Impact Factor ISSN: 1449-1907

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2013; 10(10):1301-1306. doi:10.7150/ijms.6607 This issue Cite
Research Paper
Lentivirus-based RNA Silencing of Nemo-like Kinase (NLK) Inhibits the CAL 27 Human Adenosquamos Carcinoma Cells Proliferation and Blocks G0/G1 Phase to S Phase
Bin Zhang1,2, Ke Yi Li2, Hai Ying Chen3, Shao Dong Pan3, Shuang Feng Chen3, Wei Feng Zhang2, Chun Peng Xia2, Li Cheng Jiang2, Xian Bin Liu2, Feng Jun Zhao3, Dao Ying Yuan2, Le Xin Wang2,4, Ya Ping Wu3,5 ![]() , Shu Wei Liu1
, Shu Wei Liu1 ![]()
1. Department of Anatomy Shandong University, School of Medicine, Jinan Shandong, 250012, P. R. China.
2. Department of Oral and Maxillofacial Surgery, Liaocheng People's Hospital, Liaocheng Shandong, 252000, P. R. China
3. Key Laboratory of Oral and Maxillofacial-Head and Neck Medicine, Central Laboratory, Liaocheng People's Hospital and Liaocheng Clinical School, Liaocheng Shandong, 252000, P. R. China.
4. School of Biomedical Sciences, Charles Sturt University, Wagga Wagga, NSW 2650, Australia
5. Department of Clinical Chemistry and Haematology, University Medical Centre Utrecht, G03.550, Heidelberglaan 100, 3584 CX Utrecht, The Netherlands
Received 2013-5-3; Accepted 2013-7-29; Published 2013-8-8
Abstract
Background: The Nemo-like kinase (NLK) is a serine/threonine-protein kinase that involved in a number of signaling pathways regulating cell fate. Variation of NLK has been shown to be associated with the risk of cancer. However, the function of NLK in oral adenosquamous carcinoma cells line CAL-27 is unknown.
Methods: In this study, we evaluated the function of NLK in CAL-27 cells by using lentivirus-mediated RNA silence. The targeted gene expression, cell proliferation and cell cycle are investigated by RT-PCR, western-blot, MTT method, colony forming assay and flow cytometry analysis respectively.
Results: After NLK silencing, the number of colonies was significantly reduced (54±5 colonies/well compared with 262±18 colonies/well in non-infected or 226±4 colonies/well in negative control group (sequence not related to NLK sequence with mismatched bases). Using crystal violet staining, we also found that the cell number per colony was dramatically reduced. The RNA silencing of NLK blocks the G0/G1 phase to S phase progression during the cell cycle.
Conclusions: These results suggest that NLK silencing by lentivirus-mediated RNA interference would be a potential therapeutic method to control oral squamous carcinoma growth.
Keywords: Nemo-like kinase (NLK), Lentivirus, RNAi, Oral Squamous Cell Carcinoma
Introduction
Oral squamous cell carcinoma is one of the most common malignancies worldwide. This tumor can be difficult to manage by surgery excision or chemotherapy (1). However, the gene therapy methods, which have been approved for evaluation in clinical trials, might be potentially used in the future (2, 3). Recent studies have revealed that many molecules are deeply involved in the oral squamous cell carcinoma formation and metastasis (4-7), which provide the genetic therapeutic targets for treatment of this kind of cancer.
Nemo-like kinase (NLK) is an evolutionarily conserved serine/threonine protein kinase (8-11). Evidence shows that NLK is involved in a number of important signaling pathways and regulates many transcription factors important for cell fate. NLK is able to play a role in multiple processes due to its capacity to regulate a diverse array of signaling pathways, including the Wnt/β-catenin, Activin, IL-6, adenomatous polyposis coli gene product (APC) and Notch signaling pathways (12).
Prevalence survey of ovarian cancer suggests that variation of NLK is associated with risk of ovarian cancer (13). The expression level of NLK is aberrantly upregulated in hepatocellular carcinoma (14) while downregulated in the prostate cancer cells (15). In human colon cancer cells and prostate carcinoma cells, overexpression of NLK could induce apoptosis and inhibit their proliferation (15, 16). While in the hepatocellular carcinoma cells and gallbladder carcinoma cells, targeted disruption of NLK could prevent the proliferation of carcinoma cells (14, 17). In the human oral squamous carcinoma cells, the functional role of NLK is largely unknown. We only know that WNT- 1 pathway could be important in our understanding of the mechanisms of epithelial tumors, in general, and probably also of oral squamous cell carcinoma, in particular (18, 19).
In this study, targeted disruption of NLK was performed using lentivirus-mediated siRNA method (2,3). The effect of siRNA method on the proliferation and colony formation ability of CAL-27, an oral squamous cell carcinoma cells, was evaluated. Furthermore, the effect of NLK silencing on CAL-27 cell cycles was also studied.
Methods
Vector construction
The target sequence for human NLK (NM_016231) was 5'-GATAGACCTATTGGATATG-3'.
The negative control sequence was 5'-TTCTCCGAACGTGTCACGT-3'.
The NLK- short hairpin RNA (shRNA) template DNA sequence (5'-GATAGACCTATTGGATATG-3') target NLK (GenBank accession no. NM_016231.4) containing small hairpin structure was designed and synthesized. Oligonucleotides were cloned into the vector pFH-L (Shanghai Hollybio, China), and named as Lentivirus/shRNA-NLK (LV-shNLK). Sequence not related to NLK sequence with mismatched bases was designed (5'-TTCTCCGAACGTGTCACGT-3'), used as negative contro1 and called Letivirus/shRNA-Control (LV-shCon). The shRNA was confirmed by DNA sequencing.
Packaging and target cell lines
HEK293T and CAL-27 cells were purchased from the Cell Bank of Chinese Academy of Science (Shanghai, China). Cells were cultured in DMEM (Hyclone) supplemented with 10% (vol/vol) fetal calf serum (Hyclone) in the 5% CO2 incubator at 37oC. HEK293T cells were used to produce replication-incompetent lentiviral particles. CAL-27 cells were used as target for in vitro infection experiments. The cells were divided into three groups: control group (untransfected group), LV-shCon group (negative control group) and LV-shNLK group (experimental group).
Generation of lentiviral particles and target cell infection
Lentiviral particles were produced by cotransfecting 5×106 HEK293T cells in a 10 cm diameter tissue culture dishes with a combination of LV-shNLK (or LV-shCon ) vector, pVSVG-I, pRSV-Rev and pCMV delta R8.92 plasmids. Plasmid DNA transfection is carried out using lipofectamine TM 2000 (Invitrogen, Carlsbad, CA), according to the manufacturer's instructions. At 48 and 72 hours post transfection, the cell culture media is collected and filtered, through a 0.45-micron cellulose acetate filter. The viral vector supernatant is then concentrated by ultracentrifugation, and viral vector pellet is resuspended in a minimum volume of PBS containing 1% BSA. Before lentivirus was used for subsequent experiments, virus titer was determined and calibrated. For target cell infection, CAL 27 cells were grown to 70-80% confluence and infected with Lentivirus/shRNA-NLK or Letivirus/shRNA-Control at MOI of 10 in the presence of 6ug/ml polybrene (Sigma). The infection efficiency was observed by measuring the cells expressing GFP protein using fluorescence microscopy (IX71, Olympus) after 72 hours infection.
RNA extract and real-time reverse transcription-polymerase chain reaction (RT-PCR)
After CAL-27 cells were infected after,5 days, cells were harvested and total RNA was extracted by Trizol reagent (Invitrogen). Reverse transcript was performed using M-MLV reverse transcriptase (Promega). The expression level of NLK was measured by real-time polymerase chain reaction with SYBR master mixture (Takara) using a sequence detector (7300 Sequence Detection System, Applied Biosystems). Primers used to measure the expression level of NLK are as follows:
5'-ATCATCAGCACTCGCATCATC-3' (forward) and 5'-GACCAGACAACACCAAAGGC-3' (reverse).
The thermal cycling conditions for the real-time PCR was 95 ℃ for 30 s followed by 40 cycles of 95 ℃ for 5s, 60 ℃ for 30 s. Data were collected during the 60 ℃. Relative expression levels for each primer set were normalized to the expression of GAPDH by the 2-△CT method.
Western blot
After infecting with lentivirus, CAL-27 cells were collected and lysed with RIPA buffer (20 mM Tris-HCl (pH 7.5), 150 mM NaCl, 1 mM Na2EDTA, 1 mM EGTA, 1% NP-40, 1% sodium deoxycholate, 2.5 mM sodium pyrophosphate, 1 mM b-glycerophosphate, 1 mM Na3VO4, 1 µg/ml leupeptin) with PMSF (Beyotime Biotechnology, Jiangsu, China). Protein samples were then separated by SDS-PAGE and transferred to the PVDF membranes. After probing with primary antibodies overnight at 4℃ and secondary antibodies for 1 hour at room temperature, samples were visualized with LAS3000® Luminescent image analyzer. (Fuji Photo Film Co., Ltd. Minato-ku, Tokyo 106-8620, Japan) Primary antibodies used are as follows: NLK (1:500; Sigma-Aldrich, HPA018192), GAPDH (1:3000; Beyotime Inc, China, AG019).
MTT Cell proliferation assay
Lentivirus infected CAL-27 cells were suspended and seeded in 96-well plate at an initial concentration of 2000 cells/well. After 20 µl 5 mg/ml 3-(4,5-Dimethylthiazol-2-yl)-2,5-Diphenyltetrazolium Bromide (MTT) was added into each well for 3 hours, acidified isopropanol (in 0.01 M HCl) was added into each well. Then the formazan precipitate was dissolved and plates were then read on the spectrophotometer at a wavelength of 595 nm.
Cell colony formation assay
Lentivirus infected CAL-27 cells were suspended and added in 6-well plate at an initial concentration of 200 cells/well. Cells were cultured in the 5% CO2 incubator at the 37℃ until the most single colony contains more than 50 cells. After samples were fixed and crystal violet staining was performed according to the instruction. The images were then captured. All studies were performed at least three times.
Cell cycle analysis
In brief, lentivirus infected CAL-27 cells were collected, re-suspended in cold PBS and fixed with pre-cold 75% ethanol for 30 minutes at 4°C. After the staining using Propidium iodide (PI), samples were then analyzed by flow cytometry (Cell Lab Quanta, Beckman Coulter) and the data were analyzed using Multi-Cycle AV software (Phoenix Flow Systems, San Diego, CA).
Statistical analysis
PASW Statistics 18, Prism 4 for windows v 4.00 and InStat 3.00 software were used for data analysis. Numerical data were analyzed by one-way ANOVA. Data are represented as mean and standard error. Turkey's test was used for multiple comparisons between four different groups and two-difference time course. The statistical significance was accepted as p < 0.05.
Results
Knockdown endogenous NLK expression in CAL-27 cells by lentivirus-based RNA silencing
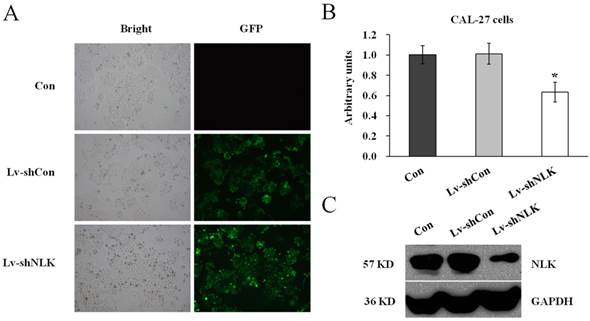
To investigate the function of NLK in oral squamous carcinoma cells, experiment to knockdown the endogenous NLK was performed. Specific shRNA against NLK was designed and cloned into the lentivirus vector (Lv-shNLK). Lentivirus expressing control shRNA (Lv-shCon) was used as negative control. As shown in Figure 1A, the infection rate was more than 90% as assessed by GFP fluorescence. RT-PCR and western blot were then performed to measure the expression level of NLK. It shows that after Lv-shNLK infection, the mRNA level was reduced about 40% compared with the non-infected or Lv-shCon infected group (Figure 1B). The endogenous protein level was also dramatically reduced after Lv-shNLK infection (Figure 1C). These results suggest that the antivirus mediated shRNA transaction could significantly suppress the expression of endogenous NLK.
Verification of NLK silencing after Lv-shNLK infecting CAL-27 cells. (A) CAL-27 cells infected with Lv-shNLK and Lv-shCon. GFP fluorescence indicated the shRNA delivery efficiency. (B) mRNA level of NLK was significantly decreased after Lv-shNLK infection*, P<0.01 versus control or Lv-shCon. (C) NLK protein level in non-infected and lentivirus infected cells as measured by Western blot. GAPDH was used as the loading control. Three independent experiments are performed.
Targeted disruption of NLK could significantly reduce the proliferation and colony formation ability of CAL-27 cells
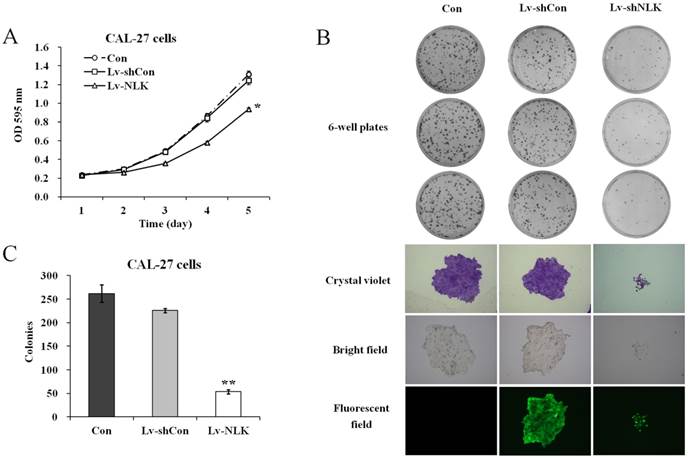
To investigate the effects of NLK knockdown in oral squamous carcinoma cells, MTT and colony formation assay were carried out after lentivirus infection. As shown in Figure 2A, Lv-shNLK infection could significantly suppress the proliferation ability of CAL-27 cells compared with non-infected or Lv-shCon infected group. Tumorigenesis of CAL-27 was then evaluated by colony formation assay. It shows that after NLK silencing by Lv-shNLK infection, the number of colonies was significantly reduced (54±5 colonies/well compared with 262±18 colonies/well in non-infected or 226±4 colonies/well in Lv-shCon infected group) (Figure 2B & 2C). Using crystal violet staining, we also found that the cell number per colony was dramatically reduced (Figure 2B). These evidences implicate that target disruption of NLK in oral adenosquamous carcinoma cells could significantly inhibit their ability of proliferation and tumorigenesis.
Effects of NLK knockdown on cell proliferation and colony formation. (A) Cell proliferation as measured by MTT assay. Lv-shNLK infection significantly suppress the proliferation ability of CAL-27 cells* compared control, P<0.01 or LV-shCon at day 5. (B) Colony formation ability as examined by the colony formation assay (upper panels). Cells in single clone are shown after crystal violet staining (lower panels). (C) Statistic results of the colony formation ability as shown. After NLK silencing by Lv-shNLK infection, the number of colonies was significantly reduced**, P<0.01 versus control and LV-shCon. Three independent experiments are performed.
NLK silencing induces G0/G1 cell cycle arrest in CAL-27 cells
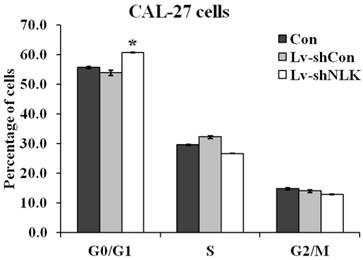
To investigate the mechanisms of NLK silencing induced proliferation and tumorigenesis inhibition, flow cytometry was carried out to identify the specific phases of the cell cycle. As shown in Figure 3, 60.67 ± 0.25% cells were at G0/G1 phase of cell cycle in the Lv-shNLK infected group, which is significantly higher than that of non-infected group (55.63±0.40%) and Lv-shCon infected group (53.80 ± 0.89%).
Statistic results of flow cytometry analysis of non-infected and Lv-shNLK or Lv-shCon infected CAL-27 cells. Cell cycle distributions as measured by flow cytometry. NLK silencing induces G0/G1 cell cycle arrest in CAL-27 cells. The cells at G0/G1 phase of cell cycle in the Lv-shNLK infected group is significantly higher than that of control group*, P<0.05 versus control or P<0.01 versus LV-shCon.
Discussion
The main cause of death in oral squamous cell carcinomas is metastasis (5). Intercellular adhesion is mediated by a family of glycoproteins called cadherins and other molecules like catenins and APC (adenomatous polyposis coli gene product). Other recent reports have revealed that many molecules are involved in the oral squamous cell carcinoma formation and metastasis (5, 6). It has been shown that the Wnt signaling pathway and adenomatous polyposis coli gene product (APC) is involved in the oral squamous cell carcinoma, as a strong neurotropic malignancy (5, 18). Compare our experiments with these reports, the novelty and importance of our results is NLK was found to be involved in the oral squamous cell carcinoma. We know that, NLK is able to play a role to regulate a diverse array of signaling pathways, including the Wnt/β-catenin, Activin, IL-6, adenomatous polyposis coli gene product (APC) and Notch signaling pathways (12). It is interested to find the link between NLK and Wnt in oral squamous cell carcinoma. Further examination is necessary. The Wnt signal stabilizes beta-catenin protein and promotes its accumulation in the cytoplasm and nucleus. In the nucleus, beta-catenin associates with T-cell factor (TCF) to form a functional transcription factor, which mediates the transactivation of target genes involved in the promotion of tumor progression, invasion, and metastasis, such as C-Myc, cyclin Dl, c-jun, fra-l, and u-PAR. There is a strong correlation between the ability of the Wnt gene to induce beta-catenin accumulation and its transforming potential in vivo, suggesting that the Wnt gene activates an intracellular signaling pathway that can induce the morphological transformation of cells (18) . In the presence of NLK, the complex of Wnt is able to promote the transcription of other genes involved in oral carcinoma proliferation or apoptosis inhibition.
Targeted silencing of the endogenous NLK in CAL-27 cells could suppress the ability of proliferation and tumorigenesis, implicated that NLK might be a therapeutic target for the genetic treatment of oral squamous cell carcinoma. NLK, an evolutionarily conserved MAP kinase-like kinase, could regulate a number of signaling pathways important for cell fate determination via its kinase activity. More and more evidences support that NLK is involved in different forms of cancer. However, the expression level and nucleofection experiments targeting NLK in different carcinoma cells are controversial. In the prostate cancer cells, the expression of NLK is downregulated (15). Overexpression NLK in this kind of carcinoma could suppress cell growth and promote apoptosis in colon and prostate carcinoma cells (15). However, in the hepatocellular carcinoma cells, NLK is found to be aberrantly upregulated and targeted disruption of NLK could inhibit the proliferation of this kind of carcinoma (14). These data suggest that proper level of NLK in the cell is very important. Aberrantly increase or decrease the expression of NLK might lead to the formation of cancer. Consistent with the results from hepatocellular and gallbladder carcinoma cells (14, 17), we found that NLK silencing in oral squamous carcinoma cells, could reduce their ability of proliferation and colony formation, implicating that NLK might be abnormally upregulated in the oral squamous carcinoma cells.
Moreover, to investigate the mechanisms of NLK silencing induced cell proliferation inhibition, we evaluated the specific phases of cell cycle after lentivirus infection. We found that NLK silencing could block the progression from the G0/G1 phase to the S phase during the cell cycle. These results suggest that signaling molecules involved in the cell cycle regulation might be modulated by NLK in oral squamous carcinoma cells.
To conclude, we found that endogenous NLK regulated oral adenosquamous carcinoma cells proliferation and colony formation. These results suggest that lentivirus-based NLK silencing is a promising gene-therapeutic method for the treatment of oral squamous cell carcinoma.
Acknowledgements
The National Natural Science Foundation of China funded project is acknowledged for the financial support for this work (81272958).
Abbreviations
NLK: Nemo-like kinase; MTT: 3-(4,5-Dimethylthiazol-2-yl)-2,5-Diphenyltetrazolium Bromide; T-cell factor: TCF; APC: adenomatous polyposis coli gene product; RT-PCR: real-time reverse transcription-polymerase chain reaction; siRNA: small interfering double stranded RNA; shRNA: short hairpin; LV-shNLK: Lentivirus/shRNA-NLK; LV-shCon: Letivirus/shRNA-Control.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Binmadi NO, Basile JR. Perineural invasion in oral squamous cell carcinoma: a discussion of significance and review of the literature. Oral Oncol. 2011;47:1005-10
2. D'Costa J, Mansfield SG, Humeau LM. Lentiviral vectors in clinical trials: Current status. Curr Opin Mol Ther. 2009;11:554-64
3. Sakuma T, Barry MA, Ikeda Y. Lentiviral vectors: basic to translational. Biochem J. 2012;443:603-18
4. Yan B, Fu Q, Lai L. et al. Downregulation of microRNA 99a in oral squamous cell carcinomas contributes to the growth and survival of oral cancer cells. Mol Med Report. 2012;6:675-81
5. Perez-Sayans M, Suarez-Penaranda JM, Herranz-Carnero M. et al. The role of the adenomatous polyposis coli (APC) in oral squamous cell carcinoma. Oral Oncol. 2012;48:56-60
6. Silva TA, Ribeiro FL, Oliveira-Neto HH. et al. Dual role of CCL3/CCR1 in oral squamous cell carcinoma: implications in tumor metastasis and local host defense. Oncol Rep. 2007;18:1107-13
7. Rosado P, Lequerica-Fernandez P, Pena I. et al. In oral squamous cell carcinoma, high FAK expression is correlated with low P53 expression. Virchows Arch. 2012;461:163-8
8. Ishitani T, Ishitani S, Matsumoto K. et al. Nemo-like kinase is involved in NGF-induced neurite outgrowth via phosphorylating MAP1B and paxillin. J Neurochem. 2009;111:1104-18
9. Ishitani T, Hirao T, Suzuki M. et al. Nemo-like kinase suppresses Notch signalling by interfering with formation of the Notch active transcriptional complex. Nat Cell Biol. 2010;12:278-85
10. Kanei-Ishii C, Ninomiya-Tsuji J. et al. Wnt-1 signal induces phosphorylation and degradation of c-Myb protein via TAK1, HIPK2, and NLK. Genes Dev. 2004;18:816-29
11. Kojima H, Sasaki T, Ishitani T. et al. STAT3 regulates Nemo-like kinase by mediating its interaction with IL-6-stimulated TGFbeta-activated kinase 1 for STAT3 Ser-727 phosphorylation. Proc Natl Acad Sci U S A. 2005;102:4524-9
12. Ishitani T, Ishitani S. Nemo-like kinase, a multifaceted cell signaling regulator. Cell Signal. 2013;25:190-7
13. Stevens KN, Kelemen LE, Wang X. et al. Common variation in Nemo-like kinase is associated with risk of ovarian cancer. Cancer Epidemiol Biomarkers Prev. 2012;21:523-8
14. Jung KH, Kim JK, Noh JH. et al. Targeted disruption of Nemo-like kinase inhibits tumor cell growth by simultaneous suppression of cyclin D1 and CDK2 in human hepatocellular carcinoma. J Cell Biochem. 2010;110:687-96
15. Emami KH, Brown LG, Pitts TE. et al. Nemo-like kinase induces apoptosis and inhibits androgen receptor signaling in prostate cancer cells. Prostate. 2009;69:1481-92
16. Yasuda J, Tsuchiya A, Yamada T. et al. Nemo-like kinase induces apoptosis in DLD-1 human colon cancer cells. Biochem Biophys Res Commun. 2003;308:227-33
17. Tan Z, Li M, Wu W, Zhang L. et al. NLK is a key regulator of proliferation and migration in gallbladder carcinoma cells. Mol Cell Biochem. 2012;369:27-33
18. Lo Muzio L. A possible role for the WNT-1 pathway in oral carcinogenesis. Crit Rev Oral Biol Med. 2001;12:152-65
19. Andrade Filho PA, Letra A, Cramer A. et al. Insights from studies with oral cleft genes suggest associations between WNT-pathway genes and risk of oral cancer. J Dent Res. 2011;90:740-6
Author contact
![]() Corresponding authors: Prof Shu-Wei Liu, Department of Anatomy, Shandong University, School of Medicine, Jinan Shandong, 250012, P. R. China. Phone: +86-531-88382093; Fax: +86-531-88382093; and Prof. Ya Ping. Wu, Central Laboratory, Liaocheng People's Hospital and Liaocheng Clinical School, Liaocheng Shandong, 252000, P. R. China. Tel: 86-531-88382093, Fax: 86-531-88382093; and Department of Clinical Chemistry and Haematology, University Medical Centre Utrecht, G03.550, Heidelberglaan 100, 3584 CX Utrecht, The Netherlands. Tel: 31-0618861111, Fax: 31-88-7557769; E-mail: ywunl
Corresponding authors: Prof Shu-Wei Liu, Department of Anatomy, Shandong University, School of Medicine, Jinan Shandong, 250012, P. R. China. Phone: +86-531-88382093; Fax: +86-531-88382093; and Prof. Ya Ping. Wu, Central Laboratory, Liaocheng People's Hospital and Liaocheng Clinical School, Liaocheng Shandong, 252000, P. R. China. Tel: 86-531-88382093, Fax: 86-531-88382093; and Department of Clinical Chemistry and Haematology, University Medical Centre Utrecht, G03.550, Heidelberglaan 100, 3584 CX Utrecht, The Netherlands. Tel: 31-0618861111, Fax: 31-88-7557769; E-mail: ywunl