L, Peter J, Ehemann V, Semmler W, Lammers T, Braun K. Theranostic cRGD-BioShuttle Constructs Containing Temozolomide- and Cy7 For NIR-Imaging and Therapy. Theranostics. 2011;1:381-94
78. Thalhammer F, Wallfahrer U, Sauer J. Reactivity of Simple Open-Chain and Cyclic Dienophiles in Inverse-Type Diels-Alder Reactions. Tetrahedron Letters. 1990;31:6851-4
79. Chen X. Integrin Targeted Imaging an Therapy. Theranostics. 2011;1:28-9
80. Cheng KT. Cy7-Tetrameric arginine-glycine-aspartic acid peptide. Molecular Imaging and Contrast Agent Database (MICAD). 2004
81. Danson SJ, Middleton MR. Temozolomide: a novel oral alkylating agent. Expert Rev Anticancer Ther. 2001;1:13-9
82. Warner S. Diagnostics + Therapy = Theranostics. The Scientist. 2004;18:38
83. Xie J, Lee S, Chen X. Nanoparticle-based theranostic agents. Adv Drug Deliv Rev. 2010;62:1064-79
84. Kahn S. Bioinformatics: a holistic approach to drug discovery. Drug Discov Today. 2002;7:633-4
Footnotes
The synthesis of the peptide with DDZ (dimethyl-3,5-dimethoxybenzyloxycarbonyl) protected-amino acid was carried out in solution. The peptides were synthesized twice on a 0.5% cross-linked polystyrene gel. All the synthetic steps were functionally controlled photometrically. Instead of Gln and Asn, Glu(OBz1) and Asp(OBz1) were used in order to avoid nitrile formation at the amide side by the condensation reagent dicyclohexylcarbodiimide (DCC). Asp was linked to the carrier with its β-carboxyl function via an electrophilic 2-oxoethyl ester bond. In this way all the later amide sides could be incorporated simultaneously at the end of the synthesis by ammonolysis of the benzyl ester and 2-oxoethyl ester bonds on release from the carrier. Proof of the stability of the benzyl ester in position A under the cleavage conditions was provided by the mass spectrum.
The peptides were synthesized by standard Fmoc-methodology on an Applied Biosystems ABI 433 automated synthesizer employing HBTU [2-(1Hbenzotriazole-yl)-1,1,3,3,-tetramethyluronium hexafluorophosphate] activation. For peptide acid synthesis preloaded Fmoc-Lys(Boc)-TCP-resin (amino-methylpolystyrol with trityl-linker, 0.55 mmol/g, PepChem, Tübingen, Germany) was used. Fmoc-Ser(tBu)-OH, Fmoc-Lys(Boc)-OH, Fmoc-Gln(Trt)-OH, Fmoc-Glu(OtBu)-OH, Fmoc-Ala-OH, Fmoc-Val-OH, and Fmoc-Gly-OH were used as amino acid building blocks. Fmoc-Asn(Trt)-OH, Fmoc-Asp(OtBu)-OH, or Fmoc-Asp-(OH)-OtBu (for the synthesis of isoAsp-peptides) were used as building blocks in position 2. Fmoc-Ser[PO(OBzl)OH]-OH (22), was used as a building block for phosphopeptide synthesis. We observed no significant α-elimination of the phosphoryl group upon exposure to base during peptide synthesis. Ac-Gly-OH was used to introduce N-terminal acetyl group, N-decanoyl-glycine and Myr-Gly-OH (Nova-Biochem, Bad Soden, Germany) were used to introduce the N-terminal C10-fatty acid and the N-terminal myristic acid, respectively. The synthesis of C6-Gly-OH is described below. Cleavage of the peptides was performed with trifluoro acetic acid (TFA)/triisopropylsilane/water (90:8:2, v:v:v) for 2 h at room temperature. Peptides were purified by quantitative reversed-phase HPLC (Kromasil, C18, 5 ím, 100 Å) and analyzed by reversed-phase HPLC (Kromasil, C18, 100 Å, 0.75 mL/min, 1 min, isocratic 0.1% TFA in water, then in 40 min linear gradient to 60% acetonitrile/0.085% TFA in water), MALDI-MS, and MS (ES-MS). Prior to the preparation of stock solutions, the peptides were lyophilized several times. Synthesis of C6-Gly-OH: Fmoc-Gly-TCP-resin was deprotected following standard procedures. Ten equivalents (calculated on the base of the amount of glycine on the resin) of capronic acid was solved in DMF, and 10.5 equiv of diisopropylcarbodiimide and 10 equiv of 1-hydroxy-benzotriazole (HOBt) were added, and the mixture was stirred for 1 h at room temperature. The preactivated capronic acid was added to the resin. After gently shaking for 2 h at room temperature, the resin was washed with DMF, chloroform, and diethyl ether and dried by air suction. The C6-glycine was cleaved from the resin with 95% TFA/5% water (1 h). After removal of the solvent under reduced pressure, the oily residue was dissolved in water and lyophilized several times. The resulting white powder 173.1 g/mol) was readily soluble in DMF and was used.
The SPPS in a fully automated synthesizer was used for preparing the peptide TQVKIWFQNRRMKQKK-Cys-NH2 and the c-myc specific PNA ATGCCCCTCAACGT-Cys-NH2. The sequences derived from human-HSMMYCC (GenBank accession no. X00364), rat-RNCMYC (GenBank Ac.No.: Y00396. The syntheses of control random PNA GCCTAGACAATCTG-Cys-NH2 peptides were carried out identically. Details synthesis procedures of the Gd-complex formation and fluorescence dye labelling are documented [60].
To perform the synthesis of peptide modules we used the N-(9-fluoenyl)methoxycarbonyl strategy in a fully automated synthesizer. The syntheses of the CPP, the NLS, and the random NLS were performed with an identical procedure. Stochiometric amounts of NLS-K2-DTPH-peptide and Gd3+ were dissolved in an aqueous NaCl solution. The complex formation of the random-K2-DTPH-peptide and Gd3+ was performed with an identical procedure. A disulfide bond enables the cleavable connection between the TPU and the NLS [61].
For SPPS of the Lys(Tct)-Lys-Lys-Pro-Lys-Lys-Arg-Lys-Val-Cys-OH [Lys(Tct)-NLS(SV40-T)-Cys] and the Cys-Arg-Gln-Ile-Lys-Ile-Trp-Phe-Gln-Asn-Arg-Arg-Met-Lys-Trp-Lys-Lys-OH (Cys-pAnt43-58), the Fmoc-strategy was applied in a fully automated multiple synthesizer. The synthesis was carried out in a 0.05 mmol scale on a Fmoc-Lys(Boc)-polystyrene resin (1% cross-linked) with 0.053 mmol/g loading and Fmoc-Cys(Trt)-polystyrene resin (1% cross-linked) with 0.005 mmol/g loading. As coupling reagents, 2-(1H-benzotriazol-1-yl)-1,1,3,3-tetramethyl uronium hexafluorophosphate (HBTU)/HOBt/DIPEA (1:1:1) was used. Cleavage/deprotection of the peptide-resin was performed with TFA/ethane dithiol/thioanisole/phenol for 2.5 h at room temperature. The products were precipitated with ether. The crude products were purified by preparative HPLC on a Kromasil 300-5C18 reverse phase column (20 × 150 mm) using as eluents 0.1% TFA in water (A) and 60% acetonitrile in water.
The PNA-sequences for hybridization to the different ORI-target sequences of the phNIS-IRES-EGFP were identified. The syntheses of the peptide modules and the PNA were carried out by Solid Phase Peptide Synthesis in a fully automated synthesizer. For PNA synthesis, we used fluorenylmethoxycarbonyl (Fmoc)-protected monomers with the exocyclic amino groups of A, G, and C bases blocked by a benzhydroxyl (Bhoc) group. Sequences of single modules as well as the complete modular construct were characterized with analytical HPLC and laser desorption mass spectrometry. Myristic acid was coupled with tetramethylfluoroformamidium-hexafluorophosphate (TFFH) in dimethylformamide/dichloromethane for one hour at the N terminus of PNA. Cysteine groups were attached via one of the COOH-terminal lysine residue of pAntp(43-58)-Cys and at the NH2-terminus of (NLS[SV40-T]). Molecules were oxidized in an aqueous solution of 2mg/ml in 20% DMSO for about five hours. The oxidation progress was monitored by analytical C18 reverse-phase HPLC. Peptide nucleic acids as well as the address peptide (NLS) carried one lysine-lysine spacer at the COOH terminus, which enabled linkage of peptide nucleic acids with identical sequence via a succinimidyl ester in a molar ratio of 1:1.
For synthesis of the FITCVKRKKKP-KD-GFGRK(Dabcyl)-RQIKIWFQNRRM-KWKK [BioShuttle-CBCSD-complex] and FITCVKRKKKP-KL-GFGRK-(Dabcyl)-RQIKIWFQNRRMKWKK [BioShuttle-CBCSL-complex] we employed the Fmoc (9-fluorenyl methyl-oxycarbonyl) methodology in a fully automated multiple synthesizer. As coupling agent 2-(1H-Benzotriazole-1-yl)-1,1,3,3-tetramethyluroniumhexafluorophosphate (HBTU) was used. The following side chain protecting groups were employed: Boc(tert-butyloxycarbonyl) for Lys and Trp, Trityl(triphenylmethane) for Gln and Asn and Pbf (2,2,4,6,7-Pentamethyl dihydrobenzofurane-5-sulfonyl) for Arg. Fmoc-Lys(Dabcyl) was purchased from Merck Biosciences. The synthesized peptides were cleaved and deprotected from the solid support by treatment with 90 % trifluoro acetic acid, 8 % tri-isopropyl silane, and 2% water for 2.5 h at room temperature. The products were precipitated in ether. The crude material was purified by preparativeHPLConaKromasil100-10C 18μm reverse phase column (30×250mm) using an eluent of 0.1 % trifluoro acetic acid in water (A) and 80 % acetonitrile in water (B). The fractions corresponding to the purified protein were lyophilized. The purified material was characterized with analytical HPLC and matrix assisted laser desorption mass spectrometry (MALDI-MS).
Author contact
![]() Corresponding author: Dr. Rüdiger Pipkorn, German Cancer Research Center (DKFZ), Dept. of Translational Immunology, Im Neuenheimer Feld 410, D-69120 Heidelberg, Germany. E-mail: r.pipkornde.
Corresponding author: Dr. Rüdiger Pipkorn, German Cancer Research Center (DKFZ), Dept. of Translational Immunology, Im Neuenheimer Feld 410, D-69120 Heidelberg, Germany. E-mail: r.pipkornde.