Impact Factor ISSN: 1449-1907

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2014; 11(4):365-372. doi:10.7150/ijms.7696 This issue Cite
Research Paper
Decreased Activities of Apolipoprotein M Promoter Are Associated with the Susceptibility to Coronary Artery Diseases
Lu Zheng1, Guanghua Luo1, Jun Zhang1, Qinfeng Mu1, Yuanping Shi1, Maria Berggren-Söderlund2, Peter Nilsson-Ehle2, Xiaoying Zhang1 ![]() , Ning Xu2
, Ning Xu2 ![]()
1. Comprehensive Laboratory, Third Affiliated Hospital of Soochow University, Changzhou 213003, China.
2. Division of Clinical Chemistry and Pharmacology, Department of Laboratory Medicine, Lund University, S-221 85 Lund, Sweden.
Received 2013-9-18; Accepted 2014-1-20; Published 2014-2-20
Abstract
The present study investigated the correlation among genetic polymorphisms of the proximal promoter region of apolipoprotein M (apoM) gene, the polymorphisms in relation to apoM expressions and the susceptibility to coronary artery diseases (CAD) in a Han Chinese population. Four common polymorphic sites, i.e., T-1628G, C-1065A, T-855C and T-778C, were confirmed, and a new deletion mutation C-724del was found, in 206 CAD patients and 209 non-CAD patients using direct DNA sequencing analyses. Occurrences of alleles T-1628G, T-855C and C-724del were significantly higher in CAD patients compared to non-CAD patients. Moreover we examined all these polymorphisms in relation to apoM expression by applying luciferase reporter assay. It demonstrated that constructs -855C and 724del showed obvious decreased luciferase activities, i.e., (0.93±0.15 vs. 2.11±0.15; P=0.012) and (1.13±0.25 vs. 2.11±0.15; P=0.009) respectively, which indicates these two polymorphisms could confer decreased apoM expressions. Meanwhile the occurrences of these two SNP were also significantly higher in the CAD patients than in non-CAD patients. It is therefore reasonable to speculate that down-regulated apoM expressions in relation to these polymorphisms may affect HDL and cholesterol metabolism in vivo and further influence the susceptibility to CAD, although the underlying mechanisms need further investigation.
Keywords: apolipoprotein M, promoter, polymorphism, expression, coronary artery diseases
Introduction
Coronary artery diseases (CAD) are predicted being the most common form of heart diseases worldwide [1]. Many factors, including age, gender, dyslipidemia, hypertension, diabetes mellitus and smoking may contribute to the initiation and progress of CAD. Recent years increasing evidence suggests that CAD could be also influenced by multi-genetic variations in different individuals, which may be considered predisposition for the risk factors of CAD as well, including the polymorphisms found in the genes of angiotensin-converting enzyme (ACE), angiotensinogen (AGT), leptin receptor (LEPR) and neuropeptide Y receptor Y5 [2, 3]. Some of these genes are leading to the clinical translation, and the advances in genomic research and technology have opened new avenues of investigation on the pathogenesis of CAD [4]. Early detection of individuals with the high-risk factors of genetic alterations of CAD and proper clinical intervention may prevent or delay the vascular events and offer an increased quality of life and life expectancy in CAD patients.
It is well known that lower serum levels of high density lipoproteins (HDL) and higher serum levels of low density lipoproteins (LDL) have been assessed as the CAD risk factors and they are also used as the clinical markers for the preventive therapy in the CAD patients. Apolipoproteins, such as apolipoprotein (apo) A1 is the major apolipoprotein in HDL and apolipoprotein B100, a structure apolipoproein in LDL [5, 6]. ApoA1 gene single nucleotide peptide alteration T-319C was found to be associated with the metabolic syndrome and increase CAD risk [7]. It has been reported that apoA5 -1131C allele could be associated with reduced plasma apoA5 concentration and an increase in CAD risk [8, 9].
ApoM is one of the latest additions to the apolipoprotein family, first identified and characterized in 1999 [10]. The apoM gene codes for a 26 kDa protein which structurally belongs to the lipocalin superfamily. It is located in a highly conserved segment in the major histocompatibility complex (MHC) class III locus on chromosome 6 in humans [11]. ApoM is predominantly present in HDL particles, and is also found in other lipoprotein subclasses, e.g., triglyceride-rich lipoprotein particles after fat intake in human plasma [10, 12]. Wolfrum, et al., have determined that apoM is critical for the formation of HDL particles, notably pre-β-HDL [13]. ApoM polymorphisms were identified to be associated with some metabolic diseases. In T2D patients, a subset of apoM SNPs were associated with disease duration and metabolic traits. SNP rs707922, one of the metabolic trait-associated SNP, led to the discovery of a novel apoM transcript at molecular level and SNP-dependent effect on cellular cholesterol content [14]. Some studies have investigated promoter variants of apoM. Allele C of SNP T-778C may increase promoter activity and confer the risk susceptibility in the development of T1D [15]. Xu, et al. and Jiao, et al. have reported that apoM T-855C and T-778C polymorphism carries an increased risk for CAD in Chinese populations [16, 17]. Although in our previous study T-778C showed no effects on CAD [18]. However, the mechanism by which mutant apoM alleles contribute to CAD susceptibility is unknown yet. One conceivable explanation might be speculated in that alleles of apoM in the proximal promoter region could influence apoM gene expression and further influence lipoprotein metabolism in vivo. In the present study, we investigated the genetic polymorphisms of the proximal promoter region of the apoM gene in CAD patients and control subjects chosen from a Han Chinese population. Furthermore we examined SNPs in relation to the apoM gene expression as well as the susceptibility of CAD.
2. Materials and methods
2.1 Subjects
Two hundred and six patients (41 females and 165 males, mean age 61.86 ± 9.20 years old) who had obtained coronary angiography and were diagnosed with CAD according to the results of angiography (a lesion was classed as being significant when stenosis was more than 50%) [19], were subjected in the present study. Subjects with a history of hematologic, neoplastic, renal, hepatic or thyroid diseases were excluded. Patients with congenital heart disease, cardiomyopathy, valvular diseases, or autoimmune diseases were also excluded. 209 age- and gender-matched patients (52 females and 157 males, mean age 60.39 ± 9.06 years old) were selected as the control group in the present study. All control subjects were confirmed free from coronary artery disease by either angiography or clinical symptom together with ECG examinations. All CAD patients and control subjects were from the third affiliated hospital of Soochow University and were informed by written consent when they interview in the study.
2.2 Amplifying DNA of apoM proximal promoter region by real-time PCR and amplicons sequencing
DNA was extracted from a 250μl peripheral blood sample of each subject with a commercial DNA extract Kit (Shenerg Biocolor, Shanghai, China). The SNP ID number and detail sequence information of apoM is publicly available (http://www.ncbi.nlm.nih.gov/SNP). Up to now there are twenty-three SNPs being reported publicly (Table 1). According to the sequence (GenBank accession nos. EU030444.1) two pairs of primers were designed as follows: forward primer 1: AGCCTGGTCTTGCAGACTTG; reverse primer 1: CAAATAGGTTGTCCTTGGATAGC; forward primer 2: AATCTATCGAGGAAGATCCATCTCT; reverse primer 2: AACCCAAACCTGGATTACTTAGTG. All specific primers were synthesized by Sangon Biotech Company (Shanghai, China). Taq DNA polymerase, 4×dNTPs, 10×polymerase chain reaction (PCR) buffer, and MgCl2 were purchased from Shenerg Biocolor (Shanghai, China). In brief, PCR was performed as follows: 40 to 80 ng of genomic DNA template, 2.5μl of 10×PCR buffer, 1.5mM MgCl2, 0.5μl of 4×dNTPs, 1.25 U Taq DNA polymerase, 10 pmol of each primer in a final reaction volume of 25μl. Thermal cycling was performed in a LightCycler(Roche, Swaziland). The cycling program consisted of 1 min of initial denaturation at 95°C, followed by 40 cycles at 95°C for 0s (temperature transition rate 20°C/s), 61°C for 10s (temperature transition rate 20°C/s), and 72°C for 10s (temperature transition rate 20°C/s). The PCR products of apoM proximal promoter region were directly sequenced on an automatic sequencer from Applied Biosystems (model 3730, Invitrogen, Shanghai, China).
Available apoM SNPs in proximal promoter region (http://www.ncbi.nlm.nih.gov/SNP)
| SNP | Allele | Region | |
|---|---|---|---|
| 1 | rs79177639 | G-1904A | Promoter |
| 2 | rs12525463 | G-1886A | Promoter |
| 3 | rs6921907 | C-1692T | Promoter |
| 4 | rs12525471 | C-1687A | Promoter |
| 5 | rs34260741 | A-1647- | Promoter |
| 6 | rs1266078 | G-1628T | Promoter |
| 7 | rs58368425 | C-1590G | Promoter |
| 8 | rs9267528 | C-1530T | Promoter |
| 9 | rs114106481 | G-1529A | Promoter |
| 10 | rs115826764 | G-1323A | Promoter |
| 11 | rs55880811 | T-1140C | Promoter |
| 12 | rs114288601 | C-1134A | Promoter |
| 13 | rs805297 | C-1065A | Promoter |
| 14 | rs77322555 | A-1041C | Promoter |
| 15 | rs4947251 | A-1009G | Promoter |
| 16 | rs9404941 | T-885C | Promoter |
| 17 | rs805296 | T-778C | Promoter |
| 18 | rs74485832 | G-723- | Promoter |
| 19 | rs62395806 | G-599A | Promoter |
| 20 | rs76611345 | C-597A | Promoter |
| 21 | rs115239580 | G-358T | Promoter |
| 22 | rs73398248 | C-319A | Promoter |
| 23 | rs114269338 | G-255A | Promoter |
2.3 Construction of plasmid containing apoM promoter region
The fragment of apoM promoter region, -2165 to -29, was amplified with primers containing restriction endonuclease sites. This fragment contains one of the four SNPs which were detectable in our study, C-1065A, T-855C, T-778C and C-724-, respectively. The primers used to amplify the promoter region were as follows: 5'- ggggtaccGTGGCGCAATCACAACTC-3' (forward) and 5'-ccaagct GACCCTTTCACCTGCTAATG-3' (reverse). To facilitate plasmid construction, two endonuclease sites, KpnI and HindIII were inserted to both ends of the amplicon (Underlined sequences). PCR was performed as follows: 40 to 80 ng of genomic DNA template, 2.5μl of 10× PCR buffer, 1.5mM MgCl2, 0.5μl of 4× dNTPs, 1.25U Taq DNA polymerase, and 10pmol of each primer in a final reaction volume of 25μl. The PCR cycle was as follows: 1 min of initial denaturation at 95°C, followed by 40 cycles at 95°C for 10s, 60°C for 90s and 72°C for 2min. The amplified fragment was digested with KpnI and HindIII, and then cloned into the luciferase expression vector pGL3 (Promega) with T4 ligase.
2.4 Site-directed mutagenesis
Four polymorphic sites were obtained by using the Site-Directed Mutagenesis Kit (Tiandz, Inc). The primers for mutation were showed in Table 2. The PCR cycle was as follows: 1 min of initial denaturation at 95°C, followed by 15 cycles at 95°C for 40s, 58°C for 30s and 68°C for 25min. Products were extended in 68°C for 2min and stored at 4°C. All plasmids used in this study were verified by sequencing, and orientations were confirmed being correct.
Primers for the site-directed mutagenesis of apoM promoter
| SNP | 5'-3' | |
|---|---|---|
| C-1065A | Sense | TTATAGCAAATATATACTTTGGAATTACCATGTGCAAGTCTTTGCT |
| Anti-sense | AGCAAAGACTTGCACATGGTAATTCCAAAGTATATATTTGCTATAA | |
| T-855C | Sense | CTCGACATCCCAGGCCCAAGCAATCCTCCCT |
| Anti-sense | AGGGAGGATTGCTTGGGCCTGGGATGTCGAG | |
| T-778C | Sense | ACCACATCGGCTAATTTTTGTACTTTTTGTAGAGACAGAGTTTTG |
| Anti-sense | CAAAACTCTGTCTCTACAAAAAGTACAAAAATTAGCCGATGTGGT | |
| C-724- | Sense | GTTTTGAACTCCTGGGTCAAGCCATCCGCCCA |
| Anti-sense | TGGGCGGATGGCTTGACCCAGGAGTTCAAAAC |
The mutant sites were marked in bold and underlined.
2.5 Luciferase reporter assay
293HEK was cultured in the RPMI1640 supplemented with 10% fetal bovine serum (Gibco). Cultured cells (1×106/well) were plated in 6-well plates and transfected with 1μg of pGL3-pApoM constructs by using Lipofectin 2000. At the same time 0.2μg of pRL-TK vectors (Promega) were transfected as the internal control. After 12hrs, the transfected cells were incubated with RPMI1640 containing 10% fetal bovine serum. All transfected cells were then washed once with PBS (pH 7.4) and lysed. Luciferase activities were performed using the Dual-Glo Luciferase Assay System (Promega) according to manufacturer's protocol. The pGL3-control and pGL3-Basic (without promoter) vectors (Promega) were used as positive and negative controls, respectively. Luciferase activity was measured using a Lumat LB 9507 luminometer (EG &G Berthhold, Bad Wildbad, Germany). The activity of firefly luciferase was normalized to that of renilla luciferase. Each experiment was performed in triplicates. The results were represented graphically as a ratio of pGL3-Basic vector activity to controls.
2.6 Statistical analysis
Results are expressed as means±SD. Comparisons of the general characteristics in two groups between different genotypes were statistically evaluated by the unpaired t-test (Prism software, version 4; GraphPad Inc., La Jolla, CA, USA). Significance was established at a P value less than 0.05. Binary logistic regression analyses were performed by the SPSS statistical package (version 10.0, SPSS Inc., Chicago, IL, USA). Genotype distributions were tested for the Hardy-Weinberg equilibrium using a chi square test. Allele frequencies, genotype frequencies, odds ratios (ORs) and 95% confidence intervals (CIs) were estimated by the Chi-squared analyses. Estimates of linkage disequilibrium (LD) between SNPs were determined by calculating pair-wise D' and r2 statistics in unrelated individuals using the SHEsis software[20]. Power calculations were performed by PS software (power and sample size calculations, version 3.0.43). Results from luciferase analyses were analyzed by Student's t-test for two-group comparison.
3. Results
3.1 General characteristics of CAD and non-CAD patients
Mean age of CAD patients and non CAD patients were 61.86 ± 9.20 years old and 60.39 ± 9.06 years old, respectively. The age and gender were matched in the CAD patients and the non-CAD patients (Table 3). The CAD patients had higher serum triglyceride (TG) levels (2.43 ± 2.10 mmol/L; P= 0.000) and higher serum cholesterol (TC) levels (5.11 ± 1.06 mmol/L; P= 0.009) compared to the non-CAD patients. Lower HDL-C and higher fasting blood glucose (FBG) were also seen in the CAD patients (Table 3).
Clinical characteristics of CAD and non-CAD patients
| CAD patients N=206 | Non-CAD patients N=209 | P-value | |
|---|---|---|---|
| Male/Female | 165/41 | 157/52 | 0.965 |
| Age (means±SD) | 61.86 ± 9.20 | 60.39 ± 9.06 | 0.125 |
| TG (mmol/L) | 2.43±2.10** | 1.76±1.01 | 0.000 |
| TC (mmol/L) | 5.11±1.06* | 4.77±1.22 | 0.009 |
| LDL-C (mmol/L) | 2.54±1.01 | 2.70±0.74 | 0.070 |
| HDL-C (mmol/L) | 1.11±0.23** | 1.29±0.32 | 0.000 |
| BMI | 23.71±1.70 | 23.45±1.39 | 0.220 |
| FBG (mmol/L) | 6.64±2.54* | 6.05±1.19 | 0.002 |
Notes: Data are means ± SD. *P<0.05, **P<0.01 vs. control group. P-Value <0.05 was shown in bold.
Abbreviations: CAD, coronary artery disease; TG, triglycerides; TC, total cholesterol; LDL-C, low-density lipoprotein cholesterol; HDL-C, high-density lipoprotein cholesterol; BMI, Body Mass Index; FBG, fasting blood glucose.
3.2 Allele frequencies and genotype distributions of apoM promoter in CAD and non-CAD patients
As shown in Table 4, four common polymorphic sites, i.e., T-1628G, C-1065A, T-855C and T-778C, were confirmed, and a new deletion mutation in site -724C was found in the present study. Figure 1 shown three genotypes detected from subjects caused by -724 deletion mutation. Figure 1A and 1C represent the homozygote CC and homozygous deletion, respectively. Heterozygote appears as a series of double peaks following the point of deletion (red arrows) (Figure 1B). Genotype distributions of the five SNPs in these two populations followed Hardy-Weinberg equilibrium (HWE, P>0.05). The allelic frequencies and genotype distributions of these five polymorphisms in CAD and non-CAD patients were summarized in Table 4. T-778C and C-1065A showed no statistical significant difference in allele frequencies and genotype distributions between CAD and non-CAD patients. While occurrences of alleles of T-1628G, T-855C and C-724del were statistically significantly higher in the CAD patients than in non-CAD patients. In SNP T-1628G (rs1266078), the frequency of G allele was 19.20% in CAD patients vs. 12.7% in non-CAD controls (P=0.011). In CAD patients, 159 patients (76.1%) had the T/T genotype, 47 patients (22.5%) had the T/G genotype and three patients (1.4%) had the G/G genotype. The genotype distribution of rs1266078 was statistically significant different between CAD patients and non-CAD patients (OR = 1.711, P= 0.012). The minor C allele frequency of SNP T-885C in the CAD group was significantly higher than that in the control group (22.8% vs. 15.6%, P=0.008), indicating that T-885C was associated with a high risk for CAD (OR= 1.711, P= 0.009). In SNP C-724del, the frequency of del C allele was 8.0% in CAD patients and only 4.1% in non-CAD controls (P=0.017). The genotype distribution was also significantly different between CAD and non-CAD patients (OR = 1.904, P= 0.023). And the power calculation showed that the study had 75.9% and 66.9% power to detect the differences of T-855C and C-724del between case and control subjects at a significance level of 0.05, respectively.
Allele frequencies and genotype distributions of apoM proximal promoter region in CAD and non-CAD patients
| SNP ID | Allele | Allele Frequency | Allele | Genotype | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CAD (%) | Non-CAD (%) | OR (95% CI) | P-value | Non-CAD (% frequencies) | CAD (% frequencies) | P-value of HWE | Common odds ratio | P-value | |||||||||
| 11 | 12 | 22 | 11 | 12 | 22 | Non-CAD | CAD | ||||||||||
| T-1628G | T | 80.8 | 87.3 | 1.634 | 0.011 | 159 | 47 | 3 | 137 | 59 | 10 | 0.822 | 0.275 | 1.711 | 0.012 | ||
| rs1266078 | G | 19.2 | 12.7 | 1.119-2.385 | (76.1) | (22.5) | (1.4) | (66.5) | (28.6) | (4.9) | |||||||
| C-1065A | C | 66.0 | 69.9 | 1.193 | 0.236 | 100 | 92 | 17 | 87 | 98 | 21 | 0.513 | 0.386 | 1.199 | 0.224 | ||
| rs805297 | A | 34.0 | 30.1 | 0.891-1.597 | (47.9) | (44.0) | (8.1) | (42.2) | (47.6) | (10.2) | |||||||
| T-855C | T | 77.2 | 84.5 | 1.605 | 0.008 | 148 | 57 | 4 | 125 | 68 | 3 | 0.579 | 0.568 | 1.711 | 0.009 | ||
| rs9404941 | C | 22.8 | 15.6 | 1.131-2.279 | (70.8) | (27.3) | (1.9) | (60.7) | (33.0) | (6.3) | |||||||
| T-778C | T | 89.6 | 89.7 | 1.016 | 0.944 | 170 | 35 | 4 | 165 | 39 | 2 | 0.180 | 0.856 | 0.974 | 0.945 | ||
| (rs805296 | C | 10.4 | 10.3 | 0.650-1.588 | (81.3) | (16.8) | (1.9) | (80.1) | (18.9) | (1.0) | |||||||
| C-724del | C | 92.0 | 95.9 | 2.054 | 0.017 | 193 | 15 | 1 | 176 | 27 | 3 | 0.246 | 0.112 | 1.904 | 0.023 | ||
| del | 8.0 | 4.1 | 1.125-3.749 | (92.3) | (7.2) | (0.5) | (85.4) | (13.1) | (1.5) | ||||||||
P-Value <0.05 was shown in bold.
A new deletion mutation in site -724 in apoM promoter gene. ApoM promoter gene was amplified by the polymerase chain reaction (PCR) from subjects. The sequence data shows a single cytidine deletion at nucleotide -724 (-C724del) resulting three genotypes. Figure 1B was a compound heterozygote for this mutation. In heterozygote the deletion mutation causes the appearance of a series of double peaks starting from the -724 position (in-box). Figure 1A and 1C were the sequence data of homozygotes for -724C and -724del, respectively. The red arrow presented the -724 mutation site.
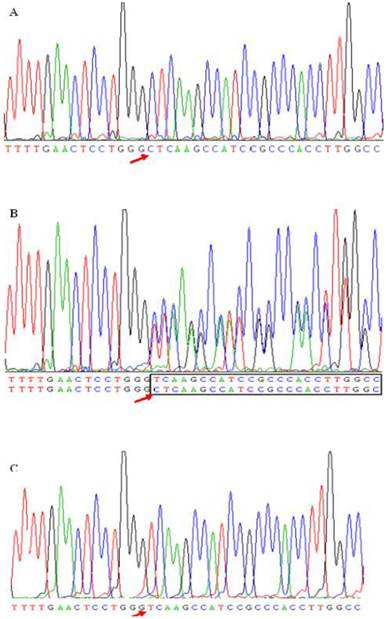
3.3 Correlation between clinical characteristics and SNP T-1628G, T-855C and C-724del in CAD patients and controls
Table 5 showed lipid levels of CAD and non-CAD patients in relation to the genotypes of SNP T-1628G, T-855C and C-724del, respectively. In SNP C-724del and T-855C, the mean TC level was lower in groups with wide-type homozygotes compared to the mutant allele carriers (P=0.000). In CAD group the HDL level was lower in the C-724del allele carriers than in patients with CC genotype (1.03 ± 0.28 vs. 1.13 ± 0.22 mmol/L; P=0.044).
3.4 Haplotype analysis
Using the SHEsis software, we estimated all possible haplotypes from the observed genotypes of three SNPs, T-1628G, T-855C and C-724del. The linkage disequilibrium analysis for the three SNPs showed no obvious LD between any of two SNPs (data not shown). We then compared haplotype frequency of apoM between CAD and controls. Five common haplotypes (frequency>3%) derived from the three SNPs accounted for about 95% of haplotype variations (ORs and P-values for the associations are listed in Table 6). Among these five common haplotypes, the haplotype G-T-C and T-C-Del (-1628-855-724) were found to be associated with an increased risk of CAD (OR = 2.421 (95% CI =1.072~2.503); P=0.022 and OR = 4.376 (95% CI =1.529~12.527); P=0.003, respectively). In addition, the haplotype T-T-C could decrease the risk of CAD (OR=0.531 (95%CI 0.392~0.717), P=0.000).
C-724del. Genotype and clinical characteristics of CAD patients and non-diabetic controls
| CAD patients | Non-CAD patients | P-value | ||||||
|---|---|---|---|---|---|---|---|---|
| CC | C/- + -/- | CC | C/- + -/- | CC vs. (C/- + -/-) in CAD patients | CC vs. (C/- + -/-) in non-CAD patients | |||
| Age (years) | 60.2±9.0 | 62.2±9.4 | 62.0±9.2 | 61.2±9.4 | 0.410 | 0.655 | ||
| TGmmol/L | 1.80±1.03 | 1.32±0.40 | 2.25±1.58 | 3.48±3.84 | 0.072 | 0.003 | ||
| TCmmol/L | 4.65±1.14 | 6.26±1.12 | 4.95±1.00 | 6.04±0.90 | <0.001 | <0.001 | ||
| HDLmmol/L | 1.30±0.32 | 1.22±0.24 | 1.13±0.22 | 1.03±0.28 | 0.361 | 0.044 | ||
| LDLmmol/L | 2.70±0.75 | 2.66±0.49 | 2.54±1.02 | 2.53±0.97 | 0.812 | 0.949 | ||
| FBGmmol/L | 6.04±1.20 | 6.12±1.24 | 6.60±2.42 | 6.84±2.63 | 0.810 | 0.626 | ||
| BMI | 23.43±1.40 | 23.67±1.34 | 23.75±1.69 | 23.47±1.71 | 0.523 | 0.406 | ||
T-855C. Genotype and clinical characteristics of CAD patients and non-diabetic controls
| CAD patients | Non-CAD patients | P-value | ||||||
|---|---|---|---|---|---|---|---|---|
| TT | TC+CC | TT | TC+CC | TT vs. (TC+CC) in CAD patients | TT vs. (TC+CC) in non-CAD patients | |||
| Age (years) | 59.6±9.0 | 62.3±9.0 | 61.9±9.3 | 61.8±9.1 | 0.045 | 0.902 | ||
| TGmmol/L | 1.70±0.79 | 1.90±1.40 | 2.29±1.61 | 2.64±2.68 | 0.205 | 0.252 | ||
| TCmmol/L | 4.55±1.04 | 5.33±1.42 | 4.82±0.96 | 5.55±1.06 | 0.000 | 0.000 | ||
| HDLmmol/L | 1.30±0.32 | 1.27±0.30 | 1.11±0.23 | 1.13±0.24 | 0.459 | 0.386 | ||
| LDLmmol/L | 2.72±0.76 | 2.65±0.69 | 2.54±0.97 | 2.53±1.08 | 0.572 | 0.956 | ||
| FBGmmol/L | 6.02±1.12 | 6.13±1.36 | 6.75±2.32 | 6.46±2.64 | 0.542 | 0.409 | ||
| BMI | 23.41±1.37 | 23.53±1.46 | 23.70±1.67 | 23.73±1.75 | 0.583 | 0.912 | ||
T-1628G. Genotype and clinical characteristics of CAD patients and non-diabetic controls
| CAD patients | Non-CAD patients | P-value | ||||||
|---|---|---|---|---|---|---|---|---|
| TT | TG+GG | TT | TG+GG | TT vs. (TG+GG) in CAD patients | TT vs. (TG+GG) in non-CAD patients | |||
| Age (years) | 60.5±9.0 | 59.9±9.3 | 61.4±9.4 | 62.8±8.7 | 0.664 | 0.295 | ||
| TGmmol/L | 1.81±1.08 | 1.59±0.71 | 2.52±2.32 | 2.24±1.55 | 0.162 | 0.355 | ||
| TCmmol/L | 4.82±1.19 | 4.62±1.31 | 5.16±1.07 | 5.01±1.04 | 0.307 | 0.337 | ||
| HDLmmol/L | 1.31±0.32 | 1.26±0.30 | 1.10±0.24 | 1.15±0.23 | 0.365 | 0.154 | ||
| LDLmmol/L | 2.69±0.76 | 2.73±0.66 | 2.52±1.03 | 2.57±0.98 | 0.685 | 0.770 | ||
| FBGmmol/L | 6.04±1.14 | 6.07±1.37 | 6.80±2.71 | 6.32±1.80 | 0.895 | 0.182 | ||
| BMI | 23.50±1.38 | 23.28±1.44 | 23.77±1.75 | 23.59±1.59 | 0.329 | 0.468 | ||
Notes: Data are means ± SD. P-Value <0.05 was shown in bold.
Association of apoM promoter haplotypes with CAD
| Haplotype* | Haplotype frequency | OR | P^ | |||
|---|---|---|---|---|---|---|
| -1628-855-724 | Cases | Controls | (95%CI) | |||
| H1 | G C C | 0.037 | 0.016 | 2.421 (0.954~6.142) | 0.055 | |
| H2 | G T C | 0.148 | 0.097 | 1.638 (1.072~2.503) | 0.022 | |
| H3 | T C C | 0.146 | 0.122 | 1.242 (0.831~1.856) | 0.290 | |
| H4 | T C D | 0.044 | 0.011 | 4.376 (1.529~12.527) | 0.003 | |
| H5 | T T C | 0.589 | 0.725 | 0.531 (0.392~0.717) | 0.000 | |
^ P-Value <0.05 is shown in bold.
* Haplotype are arranged in the order T-1628G, T-855C and C-724del. Haplotype with frequency less than 3% was pooled and not analyzed.
3.5 Effects of apoM polymorphisms on gene promoter activities
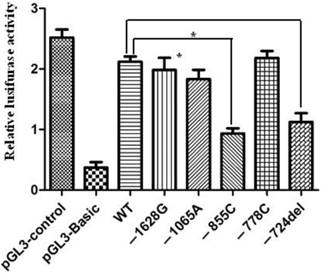
In order to assess the relationship between the promoter polymorphisms and the expression of apoM, luciferase reporter gene assay was used to measure the alteration of apoM promoter activity. It demonstrated that constructs -855C and -724del showed significantly decreased luciferase activities, (0.93±0.15 vs. 2.11±0.15; P=0.012) and (1.13±0.25 vs. 2.11±0.15; P=0.009), respectively. However, the promoter activity had no significant difference between wild-type and mutant in -778, -1065 and -1628 sites (Fig. 2).
Relative luciferase activities regulated by mutations in the proximal promoter region of apoM. 293HEK were transfected with different pGL3-pAPOM constructs and luciferase activities were measured by using the Luciferase Assay System as described in the material and methods. Significantly lower luciferase activities were generated by the constructs of pGL3-855C and pGL3-724del compared to the wide-type (WT). Results are represented as mean ± SD (n = 3 for each group). *P<0.05 vs. WT.
4. Discussion
In the present study we performed genetic association analysis on SNPs in the human apoM promoter gene in 206 CAD patients and 209 healthy controls from a Chinese Han population. Four common polymorphic sites, i.e., T-1628G, C-1065A, T-855C and T-778C, were confirmed and a new deletion mutation C-724del was found in both CAD and non-CAD patients. It clearly demonstrated that T-1628G, T-855C and C-724del polymorphism in the promoter region appeared to increase susceptibility to CAD. However, T-778C and C-1065A have no significant difference in allele frequencies and genotype distributions between CAD and non-CAD patients. These results were consistent with our previous findings [18]. Consistently, the haplotype analysis showed that the haplotype G-T-C and T-C-Del (-1628-855-724) was associated with an increased risk for CAD (OR = 2.421 (95% CI =1.072~2.503); P=0.022 and OR = 4.376 (95% CI =1.529~12.527) in the same population. In addition, the haplotype T-T-C could decrease the risk for CAD (OR=0.531 (95%CI 0.392~0.717), P=0.000).
ApoM is a plasma apolipoprotein that is particularly abundant in HDL particles [12]. The expression of ApoM and its concentration in plasma are dependent upon a number of nuclear transcription factors in vivo and in vitro, such as hepatic nuclear factor-1α (HNF-1α)[21], liver receptor homolog-1 (LRH-1)[22], forkhead box A2 (Foxa2)[23], and liver X receptor (LXR) [24], all of which contribute to hepatic lipid and glucose metabolism. By contrast, neither peroxisome proliferators activated receptor-α (PPAR-α) agonist (GW7647) nor PPAR-γ agonist (GW1929) influences APOM expression in HepG2 cells, indicating that these factors are not involved in regulating hepatic expression of the gene[25]. Certain hormones, such as leptin and insulin, are also involved in the regulation of apoM transcription and secretion [26]. Lacking apoM led to the disappearance of pre-β-HDL in plasma and showed significant decreased HDL-C (HDL cholesterol) levels [13, 27]. HDL is well known being a protective factor of CAD ascribing to its role of the “reverse cholesterol transport” [28]. In a recent study, Plomgaard, et al., showed that apoM is associated with pre-β-HDL formation in type 2 diabetes (T2D) [29], which may directly affect plasma HDL levels. Christoffersson, et al., [30, 31] demonstrated that apoM+ HDL particles showed more efficiency in stimulating cholesterol efflux and reducing total levels of intracellular cholesterol than total HDL or apoM- HDL particles. ApoM+ HDL was also found to induce endothelial cell migration and formation of endothelial adhesion junctions by the S1P1 receptor, which suggests that apoM has a critical role in the reverse cholesterol transport and is also a candidate cardiovascular risk factor [31]. Su et al. confirmed that apoM could not be an independent risk factor but a biomarker of CAD [32]. But Ahnstrom J et al. [33] showed in two independent case-control studies with large populations that serum concentrations of apoM do not influence CAD susceptibility. It is interesting to investigate whether apoM is a predictor of cardiovascular disease.
In genetics, a promoter is a region of initiation transcription of a particular gene. SNPs in promoter region could affect the gene transcription as well as protein expression. Moreover we detected the activation of apoM promoter with different mutants. It demonstrated that constructs -855C and -724del showed significant decreased luciferase activities (P=0.012 and P=0.009, respectively), which indicates that these polymorphisms may depress apoM expression. Our results also showed that polymorphism -855C and -724del carriers had significantly higher total cholesterol (TC) levels compared to groups with wild-type homozygotes. It suggests that these mutations in promoter region could influence HDL metabolism in vivo by down-regulating the expression of apoM. Subsequently, the process of reverse cholesterol transport was interfered resulting in the increase of TC level in peripheral blood.
The present study has several strengths along with certain limitations. The case and control groups were from a Chinese Han population, which may help to eliminate false positive association due to population admixture. Moreover, we incorporated a haplotype-based analysis across the candidate gene region, which may increase statistic power and reduce the problem of multiple testing. Furthermore, the functionality data provides evidence supporting our hypothesis. Limitations, which require further investigation, include a lack of serum samples preventing analysis of serum apoM levels. Instead we functionally examined activities of the promoter variants by the luciferase assay in vitro in order to reveal these SNPs in relation to the apoM expressions. Second, the total number of the patient and control cases are insufficient at present (<500) with collected continually. The results of power calculation performed with software PS (Power and sample size program, vision 3.0.43) showed that the study had 75.9% and 66.9% power to detect the differences of T-855C and C-724del between case and control subjects at a significance level of 0.05. It should be mentioned that the observed associations need further replications to avoid spurious associations which are common in genetic association studies.
In summary, we report a new polymorphism, C-724del, in region of apoM promoter. Additionally, we showed that the polymorphism C-724del and T-855C have significant associations with CAD in a Chinese Han population, and that these two genetic variations could impair apoM gene expression. Based on the findings above, we therefore speculate that the down-regulation of apoM expression could affect HDL and total cholesterol metabolism in vivo and further influence the susceptibility to CAD, which may be important for clarifying the role of apoM in vivo, although the detail mechanism remains to be elucidated.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (NSFC) (81201352), the research grant of Jiangsu province (BK2012154), the research grant of the Changzhou Health Bureau (ZD201104) and the research grant of the Changzhou Science and Technology Bureau (CM20113007).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Padmanabhan S, Hastie C, Prabhakaran D, Dominczak AF. Genomic approaches to coronary artery disease. Indian J Med Res. 2010;132:567-578
2. Windemuth A, de Leon J, Goethe JW, Schwartz HI, Woolley S, Susce M, Kocherla M, Bogaard K, Holford TR, Seip RL. et al. Validation of candidate genes associated with cardiovascular risk factors in psychiatric patients. Prog Neuropsychopharmacol Biol Psychiatry. 2012;36(2):213-219
3. Visvikis-Siest S, Marteau JB. Genetic variants predisposing to cardiovascular disease. Curr Opin Lipidol. 2006;17(2):139-151
4. Swerdlow DI, Holmes MV, Harrison S, Humphries SE. The genetics of coronary heart disease. Br Med Bull. 2010;102:59-77
5. Walldius G, Jungner I. Apolipoprotein B and apolipoprotein A-I: risk indicators of coronary heart disease and targets for lipid-modifying therapy. J Intern Med. 2004;255(2):188-205
6. Walldius G, Jungner I. Rationale for using apolipoprotein B and apolipoprotein A-I as indicators of cardiac risk and as targets for lipid-lowering therapy. Eur Heart J. 2005;26(3):210-212
7. Dodani S, Henkhaus R, Dong L, Butler MG. Apo lipoprotein A1 gene polymorphisms predict cardio-metabolic risk in South Asian immigrants. Dis Markers. 2012;32(1):9-19
8. Jang Y, Paik JK, Hyun YJ, Chae JS, Kim JY, Choi JR, Lee SH, Shin DJ, Ordovas JM, Lee JH. The apolipoprotein A5 -1131T>C promoter polymorphism in Koreans: association with plasma APOA5 and serum triglyceride concentrations, LDL particle size and coronary artery disease. Clin Chim Acta. 2009;402(1-2):83-87
9. Cardona F, Guardiola M, Queipo-Ortuno MI, Murri M, Ribalta J, Tinahones FJ. The -1131T>C SNP of the APOA5 gene modulates response to fenofibrate treatment in patients with the metabolic syndrome: a postprandial study. Atherosclerosis. 2009;206(1):148-152
10. Xu N, Dahlback B. A novel human apolipoprotein (apoM). J Biol Chem. 1999;274(44):31286-31290
11. Xie T, Rowen L, Aguado B, Ahearn ME, Madan A, Qin S, Campbell RD, Hood L. Analysis of the gene-dense major histocompatibility complex class III region and its comparison to mouse. Genome Res. 2003;13(12):2621-2636
12. Dahlback B, Nielsen LB. Apolipoprotein M--a novel player in high-density lipoprotein metabolism and atherosclerosis. Curr Opin Lipidol. 2006;17(3):291-295
13. Wolfrum C, Poy MN, Stoffel M. Apolipoprotein M is required for prebeta-HDL formation and cholesterol efflux to HDL and protects against atherosclerosis. Nat Med. 2005;11(4):418-422
14. Zhou JW, Tsui SK, Ng MC, Geng H, Li SK, So WY, Ma RC, Wang Y, Tao Q, Chen ZY. et al. Apolipoprotein M gene (APOM) polymorphism modifies metabolic and disease traits in type 2 diabetes. PLoS One. 2011;6(2):e17324
15. Wu X, Niu N, Brismar K, Zhu X, Wang X, Efendic S, Du T, Liu Y, Gu HF, Liu Y. Apolipoprotein M promoter polymorphisms alter promoter activity and confer the susceptibility to the development of type 1 diabetes. Clin Biochem. 2009;42(1-2):17-21
16. Xu WW, Zhang Y, Tang YB, Xu YL, Zhu HZ, Ferro A, Ji Y, Chen Q, Fan LM. A genetic variant of apolipoprotein M increases susceptibility to coronary artery disease in a Chinese population. Clin Exp Pharmacol Physiol. 2008;35(5-6):546-551
17. Jiao GQ, Yuan ZX, Xue YS, Yang CJ, Lu CB, Lu ZQ, Xiao MD. A prospective evaluation of apolipoprotein M gene T-778C polymorphism in relation to coronary artery disease in Han Chinese. Clin Biochem. 2007;40(15):1108-1112
18. Zheng L, Luo G, Zhang X, Zhang J, Zhu J, Wei J, Mu Q, Chen L, Nilsson-Ehle P, Xu N. Determination of single-nucleotide polymorphism in the proximal promoter region of apolipoprotein M gene in coronary artery diseases. Int J Gen Med. 2009;2:177-182
19. Chen Q, Reis SE, Kammerer CM, McNamara DM, Holubkov R, Sharaf BL, Sopko G, Pauly DF, Merz CN, Kamboh MI. et al. Association between the severity of angiographic coronary artery disease and paraoxonase gene polymorphisms in the National Heart, Lung, and Blood Institute-sponsored Women's Ischemia Syndrome Evaluation (WISE) study. Am J Hum Genet. 2003;72(1):13-22
20. Shi YY, He L. SHEsis, a powerful software platform for analyses of linkage disequilibrium, haplotype construction, and genetic association at polymorphism loci. Cell Res. 2005;15(2):97-98
21. Skupien J, Kepka G, Gorczynska-Kosiorz S, Gebska A, Klupa T, Wanic K, Nowak N, Borowiec M, Sieradzki J, Malecki MT. Evaluation of Apolipoprotein M Serum Concentration as a Biomarker of HNF-1alpha MODY. Rev Diabet Stud. 2007;4(4):231-235
22. Venteclef N, Haroniti A, Tousaint JJ, Talianidis I, Delerive P. Regulation of anti-atherogenic apolipoprotein M gene expression by the orphan nuclear receptor LRH-1. J Biol Chem. 2008;283(7):3694-3701
23. Hu YW, Zheng L, Wang Q, Zhong TY, Yu X, Bao J, Cao NN, Li B, Si-Tu B. Vascular endothelial growth factor downregulates apolipoprotein M expression by inhibiting Foxa2 in a Nur77-dependent manner. Rejuvenation Res. 2012;15(4):423-434
24. Zhang X, Zhu Z, Luo G, Zheng L, Nilsson-Ehle P, Xu N. Liver X receptor agonist downregulates hepatic apoM expression in vivo and in vitro. Biochem Biophys Res Commun. 2008;371(1):114-117
25. Xu N, Ahren B, Jiang J, Nilsson-Ehle P. Down-regulation of apolipoprotein M expression is mediated by phosphatidylinositol 3-kinase in HepG2 cells. Biochim Biophys Acta. 2006;1761(2):256-260
26. Xu N, Nilsson-Ehle P, Hurtig M, Ahren B. Both leptin and leptin-receptor are essential for apolipoprotein M expression in vivo. Biochem Biophys Res Commun. 2004;321(4):916-921
27. Wroblewska M. The origin and metabolism of a nascent pre-beta high density lipoprotein involved in cellular cholesterol efflux. Acta Biochim Pol. 2011;58(3):275-285
28. Weissglas-Volkov D, Pajukanta P. Genetic causes of high and low serum HDL-cholesterol. J Lipid Res. 2010;51(8):2032-2057
29. Plomgaard P, Dullaart RP, de Vries R, Groen AK, Dahlback B, Nielsen LB. Apolipoprotein M predicts pre-beta-HDL formation: studies in type 2 diabetic and nondiabetic subjects. J Intern Med. 2009;266(3):258-267
30. Christoffersen C, Nielsen LB, Axler O, Andersson A, Johnsen AH, Dahlback B. Isolation and characterization of human apolipoprotein M-containing lipoproteins. J Lipid Res. 2006;47(8):1833-1843
31. Christoffersen C, Obinata H, Kumaraswamy SB, Galvani S, Ahnstrom J, Sevvana M, Egerer-Sieber C, Muller YA, Hla T, Nielsen LB. et al. Endothelium-protective sphingosine-1-phosphate provided by HDL-associated apolipoprotein M. Proc Natl Acad Sci U S A. 2011;108(23):9613-9618
32. Su W, Jiao G, Yang C, Ye Y. Evaluation of apolipoprotein M as a biomarker of coronary artery disease. Clin Biochem. 2009;42(4-5):365-370
33. Ahnstrom J, Axler O, Jauhiainen M, Salomaa V, Havulinna AS, Ehnholm C, Frikke-Schmidt R, Tybjaerg-Hansen A, Dahlback B. Levels of apolipoprotein M are not associated with the risk of coronary heart disease in two independent case-control studies. J Lipid Res. 2008;49(9):1912-1917
Author contact
![]() Corresponding author: Ning Xu, Division of Clinical Chemistry and Pharmacology, Department of Laboratory Medicine, Lund University, S-221 85 Lund, Sweden, Phone: (46)736395462: e-mail address: ning.xulu.se or Xiaoying Zhang, Comprehensive Laboratory, Third Affiliated Hospital of Soochow University, Changzhou 213003, China. Phone: (86)519-68871278; e-mail address: zhangxy6689996com
Corresponding author: Ning Xu, Division of Clinical Chemistry and Pharmacology, Department of Laboratory Medicine, Lund University, S-221 85 Lund, Sweden, Phone: (46)736395462: e-mail address: ning.xulu.se or Xiaoying Zhang, Comprehensive Laboratory, Third Affiliated Hospital of Soochow University, Changzhou 213003, China. Phone: (86)519-68871278; e-mail address: zhangxy6689996com