Impact Factor ISSN: 1449-1907

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2013; 10(13):1888-1898. doi:10.7150/ijms.6019 This issue Cite
Research Paper
Modulation of β-Catenin Signaling by the Inhibitors of MAP Kinase, Tyrosine Kinase, and PI3-Kinase Pathways
Wenwen Zhang1,2,3, Hongyu Zhang1,2, Ning Wang2,4, Chen Zhao2,4, Hongmei Zhang1,2, Fang Deng1,4, Ningning Wu1,2, Yunfeng He1,2, Xian Chen1,2, Junhui Zhang1,2, Sheng Wen1,2, Zhan Liao2, Qian Zhang1,2, Zhonglin Zhang2, Wei Liu1,2, Zhengjian Yan1,2, Hue H. Luu2, Rex C. Haydon2, Lan Zhou1, Tong-Chuan He1,2 ![]()
1. Ministry of Education Key Laboratory of Diagnostic Medicine and School of Clinical Diagnostic Medicine, and the Affiliated Hospitals of Chongqing Medical University, Chongqing 400016, China;
2. Molecular Oncology Laboratory, Department of Orthopaedic Surgery, The University of Chicago Medical Center, 5841 South Maryland Avenue, MC 3079, Chicago, IL 60637, USA;
3. Department of Laboratory Medicine of the Affiliated Hospital, Bingzhou Medical University, Yantai, Shandong, China;
4. School of Laboratory Medicine and Departments of Cell Biology and Oncology of the Affiliated Southwest Hospital, the Third Military Medical University, Chongqing 400038, China
Received 2013-2-2; Accepted 2013-10-17; Published 2013-11-25
Abstract
Aberrant activation of β-catenin signaling plays an important role in human tumorigenesis. However, molecular mechanisms behind the β-catenin signaling deregulation are mostly unknown because genetic alterations in this pathway only account for a small fraction of tumors. Here, we investigator if other major pathways can regulate β-catenin signaling activity. By employing a panel of chemical activators and/or inhibitors of several cellular signaling pathways, we assess these modulators' effects on luciferase reporter driven by β-catenin/TCF4-responsive elements. We find that lithium-stimulated β-catenin activity is synergistically enhanced by protein kinase C activator PMA. However, β-catenin-regulated transcriptional (CRT) activity is significantly inhibited by casein kinase II inhibitor DRB, MEK inhibitor PD98059, G-proteins and their receptor uncoupling agent suramin, protein tyrosine kinase inhibitor genistein, and PI-3 kinase inhibitor wortmannin, suggesting that these cellular pathways may participate in regulating β-catenin signaling. Interestingly, the Ca++/calmodulin kinase II inhibitor HDBA is shown to activate β-catenin activity at low doses. Furthermore, Wnt3A-stimulated and constitutively activated CRT activities, as well as the intracellular accumulation of β-catenin protein in human colon cancer cells, are effectively suppressed by PD98059, genistein, and wortmannin. We further demonstrate that EGF can activate TCF4/β-catenin activity and induce the tyrosine phosphorylation of β-catenin protein. Thus, our results should provide important insights into the molecular mechanisms underlying Wnt/β-catenin activation. This knowledge should facilitate our efforts to develop efficacious and novel therapeutics by targeting these pathways.
Keywords: β-catenin, G protein, MEK kinase, PI3 kinase, protein kinase C, tumorigenesis, tyrosine phosphorylation, Wnt signaling.
Introduction
Originally identified as a cytoplasmic protein that interacts with cell adhesion molecules, such as E-cadherin, β-catenin was found to be the mammalian homologue of armadillo, a segment polarity gene involved in the Wingless pathway in Drosophilla [1-7] . The Wnt/Wingless signaling pathway is involved in many developmental processes, and many components of this pathway are evolutionarily conserved among Drosophila, Dictyostelium, C. elegans, Xenopus, and mammals [1-5]. The Wnt ligands initiate the signaling pathway by binding to the frizzled receptors, leading to phosphorylation of the dishevelled protein, which, through its association with Axin and the APC tumor suppressor [8, 9], prevents glycogen synthase kinase 3β (GSK3β) from phosphorylating β-catenin [1]. Unphosphorylated β-catenin is stabilized via escaping the recognition by β-TrCP, a component of an E3 ubiquitin ligase, and eventually translocates to the nucleus where it engages transcription factors LEF/TCF-4 to activate expression of downstream genes. In normal and unstimulated cells, the majority of β-catenin protein is present in cell-cell junctions with very little in cytoplasmic or nuclear fractions, due to the rapid turnover of β-catenin promoted by the complexes containing APC, GSK3β, and Axin. However, in the presence of Wnt signal, GSK3β activity is inactivated, leading to the accumulation of cytoplasmic and, subsequently, nuclear β-catenin, and the activation of β-catenin/TCF-4 downstream target genes, such as c-Myc, cyclin D1, and PPARδ [10-13] . The β-catenin activity is negatively regulated by many cellular factors, including TCF1, Grouch, ICAT, Idax, Duplin, Axam [1, 6, 7, 14], clearly indicating that β-catenin signaling is tightly regulated in normal cells.
Activation of the β-catenin signaling plays an important role in tumorigenesis [5-7, 15]. Elucidation of molecular mechanisms behind its activation should help to define the molecular basis of tumor development. Although the involvement of β-catenin in tumorigenesis was first established in colorectal cancer, where β-catenin was found to form a complex with the APC tumor suppressor gene product [16, 17], the importance of β-catenin in regulating cell proliferation has been highlighted by the discovery of oncogenic mutations of the β-catenin gene in colon cancers containing the wild-type APC gene [18]. Mutant β-catenin protein becomes more stable because of its capability of bypassing APC-targeted degradation. Although at a much lower frequency, oncogenic β-catenin mutations have been uncovered in a variety of human tumors [6, 7, 18]. The collective genetic evidence is highly indicative that deregulation of β-catenin signaling may be involved in the development of a broad range of human malignancies, which is further supported by a long-standing observation that over-expression of β-catenin downstream targets, such as c-Myc and cyclin D1, has been extensively documented in many human tumors [5-7, 14, 19]. Furthermore, abundant immunohistochemical studies have demonstrated that the cytoplasmic and/or nuclear level of β-catenin is frequently elevated in most human tumors [5-7, 20]. Although Wnts are considered bona fide regulators of β-catenin signaling, with an exception of colorectal cancer, in which β-catenin signaling is activated by either loss-of-function mutations of the APC tumor suppressor gene or gain-of-function mutations of the β-catenin gene, causes of β-catenin signaling deregulation in most human tumors remain to be determined. In order to search for alternative cellular pathways that may regulate β-catenin signaling, we analyze a panel of activators and inhibitors of various signaling pathways for their effect on β-catenin-regulated transcription (CRT). We find that lithium-stimulated β-catenin/TCF4 activity is synergistically enhanced by protein kinase C activator PMA. However, the CRT activity is effectively inhibited by the casein kinase II inhibitor DRB, the MEK inhibitor PD98059, the G-proteins and their receptor uncoupling agent suramin sodium, the protein tyrosine kinase inhibitor genistein, and the PI3 kinase inhibitor wortmannin, respectively. Furthermore, Wnt3A-stimulated and constitutively activated CRT activities, as well as the intracellular accumulation of β-catenin protein in human colon cancer cells, are effectively suppressed by PD98059, genistein, and wortmannin. These results strongly suggest that these cellular pathways may participate in regulating β-catenin signaling. Interestingly, the Ca++/calmodulin kinase II inhibitor HDBA can activate β-catenin activity at low doses. Moreover, EGF is shown to activate TCF/β-catenin activity and induce the tyrosine phosphorylation of β-catenin protein. Taken together, our findings should provide important insights into the molecular mechanisms underlying Wnt/β-catenin signaling activation, particularly in the context of tumorigenesis.
Materials and Methods
Cell Lines and Culture Medium
Human embryonic kidney cell line HEK 293, human osteosarcoma lines 143B and HOS, and human colon cancer line SW480 were purchased from American Type Culture Collection (ATCC, Manassas, VA) and maintained in complete DMEM or McCoy 5A (i.e., supplemented with 10% fetal calf serum, 100 units/ml of penicillin, and 100 μg/ml streptomycin) at 370C in 5% CO2 incubators.
Chemicals
Lithium chloride was purchased from Sigma-Aldrich (St. Louis, MO). PMA (phorbol 12-myristate 13-acetate; or TPA, 12-tetradecanoyl phorbol 13-acetate), suramin sodium, wortmannin, HDBA (2-hyroxyl-5-(2,5-dihydroxybenzylamino) benzoic acid), DRB (5,6-dichloro-1-β-D-ribofuranosylbenzimidazole), PD-98059, and genistein (4',5,7-trihydroxyisoflavone) were purchased from BIOMOL (Plymouth Meeting, PA). Stock solutions of above chemicals were prepared according to manufacturer's instructions. Recombinant human EGF protein (rhEGF) was purchased from Sigma-Aldrich (St. Louis, MO). Unless indicated otherwise, all chemicals were obtained from Fisher Scientific (Pittsburgh, PA) or Sigma-Aldrich.
Reporter Plasmids, Transfections, and Luciferase Assays
Construction and use of TCF-4 responsive luciferase reporter, pTOP-Luc, was described previously [10, 11, 21, 22]. The control reporter, pGL3-control, was purchased from Promega (Madison, WI). For lithium-stimulated CRT assays, exponentially growing HEK293 cells were transfected with pTOP-Luc or pGL3-control plasmid DNA (3 μg per 25 cm2 flask) by using LipofectAmine (Life Technologies, Gaithersburg, MD). At 24h after transfection, cells were trypsinized and replated to 24-well plates in the absence of fetal calf serum. After 15h of starvation, cells were treated in lithium-containing complete DMEM, with various concentrations of chemicals for five hours, and cell lysates were prepared for luciferase assays using Promega's Luciferase Assay kit. Each assay condition was performed in triplicate.
For Wnt3a-stimulated CRT activity assays, subconfluent HEK-293 cells were co-transfected with the Wnt3a expression vector pCMV-Wnt3a and GL3-control or TOP-Luc reporter for 16h. The transfected cells were replated and maintained in serum-free medium for additional 16h and then treated with PD98059 (10μM), genistein (150μM) and wortmannin (150nM), respectively, for 5h. The cells were lysed for luciferase assays. Each assay condition was done in triplicate.
For assessing the constitutive CRT activity in human colon cancer cells, subconfluent SW480 cells were transfected with GL3-Control or TOP-Luc reporter for 16h, replated, and treated with PD98059 (10μM), genistein (150μM), wortmannin (150nM) or DMSO control for 5h. Cells were lysed for luciferease assays. Each assay condition was done in triplicate.
RNA Isolation and Real-Time Quantitative-PCR (qPCR)
Subconfluent HEK-293 cells were transfected with Wnt3A expression vector pCMV-Wnt3A or the empty vector control. The transfected cells were maintained in 1% FBS DMEM and treated with respective inhibitors. At 30h after treatment, total RNA was isolated using TRIzol Reagents (Invitrogen), according to the manufacturer's instructions. Ten micrograms of total RNA were used to generate cDNA templates by reverse transcription with hexamer and Superscript II reverse transcriptase (Invitrogen). The first strand cDNA products were further diluted 5- to 10-fold and used as qPCR templates. The qPCR primers were 18-20-mers and designed by using the Primer3 program to amplify the 3'-end (approximately 150 bps) of the gene of interest. The primer sequences are, for human c-Myc: forward 5'-CGT CCT GGG AAG GGA GAT-3' and reverse 5'-CGC TGC TAT GGG CAA AGT-3'; for human cyclin D1: forward 5'-TGT TTG CAA GCA GGA CTT TG-3' and reverse 5'-TGG CAC CAA AGG ATT CCT AA-3'; and for human GAPDH: forward 5'-CAA CGA ATT TGG CTA CAG CA-3' and reverse 5'-AGG GGA GAT TCA GTG TGG TG-3'. SYBR Green-based qPCR analysis was carried out using the Opticon DNA Engine (M J Research). The specificity of each qPCR reaction was verified by melting curve analysis and further confirmed by resolving the PCR products on 1.5% agarose gels. Five-fold serially diluted pUC19 was used as a standard. Duplicate reactions were carried out for each sample. All samples were normalized by the expression level of GAPDH.
Immunofluorescence Staining
The immunostaining assays were carried out as previously described [23-33]. Briefly, subconfluent SW480 cells were treated with PD98059 (10μM), genistein (150μM), wortmannin (150nM) or DMSO control for 20h. Cells were fixed and stained with β-catenin antibody (Santa Cruz Biotechnology). Isotype IgG and minus primary antibody were used as negative controls.
Immunoprecipitation and Western Blotting
Subconfluent HEK-293 and HOS cells were maintained in serum-free medium overnight and stimulated with or without rhEGF (50ng/ml) for 2h. Cells were lysed in a cell lysis buffer (50mM Tris-HCl, pH7.5, 100mM NaCl, and 0.5% NP-40) containing protease inhibitors (Roche Molecular Biochemicals, Indianapolis, IN). The cleared cell lysate was incubated with phosphor-tyrosine antibody 4G10 (Upstate Inc) or β-catenin antibody (Pharmingen) at 4ºC for 60 minutes. 100 μl of 50% slurry protein G-Agarose 4B was added to the cell lysate mixtures for additional 30min at 4ºC. The protein/antibody-bound Sepharose 4B was washed with PBS three times. Bound proteins were eluted by boiling the samples in Laemmli sample buffer and were loaded onto a 4-20% gradient SDS-polyacrylamide gel. After being resolved by electrophoresis, proteins were transferred to an Immobilon-P membrane (Millipore) via electroblotting. The membrane was blocked with 5% nonfat milk in TBST (10mM Tris-HCl, pH8.0, 150mM NaCl, 0.05% Tween-20) at room temperature for 60min and probed with anti-β-catenin or 4G10 antibody for 60min, followed by 30min incubation with anti-mouse IgG conjugated with horseradish peroxidase (PIERCE). The presence of β-catenin protein was detected by using the SuperSignal West Pico Chemiluminescent substrate kit (PIERCE).
Statistical Analysis
All quantitative experiments were performed in triplicate and/or repeated three times. Data were expressed as mean ± SD. Statistical significances between treatment groups vs. control groups were determined by one-way analysis of variance and the two-tailed Student's t test. A p-value of < 0.05 was defined as statistically significance.
Results and Discussion
Lithium-Stimulated CRT Activity Is Synergistically Enhanced the PKC Activator PMA
To search for potential pathways that may regulate β-catenin activity, we tested a panel of synthetic or exogenous activators and inhibitors of several important cellular signaling pathways. To assess β-catenin activity upon different stimuli, we used a β-catenin-regulated luciferase reporter pTOP-Luc [21, 34]. We have previously demonstrated that this reporter exhibited low basal activity but can be specifically activated by oncogenic forms of β-catenin in HEK 293 cells [10, 11, 21, 22, 35]. As shown in Fig. 1A, the β-catenin /TCF-4 reporter was effectively activated by lithium, a ten-fold activation by LiCl at a concentration of 30mM. Clinically used as a therapeutic agent for manic depression lithium has been shown to mimic the Wnt signal in Xenopus embryonic development [36-38]..
PKC activator PMA enhances lithium-induced β-catenin-regulated transcription activity. (A) Activation of β-catenin/TCF4 signaling by lithium in HEK 293 cells. Exponentially growing 293 cells were transfected with pTOP-Luc reporter, replated into 24-well plates, and maintained in serum-free medium for 16h. The cells were treated with lithium chloride at the indicated concentrations for 5h and collected for luciferase assays. (B) Synergistic effect of PMA on lithium-activated β-catenin/TCF4 signaling. Exponentially growing 293 cells were transfected with pTOP-Luc, replated into 24-well plates, and maintained in serum-free medium for 16h. The cells were stimulated with LiCl and/or PMA at the indicated concentrations for 5h and collected for luciferase assays. Each assay condition was done in triplicate. Luciferase activity was normalized by protein concentrations of lysates. Data were representative of three independent experiments.
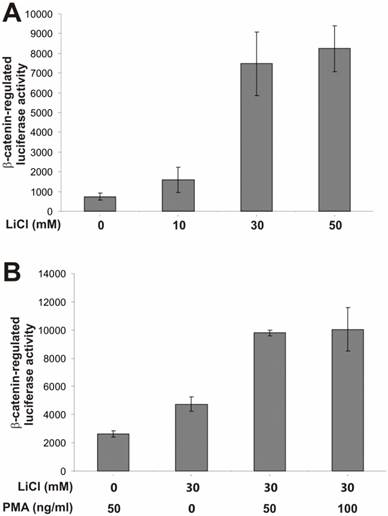
Here, we used the lithium-stimulated β-catenin activation to assess synergistic or inhibitory effects exerted by the activators/inhibitors of other signaling pathways. We first tested the effect of PMA on β-catenin signaling. PMA binds to and activates protein kinase C (PKC). It has been previously reported that PMA-sensitive isoforms of PKC may be involved in Wnt-induced inactivation of GSK3β [39]. As shown in Fig. 1B, PMA alone exhibited a marginal activation of β-catenin signaling. However, a synergistic activation was observed when both lithium (30mM) and PMA were presented, suggesting that the PMA-sensitive isoforms of PKC may regulate β-catenin signaling synergistically in the presence of Wnt signal. These findings are consistent with previous observation [39].
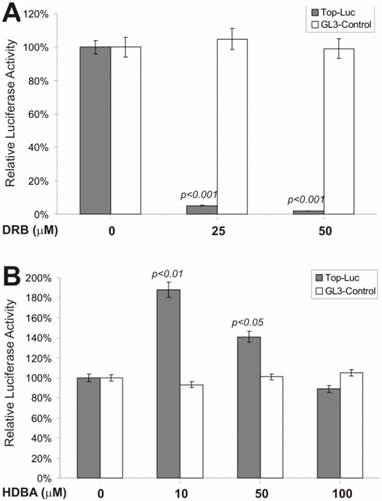
β-Catenin-Regulated Transcription Is Antagonized by a Casein Kinase II Inhibitor but Slightly Activated by a Ca++/Calmodulin Kinase II Inhibitor
We next tested if an inhibitor of casein kinase II would exhibit any effect on β-catenin signaling. Casein kinase II has been shown to phosphorylate the dishevelled, leading to inactivation of GSK3β activity [40, 41] . As shown in Fig. 2A, DRB, a potent and specific inhibitor of casein kinase II, exerted remarkable inhibition on lithium-stimulated β-catenin activity, but not on control reporter pGL3-control. This observation is consistent with the proposed role of casein kinase II in Wnt signaling [6, 7]. However, when HDBA, a Ca++/Calmodulin kinase II inhibitor, was tested, we observed a more complex effect on lithium-stimulated β-catenin activity (Fig. 2B). At lower concentrations (e.g., 10 and 50 μM), HDBA slightly potentiated β-catenin activity, while at a higher concentration (e.g., 100 μM), no activating and a rather slightly inhibitory, effect was observed. It has been suggested that Ca++/Calmodulin may be involved in Wnt signal transduction [42]. These findings may argue against that possibility although further investigation is required. It is noteworthy that at higher concentrations HDBA can also inhibit other cellular targets, including EGF receptor tyrosine kinase and pp60c-src kinase [43]. This action of HDBA may partially account for the inhibitory effects observed with HDBA at 100μM.
Effects on the β-catenin/TCF4 activity by inhibitors of casein kinase II and Ca++/calmodulin kinase II. Exponentially growing 293 cells were transfected with pTOP-Luc (filled bars) or pGL3-control (blank bars), replated and maintained in serum-free medium for 16hstarvation. Cells were stimulated with lithium (30mM) in combination with DRB (A) or HDBA (B) at the indicated concentrations for 5h and collected for luciferase assays (Promega). Each assay condition was done in triplicate. Luciferase activity was normalized by protein concentrations of lysates. Data were representative of three independent experiments.
β-Catenin Activity Is Regulated by MAP Kinase and G-Protein Pathways
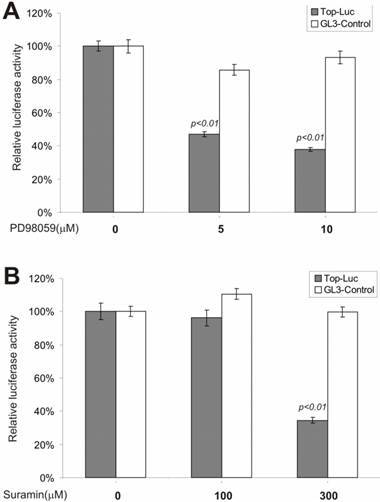
We next tested whether inhibitors of the MEK and G-protein signaling pathways exerted any regulatory effect on CRT activity. MAP kinase kinase (MEKK1) can interact with Axin [44] and the disheveled protein can activate JNK activity [45, 46], suggesting that some components of the MAP kinase pathway may cross-talk with Wnt signal and play a regulatory role on β-catenin signaling [47]. We tested PD98059, a potent inhibitor of MEK activity, for its effect on CRT activity. As shown in Fig. 3A, approximately 60% of CRT activity, but not the pGL3-control, was remarkably repressed by PD98059 at a concentration of 10 μM, confirming that MEK activity may indeed play an important role in modulating β-catenin activity. Because G protein signals can modulate PKC and MEK activities, both of which have been shown to be involved in regulating β-catenin signaling, we tested suramin sodium, a potent inhibitor of G protein signaling, for its inhibitory effect on the CRT activity. At the concentration of 300 μM, suramin sodium exerted striking repression on CRT, with no effect on the control reporter pGL3-control (Fig. 3B).
Inhibition of the β-catenin/TCF4 activity by inhibitors of MAP kinase and G protein pathways. The experiments were performed in a similar fashion to that described in Figure 2. MAP kinase inhibitor PD98059 and G protein inhibitor suramin were used in this study. Each assay condition was done in triplicate. Data were representative of three independent experiments.
β-Catenin-Regulated Transcription Is Antagonized by Tyrosine Kinase and PI3 Kinase Inhibitors
To test possible involvement of tyrosine phosphorylation in regulating β-catenin signaling, we examined genistein, a tyrosine kinase inhibitor, for its effect on lithium-stimulated β-catenin activity. genistein is known as a potent and specific inhibitor for a broad range of tyrosine kinases. As shown in Fig. 4A, at a moderate concentration (i.e., 75 μM), genistein dramatically inhibited the CRT activity, but not that of the control reporter pGL3-control. Although it has been reported that β-catenin protein was highly tyrosine phosphorylated during development, our observations are the first to demonstrate that tyrosine phosphorylation may play an important role in regulating β-catenin signaling.
Inhibition of the β-catenin/TCF4 activity by inhibitors of tyrosine kinases and PI3 kinase.. The experiments were performed in a similar fashion to that described in Figure 2. Each assay condition was done in triplicate. Tyrosine kinase inhibitor genistein and PI3-kinase inhibitor wortmannin were used in this study. Data were representative of three independent experiments.
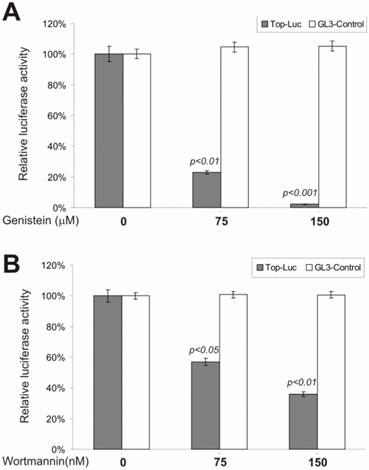
Phosphatidylinositol-3-kinase (PI3 kinase) is considered to be a nodal point for many cell growth/survival signaling pathways [48, 49]. To assess whether PI3 kinase activity exhibited any effect on β-catenin signaling, we tested a commonly used PI3 kinase inhibitor, wortmannin, for its ability to inhibit lithium-stimulated β-catenin activity. By covalently binding to PI3 kinase, the fungal metabolite wortmannin is a potent and selective inhibitor of PI3 kinase. As shown in Fig. 4B, the CRT activity was significantly inhibited by wortmannin. This inhibition was specific as no appreciable reduction in pGL3-control's activity was observed under the same conditions. Our findings demonstrate that PI3 kinase activity may regulate β-catenin signaling.
Interestingly, the possible involvement of PI3 kinase in regulating β-catenin signaling has been suggested because AKT/PKB, a downstream target of PI3 kinase, has been shown to phosphorylate and inactivate GSK3β [50], potentially leading to stabilization of the β-catenin protein. It has been reported that integrin-linked kinase (ILK) can induce nuclear translocation of the β-catenin by activation of PKB/AKT and inhibition of GSK3β in a PI3 kinase-dependent manner [51]. Microinjection or stable expression of oncogenic H-Ras in keratinocytes has been shown to promote the accumulation of cytoplasmic and nuclear β-catenin, mediated by PI3 kinase activity [52]. Moreover, in vitro binding assays demonstrated a strong association between β-catenin and the p85α subunit of the PI3 kinase [52]. However, a recent study has suggested that serum growth factors may exert their enhancement effect on the lithium-mediated accumulation of β-catenin through protein kinase C pathway, rather than through Ras or PI3 kinase signaling [39]. Thus, it is important to understand the actual role of PI3 kinase in regulating β-catenin signaling, particularly in the context of tumorigenesis.
Wnt3a-Stimulated CRT Activity and Nuclear β-catenin Accumulation Are Suppressed by the Inhibitors of MAPK, Tyrosine Kinase and PI3-Kinase Pathways
We next analyzed if the above pathway inhibitors exert a similar effect on Wnt3a-activated CRT activity. When Wnt3a-expressing HEK293 cells were treated with PD98059 (10μM), genistein (150μM) and wortmannin (150nM), the CRT activity was significantly decreased by 60.7%, 66.2%, and 72.1%, respectively, compared to that of DMSO control's (Fig. 5A), while no significant effects were observed on pGL3-control reporter activity (Fig. 5B). These results further suggest that inhibitors of these major pathways may exert relatively specific inhibitory effect on Wnt/β-catenin signaling activity.
Wnt-induced β-catenin/TCF4 activity and nuclear β-catenin accumulation are repressed by PD98059, genistein and wortmannin. (A) and (B) Subconfluent HEK-293 cells were co-transfected with the Wnt3a expression vector pCMV-Wnt3a and TOP-Luc (A) or GL3-control (B) reporter for 16h. The transfected cells were replated and maintained in serum-free medium for additional 16h and then treated with PD98059 (10μM), genistein (150μM) and wortmannin (150nM), respectively, for 5h. The cells were lysed for luciferase assays. Each assay condition was done in triplicate. Data were representative of three independent experiments. (C) Reduced cellular accumulation of β-catenin in human colon cancer cells by the inhibitors. Subconfluent SW480 cells were treated with PD98059 (10μM), genistein (150μM), wortmannin (150nM), DRB (50μM) or DMSO control for 20h. Cells were fixed and stained with β-catenin antibody (Santa Cruz Biotechnology). Isotype IgG and minus primary antibody were used as negative controls. Representative results are shown. (D) The constitutive β-catenin/TCF4 activity in human colon cancer cells is repressed by the inhibitors. Subconfluent SW480 cells were transfected with GL3-Control or TOP-Luc reporter for 16h, replated, and treated with PD98059 (10μM), genistein (150μM), wortmannin (150nM) or DMSO control for 5h. Cells were lysed for luciferease assays. Each assay condition was done in triplicate. Data were representative of three independent experiments.
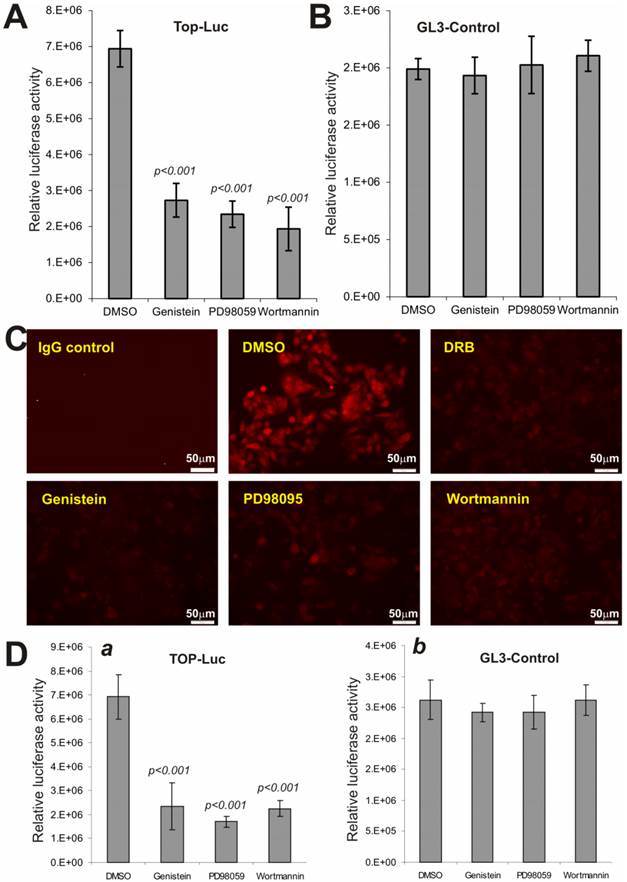
We further examined the effect of these inhibitors on the constitutive CRT activity in human colon cancer line SW480, which harbors APC mutations. When SW480 cells were treated with PD98059, genistein, and wortmannin, the staining of nuclear and/or cytoplasmic β-catenin protein was significantly inhibited in the treated cells, compared with solvent control (Fig. 5C). As expected, the casein kinase inhibitor DRB effectively inhibited β-catenin accumulation in SW480 cells, suggesting the inhibitory effect of PD98059, genistein, and wortmannin on β-catenin accumulation should be considered rather specific. Moreover, SW480 cells exhibited a high level of CRT activity, which was effectively inhibited by PD98059, genistein, and wortmannin, respectively (Fig. 5D). Collectively, the above results strongly indicate that Wnt/β-catenin signaling can be significantly suppressed by the Inhibitors of MAPK, tyrosine kinase and PI3-kinase pathways.
EGF Signaling Activates TCF4/β-Catenin Transcriptional Activity and Induces Tyrosine Phosphorylation of the β-Catenin Protein
We next sought to determine if EGF stimulation activates β-catenin signaling pathway and induces tyrosine phosphorylation. Here, we focused on HEK-293 and two human osteosarcoma lines 143B and HOS, which are known to contact intact Wnt/β-catenin pathway. We first tested if EGF activates β-catenin/TCF4-mediated transcription. When subconfluent cells were transfected with pTOP-Luc and stimulated with rhEGF, we found that EGF induced the reporter activity in a dose-dependent manner in all three tested lines (Fig. 6A). Accordingly, immunofluorescence staining experiments indicate that EGF stimulation stimulated the nuclear accumulation of β-catenin protein (Fig. 6B).
(A) EGF activates β-catenin/TCF4-mediated transcription. Subconfluent cells were transfected with pTOP-Luc and maintained in 0.5%FCS medium for 16 hrs. The transfected cells were then stimulated with rhEGF (Sigma) at the indicated concentrations for 6h, and collected for luciferase assays. Each assay condition was done in triplicate. Transfection efficiency was normalized by measuring the β-galactosidase activity of the co-transfected pCMV-β. (B) EGF induces cytoplasmic and nuclear accumulation of β-catenin in HOS cells. Cells were starved in 0.5% FCS medium for 15h, and treated with or without rhEGF for 2h. The cells were fixed for immunofluorescence staining using a β-catenin antibody or mouse IgG. The β-catenin was visualized by incubating the cells with Alexa 594-conjugated anti-mouse IgG. (C) EGF induces tyrosine phosphorylation of β-catenin protein. Subconfluent cells were maintained in serum-free medium overnight and stimulated with or without rhEGF (50ng/ml) for 2h. Cells were lysed and subjected to immunoprecipitation using phosphor-tyrosine antibody 4G10 (Upstate Inc., panel a) or β-catenin antibody (Pharmingen, panels c & d). Immunoprecipitated complexes were subjected to Western blotting analysis using β-catenin antibody (a, b and d) or 4G10 antibody (c). Panels b and d were controls for sample loading. Arrows indicate the position of β-catenin protein. (D) Wnt3A-induced expression of β-catenin target genes c-Myc and cyclin D1 activity is inhibited by the inhibitors. Subconfluent HEK-293 cells were transfected with Wnt3A expression vector pCMV-Wnt3A (Wnt3A) or the empty vector (EV) control. The transfected cells were maintained in 1% FBS DMEM and treated with PD98059 (5μM), genistein (100μM), wortmannin (100nM) or DMSO control for 30h. Total RNA was isolated for reverse transcription and qPCR analysis as described in Methods. The dotted lines indicate the basal expression level of c-Myc and cyclin D1. “*”, p<0.05; “**”, p<0.001. (E) The proposed working model of Wnt signal crosstalk with other pathways. Black lines indicate inhibitory effects. Red lines indicate activating effects. Dotted lines indicate that the mechanisms may be less well established. RTKs, receptor tyrosine kinases; TFs, transcriptional factors.
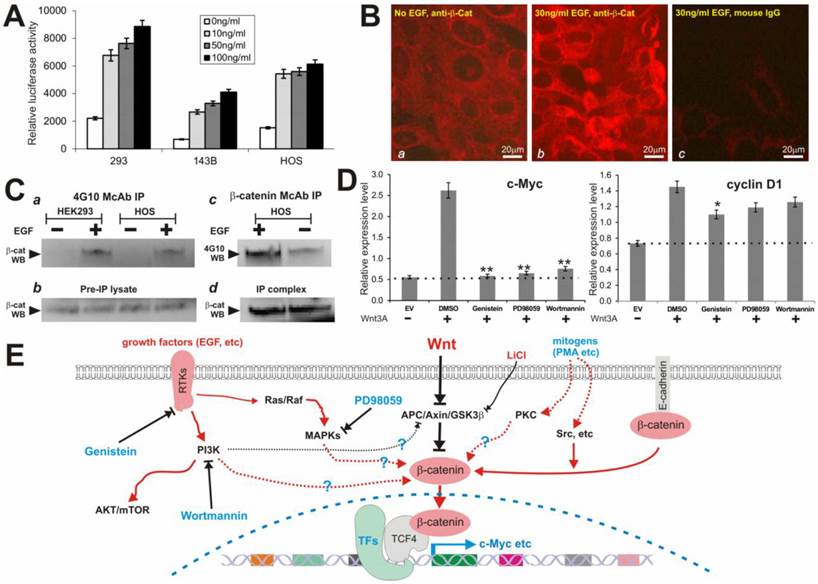
Furthermore, we found that tyrosine phosphorylated β-catenin was immunoprecipitated by using 4G10 antibody in the EGF-stimulated HEK-293 and HOS cells (but not in the unstimulated cells) (Fig. 6C, panel a), while the overall level of the β-catenin protein in these cells was independent of EGF stimulation (Fig. 6C panel b). Conversely, when the β-catenin antibody immunoprecipitated complexes were probed with 4G10 antibodies, a significantly more intense signal was detected in the HOS cells stimulated with EGF (Fig. 6C panel c) although a basal level of tyrosine phosphorylation of β-catenin was also present. Control Western blotting using β-catenin antibody indicated that β-catenin protein was efficiently immunoprecipitated and an equivalent amount of β-catenin protein was used in 4G10 Western blotting analysis (Fig. 6C panel d). Thus, these results demonstrate that EGF can effectively activate TCF4/β-catenin activityin, which may provide a molecular basis for the inhibitory effect of tyrosine kinase inhibitors on Wnt/β-catenin signal activity.
We further analyzed if these inhibitors exerted any effect on Wnt-induced expression of β-catenin target genes, such as c-Myc and cyclin D1. As expected, c-Myc and to lesser extent cyclin D1 were up-regulated in Wnt3A-stimulated HEK-293 cells (Fig. 6D). However, the Wnt3A-upregulated c-Myc expression was effectively inhibited by three inhibitors PD98059, genistein, and wortmannin (p<0.001) although only genistein was shown to significantly inhibit cyclin D1 expression (p<0.05) (Fig. 6D). These results are consistent with the fact these small molecule inhibitors can suppress Wnt/β-catenin signaling activity.
Multiple Cellular Pathways May Participate in Regulating Wnt/β-Catenin Signaling
Here we assess a panel of chemical activators and inhibitors for their abilities to modulate β-catenin-regulated transcription. We found that protein kinase C may regulate β-catenin activity synergistically in the presence of Wnt signal. CRT activity was significantly repressed by inhibitors of casein kinase II and MEK, consistent with recent studies of their possible involvement in Wnt signaling. On the contrary, an inhibitor of Ca++/calmodulin kinase II did not exhibit any significant effect on β-catenin signaling. More interestingly, we demonstrated that β-catenin signaling was dramatically inhibited by suramin sodium, genistein and wortmannin, suggesting that other important cellular signaling pathways, such as G protein signaling, tyrosine kinases and PI3 kinase, may play an important role in regulating β-catenin signaling (Fig. 6E). Although these observations are interesting, further studies employing molecular and cell biology approaches are needed to elucidate their molecular mechanisms of regulating β-catenin signaling.
Wnt signal plays a fundamental role in several developmental processes [53]. However, the following concerns raise questions about its actual role in human tumorigenesis. First, although the Wnt was initially discovered as a proto-oncogene [54], current models regarding the role of Wnt signaling in tumorigenesis are, for the most part, based upon findings of β-catenin deregulation in human tumors [5-7, 20]. Genetic alterations (such as gene amplifications, translocations and gain-of-function mutations) of the genes encoding either Wnt ligands or their receptors have yet to be uncovered. Second, although well established as a tumor suppressor and a negative regulator of β-catenin activity in mammalian cells, the APC tumor suppressor (and its homologues) almost exhibits the opposite function in other organisms. Overexpression of human or Xenopus APC in ventral cells of Xenopus embyros induces a secondary axis [55], mimicking activation of β-catenin. Inactivation of the only APC-related gene (APR-1) in C. elegans gives rise to a loss-of-function phenotype that mimics β-catenin loss in this organism [56]. Further, inactivation of a Drosophila homologue dAPC results in a β-catenin/TCF-mediated loss of photoreceptor cells by apoptosis [57], suggesting that although some components of the Wnt/Wingless pathway are structurally conserved, the regulation and biological functions of this pathway may vary drastically in different organisms. Consistent with this notion are the findings that suggest an increasing complexity in regulating the Wnt signal and β-catenin signaling. For instance, the serine/threonine phosphatase PP2A has been shown to associate with and dephosphorylate Axin [58], suggesting that PP2A may regulate β-catenin stability in response to Wnt signal. Casein kinases Iε and II have been shown to phosphorylate the dishevelled [40, 41, 59], suggesting that the casein kinases may be involved in the disheveled-mediated inhibition of β-catenin degradation.
The essence of Wnt signal is to stabilize the β-catenin protein by down-regulating GSK3β activity. This may be critical for activation of β-catenin signaling during tumorigenesis because oncogenic mutations in β-catenin gene that result in a stabilized protein are necessary and sufficient for tumor development [18]. Thus, it is conceivable that any signals capable of antagonizing the β-catenin destruction signal could lead to activation of β-catenin signaling pathway. Our findings suggest that other signaling pathways may also play an important role in modulating β-catenin activity in the presence or absence of Wnt signals (Fig. 6E). Moreover, an increasing number of reports have indicated that β-catenin signaling can be activated by Wnt-independent signals in mammalian cells, including G-protein [60], integrin-linked kinase (ILK) [61], hepatocyte growth factor (HGF) [62, 63], growth factor Gas6 [64], insulin-like growth factor-1 (IGF-1) [65] and vascular endothelial growth factor (VEGF) [66]. It has also become evident that there are cross-talks between the Wnt signaling and other pathways, including MAP kinase [44, 46, 67], Ca++/calmodulin kinase II [42], TGFβ [68], PKC [39, 69] and retinoid signaling [70].
Taken together, our results and the collective data from other studies suggest that the regulation of β-catenin signaling may not just involve a simple linear pathway, rather is more complex in mammalian cells. Thus, future investigations should be devoted to the elucidation of molecular mechanisms through which β-catenin signaling is activated during human tumorigenesis. Ultimately, efficacious treatment of human cancer can be developed by targeting the β-catenin signaling pathway.
Acknowledgements
This work was supported in part by research grants from the American Cancer Society (HHL and TCH), the National Institutes of Health (HHL, RCH, and TCH), and the 973 Program of Ministry of Science and Technology of China (#2011CB707900 to TCH and LZ).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Nusse R, Fuerer C, Ching W, Harnish K, Logan C, Zeng A. et al. Wnt signaling and stem cell control. Cold Spring Harb Symp Quant Biol. 2008;73:59-66
2. Miller JR. The Wnts. Genome Biol. 2002 3
3. Gordon MD, Nusse R. Wnt signaling: multiple pathways, multiple receptors, and multiple transcription factors. J Biol Chem. 2006;281:22429-33
4. Clevers H. Wnt/beta-catenin signaling in development and disease. Cell. 2006;127:469-80
5. Luo J, Chen J, Deng ZL, Luo X, Song WX, Sharff KA. et al. Wnt signaling and human diseases: what are the therapeutic implications? Lab Invest. 2007;87:97-103
6. Anastas JN, Moon RT. WNT signalling pathways as therapeutic targets in cancer. Nature reviews. 2013;13:11-26
7. Clevers H, Nusse R. Wnt/beta-catenin signaling and disease. Cell. 2012;149:1192-205
8. Kinzler KW, Nilbert MC, Su LK, Vogelstein B, Bryan TM, Levy DB. et al. Identification of FAP locus genes from chromosome 5q21. Science. 1991;253:661-5
9. Groden J, Thliveris A, Samowitz W, Carlson M, Gelbert L, Albertsen H. et al. Identification and characterization of the familial adenomatous polyposis coli gene. Cell. 1991;66:589-600
10. He TC, Chan TA, Vogelstein B, Kinzler KW. PPARdelta is an APC-regulated target of nonsteroidal anti-inflammatory drugs. Cell. 1999;99:335-45
11. He TC, Sparks AB, Rago C, Hermeking H, Zawel L, da Costa LT. et al. Identification of c-MYC as a target of the APC pathway [see comments]. Science. 1998;281:1509-12
12. Tetsu O, McCormick F. Beta-catenin regulates expression of cyclin D1 in colon carcinoma cells. Nature. 1999;398:422-6
13. Shtutman M, Zhurinsky J, Simcha I, Albanese C, D'Amico M, Pestell R. et al. The cyclin D1 gene is a target of the beta-catenin/LEF-1 pathway. Proc Natl Acad Sci U S A. 1999;96:5522-7
14. Wagner ER, Zhu G, Zhang BQ, Luo Q, Shi Q, Huang E. et al. The therapeutic potential of the Wnt signaling pathway in bone disorders. Curr Mol Pharmacol. 2011;4:14-25
15. Kinzler KW, Vogelstein B. Lessons from hereditary colorectal cancer. Cell. 1996;87:159-70
16. Rubinfeld B, Souza B, Albert I, Muller O, Chamberlain SH, Masiarz FR. et al. Association of the APC gene product with beta-catenin. Science. 1993;262:1731-4
17. Su LK, Vogelstein B, Kinzler KW. Association of the APC tumor suppressor protein with catenins. Science. 1993;262:1734-7
18. Morin PJ. beta-catenin signaling and cancer. Bioessays. 1999;21:1021-30
19. Kim JH, Liu X, Wang J, Chen X, Zhang H, Kim SH. et al. Wnt signaling in bone formation and its therapeutic potential for bone diseases. Therapeutic advances in musculoskeletal disease. 2013;5:13-31
20. Luu HH, Zhang R, Haydon RC, Rayburn E, Kang Q, Si W. et al. Wnt/beta-catenin signaling pathway as a novel cancer drug target. Curr Cancer Drug Targets. 2004;4:653-71
21. Morin PJ, Sparks AB, Korinek V, Barker N, Clevers H, Vogelstein B. et al. Activation of beta-catenin-Tcf signaling in colon cancer by mutations in beta-catenin or APC [see comments]. Science. 1997;275:1787-90
22. Tang N, Song WX, Luo J, Luo X, Chen J, Sharff KA. et al. BMP9-induced osteogenic differentiation of mesenchymal progenitors requires functional canonical Wnt/beta-catenin signaling. J Cell Mol Med. 2009;13:2448-64
23. Luo Q, Kang Q, Si W, Jiang W, Park JK, Peng Y. et al. Connective Tissue Growth Factor (CTGF) Is Regulated by Wnt and Bone Morphogenetic Proteins Signaling in Osteoblast Differentiation of Mesenchymal Stem Cells. J Biol Chem. 2004;279:55958-68
24. Luo X, Chen J, Song WX, Tang N, Luo J, Deng ZL. et al. Osteogenic BMPs promote tumor growth of human osteosarcomas that harbor differentiation defects. Lab Invest. 2008;88:1264-77
25. Huang E, Bi Y, Jiang W, Luo X, Yang K, Gao JL. et al. Conditionally Immortalized Mouse Embryonic Fibroblasts Retain Proliferative Activity without Compromising Multipotent Differentiation Potential. PloS one. 2012;7:e32428
26. Huang E, Zhu G, Jiang W, Yang K, Gao Y, Luo Q. et al. Growth hormone synergizes with BMP9 in osteogenic differentiation by activating the JAK/STAT/IGF1 pathway in murine multilineage cells. J Bone Miner Res. 2012;27:1566-75
27. Luther GA, Lamplot J, Chen X, Rames R, Wagner ER, Liu X. et al. IGFBP5 Domains Exert Distinct Inhibitory Effects on the Tumorigenicity and Metastasis of Human Osteosarcoma. Cancer Lett. 2013;336:222-30
28. Su Y, Wagner ER, Luo Q, Huang J, Chen L, He BC. et al. Insulin-like growth factor binding protein 5 suppresses tumor growth and metastasis of human osteosarcoma. Oncogene. 2011;30:3907-17
29. Kang Q, Song WX, Luo Q, Tang N, Luo J, Luo X. et al. A comprehensive analysis of the dual roles of BMPs in regulating adipogenic and osteogenic differentiation of mesenchymal progenitor cells. Stem Cells Dev. 2009;18:545-59
30. Chen L, Jiang W, Huang J, He BC, Zuo GW, Zhang W. et al. Insulin-like growth factor 2 (IGF-2) potentiates BMP-9-induced osteogenic differentiation and bone formation. J Bone Miner Res. 2010;25:2447-59
31. Hu N, Jiang D, Huang E, Liu X, Li R, Liang X. et al. BMP9-regulated angiogenic signaling plays an important role in the osteogenic differentiation of mesenchymal progenitor cells. J Cell Sci. 2013;126:532-41
32. Yang K, Chen J, Jiang W, Huang E, Cui J, Kim SH. et al. Conditional Immortalization Establishes a Repertoire of Mouse Melanocyte Progenitors with Distinct Melanogenic Differentiation Potential. J Invest Dermatol. 2012;132:2479-83
33. Li M, Chen Y, Bi Y, Jiang W, Luo Q, He Y. et al. Establishment and characterization of the reversibly immortalized mouse fetal heart progenitors. International journal of medical sciences. 2013;10:1035-46
34. Korinek V, Barker N, Morin PJ, van Wichen D, de Weger R, Kinzler KW. et al. Constitutive transcriptional activation by a beta-catenin-Tcf complex in APC-/- colon carcinoma. Science. 1997;275:1784-7
35. Zhou L, An N, Jiang W, Haydon R, Cheng H, Zhou Q. et al. Fluorescence-based functional assay for Wnt/beta-catenin signaling activity. Biotechniques. 2002;33:1126-8
36. Sokol S, Christian JL, Moon RT, Melton DA. Injected Wnt RNA induces a complete body axis in Xenopus embryos. Cell. 1991;67:741-52
37. He X, Saint-Jeannet JP, Woodgett JR, Varmus HE, Dawid IB. Glycogen synthase kinase-3 and dorsoventral patterning in Xenopus embryos [published erratum appears in Nature 1995 May 18;375(6528):253]. Nature. 1995;374:617-22
38. Pierce SB, Kimelman D. Regulation of Spemann organizer formation by the intracellular kinase Xgsk-3. Development. 1995;121:755-65
39. Chen RH, Ding WV, McCormick F. Wnt signaling to beta-catenin involves two interactive components. Glycogen synthase kinase-3beta inhibition and activation of protein kinase C. J Biol Chem. 2000;275:17894-9
40. Peters JM, McKay RM, McKay JP, Graff JM. Casein kinase I transduces Wnt signals. Nature. 1999;401:345-50
41. Sakanaka C, Leong P, Xu L, Harrison SD, Williams LT. Casein kinase iepsilon in the wnt pathway: regulation of beta-catenin function. Proc Natl Acad Sci U S A. 1999;96:12548-52
42. Kuhl M, Sheldahl LC, Malbon CC, Moon RT. Ca(2+)/calmodulin-dependent protein kinase II is stimulated by Wnt and Frizzled homologs and promotes ventral cell fates in Xenopus. J Biol Chem. 2000;275:12701-11
43. O'Dell TJ, Kandel ER, Grant SG. Long-term potentiation in the hippocampus is blocked by tyrosine kinase inhibitors. Nature. 1991;353:558-60
44. Zhang Y, Neo SY, Wang X, Han J, Lin SC. Axin forms a complex with MEKK1 and activates c-Jun NH(2)-terminal kinase/stress-activated protein kinase through domains distinct from Wnt signaling. J Biol Chem. 1999;274:35247-54
45. Boutros M, Paricio N, Strutt DI, Mlodzik M. Dishevelled activates JNK and discriminates between JNK pathways in planar polarity and wingless signaling. Cell. 1998;94:109-18
46. Li L, Yuan H, Xie W, Mao J, Caruso AM, McMahon A. et al. Dishevelled proteins lead to two signaling pathways. Regulation of LEF- 1 and c-Jun N-terminal kinase in mammalian cells. J Biol Chem. 1999;274:129-34
47. Behrens J. Cross-regulation of the Wnt signalling pathway: a role of MAP kinases. J Cell Sci. 2000;113:911-9
48. Fruman DA, Meyers RE, Cantley LC. Phosphoinositide kinases. Annu Rev Biochem. 1998;67:481-507
49. Stein CS, Martins I, Davidson BL. Long-term reversal of hypercholesterolemia in low density lipoprotein receptor (LDLR)-deficient mice by adenovirus-mediated LDLR gene transfer combined with CD154 blockade. J Gene Med. 2000;2:41-51
50. Stokoe D, Stephens LR, Copeland T, Gaffney PR, Reese CB, Painter GF. et al. Dual role of phosphatidylinositol-3,4,5-trisphosphate in the activation of protein kinase B [see comments]. Science. 1997;277:567-70
51. Delcommenne M, Tan C, Gray V, Rue L, Woodgett J, Dedhar S. Phosphoinositide-3-OH kinase-dependent regulation of glycogen synthase kinase 3 and protein kinase B/AKT by the integrin-linked kinase. Proc Natl Acad Sci U S A. 1998;95:11211-6
52. Espada J, Perez-Moreno M, Braga VM, Rodriguez-Viciana P, Cano A. H-Ras activation promotes cytoplasmic accumulation and phosphoinositide 3-OH kinase association of beta-catenin in epidermal keratinocytes. J Cell Biol. 1999;146:967-80
53. Wodarz A, Nusse R. Mechanisms of Wnt signaling in development. Annu Rev Cell Dev Biol. 1998;14:59-88
54. Nusse R, Varmus HE. Many tumors induced by the mouse mammary tumor virus contain a provirus integrated in the same region of the host genome. Cell. 1982;31:99-109
55. Vleminckx K, Wong E, Guger K, Rubinfeld B, Polakis P, Gumbiner BM. Adenomatous polyposis coli tumor suppressor protein has signaling activity in Xenopus laevis embryos resulting in the induction of an ectopic dorsoanterior axis. J Cell Biol. 1997;136:411-20
56. Rocheleau CE, Downs WD, Lin R, Wittmann C, Bei Y, Cha YH. et al. Wnt signaling and an APC-related gene specify endoderm in early C. elegans embryos [see comments]. Cell. 1997;90:707-16
57. Ahmed Y, Hayashi S, Levine A, Wieschaus E. Regulation of armadillo by a Drosophila APC inhibits neuronal apoptosis during retinal development. Cell. 1998;93:1171-82
58. Wang SS, Esplin ED, Li JL, Huang L, Gazdar A, Minna J. et al. Alterations of the PPP2R1B gene in human lung and colon cancer. Science. 1998;282:284-7
59. Song DH, Sussman DJ, Seldin DC. Endogenous protein kinase CK2 participates in Wnt signaling in mammary epithelial cells. J Biol Chem. 2000;275:23790-7
60. Kawasaki Y, Senda T, Ishidate T, Koyama R, Morishita T, Iwayama Y. et al. Asef, a link between the tumor suppressor APC and G-protein signaling. Science. 2000;289:1194-7
61. Novak A, Hsu SC, Leung-Hagesteijn C, Radeva G, Papkoff J, Montesano R. et al. Cell adhesion and the integrin-linked kinase regulate the LEF-1 and beta-catenin signaling pathways. Proc Natl Acad Sci U S A. 1998;95:4374-9
62. Papkoff J, Aikawa M. WNT-1 and HGF regulate GSK3 beta activity and beta-catenin signaling in mammary epithelial cells. Biochem Biophys Res Commun. 1998;247:851-8
63. Hiscox S, Jiang WG. Association of the HGF/SF receptor, c-met, with the cell-surface adhesion molecule, E-cadherin, and catenins in human tumor cells. Biochem Biophys Res Commun. 1999;261:406-11
64. Goruppi S, Chiaruttini C, Ruaro ME, Varnum B, Schneider C. Gas6 Induces Growth, beta-Catenin Stabilization, and T-Cell Factor Transcriptional Activation in Contact-Inhibited C57 Mammary Cells. Mol Cell Biol. 2001;21:902-15
65. Playford MP, Bicknell D, Bodmer WF, Macaulay VM. Insulin-like growth factor 1 regulates the location, stability, and transcriptional activity of beta -catenin [In Process Citation]. Proc Natl Acad Sci U S A. 2000;97:12103-8
66. Carmeliet P, Lampugnani MG, Moons L, Breviario F, Compernolle V, Bono F. et al. Targeted deficiency or cytosolic truncation of the VE-cadherin gene in mice impairs VEGF-mediated endothelial survival and angiogenesis. Cell. 1999;98:147-57
67. Ishitani T, Ninomiya-Tsuji J, Nagai S, Nishita M, Meneghini M, Barker N. et al. The TAK1-NLK-MAPK-related pathway antagonizes signalling between beta- catenin and transcription factor TCF. Nature. 1999;399:798-802
68. Nishita M, Hashimoto MK, Ogata S, Laurent MN, Ueno N, Shibuya H. et al. Interaction between Wnt and TGF-beta signalling pathways during formation of Spemann's organizer. Nature. 2000;403:781-5
69. Sheldahl LC, Park M, Malbon CC, Moon RT. Protein kinase C is differentially stimulated by Wnt and Frizzled homologs in a G-protein-dependent manner. Curr Biol. 1999;9:695-8
70. Easwaran V, Pishvaian M, Salimuddin, Byers S. Cross-regulation of beta-catenin-LEF/TCF and retinoid signaling pathways. Curr Biol. 1999;9:1415-8
Author contact
![]() Corresponding author: T.-C. He, MD, PhD, Molecular Oncology Laboratory, The University of Chicago Medical Center, 5841 South Maryland Avenue, MC 3079, Chicago, IL 60637, USA. Tel. (773) 702-7169; Fax (773) 834-4598; E-mail: tcheedu
Corresponding author: T.-C. He, MD, PhD, Molecular Oncology Laboratory, The University of Chicago Medical Center, 5841 South Maryland Avenue, MC 3079, Chicago, IL 60637, USA. Tel. (773) 702-7169; Fax (773) 834-4598; E-mail: tcheedu