Impact Factor ISSN: 1449-1907

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2008; 5(6):295-302. doi:10.7150/ijms.5.295 This issue Cite
Research Paper
Upregulation of Bax and Bcl-2 following prenatal cocaine exposure induces apoptosis in fetal rat brain
DaLiao Xiao ![]() , Lubo Zhang
, Lubo Zhang
Center for Perinatal Biology, Department of Physiology and Pharmacology, Loma Linda University School of Medicine, Loma Linda, California 92350, USA
Received 2008-9-30; Accepted 2008-10-16; Published 2008-10-17
Abstract
Cocaine abuse during pregnancy has been associated with numerous adverse perinatal outcomes. Aims: The present study was to determine whether prenatal cocaine exposure induced apoptosis and the possible role of Bcl-2 family genes in the programming cell death in fetal rat brain. Main methods: Pregnant rats were treated with cocaine subcutaneously (30 & 60 mg/kg/day) from day 15 to 21 of gestation. Then the fetal and maternal brains were isolated. Key findings: Cocaine produced a dose-dependent decrease in fetal brain weight and brain/body weight ratio (P<0.05). Apoptotic nuclei in fetal brain were increased from 2.6 ± 0.1 (control) to 8.1± 0.6 (low dose) and 10.4 ± 0.2% (high dose) (P<0.05). In accordance, cocaine dose dependently increased activities of caspase-3, caspase-8, and caspase-9 (% of control) in the fetal brain by 177%, 155%, 174%, respectively, at 30 mg/kg/day, and by 191%, 176%, 274%, respectively, at 60 mg/kg/day. In contrast, cocaine showed no effect on caspase activities in the maternal brain. Cocaine produced a dose-dependent increase in both Bcl-2 and Bax protein expression in the fetal brain, and increased the ratio of Bax/Bcl-2 at dose of 30 mg/kg/day (P<0.05). Significance: Our study has demonstrated that prenatal cocaine exposure induces apoptosis in the fetal brain, and suggested that up-regulating Bax/Bcl-2 gene expression may be involved in cocaine-induced apoptosis. The increased apoptosis of neuronal cells in the fetal brain is likely to play a key role in cocaine-induced neuronal defects during fetal development.
Keywords: cocaine, fetus, brain, apoptosis, caspase, Bcl-2 proteins
Introduction
Cocaine abuse among women of childbearing age is prevalent in the United States. It has been estimated that each year more than 100,000 infants who were exposed prenatally to cocaine are born in this country. Cocaine abuse during pregnancy has been associated with numerous adverse perinatal outcomes [1-5]. Although teratogenic effects of cocaine on the human fetal brain, such as destructive lesions and disturbances of the neurodevelopmental program are well documented [3, 6], the underlying mechanisms remain controversial. Several previous studies have reported that cocaine induces apoptosis in fetal cardiomyocytes [7-9], endothelium [10-12], thymocytes [13], hepatocytes [14], and testes [15]. Compelling evidence has accumulated indicating that programmed cell death (apoptosis) plays an important role in neuronal development [16-18] as well as in several brain diseases including stroke, Alzheimer, Parkinson, and Huntington diseases [19-21]. A previous study demonstrated that cocaine induced apoptosis in cultured cortical neuronal cells of fetal mice [22]. Most recent studies have further suggested that maternal cocaine exposed may increase cell death in the fetal nervous system [23-25]. Nivikova et al.[24] has detected cocaine exposure-induced changes in expression of some apoptosis-related genes in the fetal mouse cerebral wall by microarray analysis and demonstrated that maternal cocaine exposure could influence transcriptional expression levels of multiple apoptosis related genes in fetal cerebral wall. However, whether maternal cocaine exposure causes a typical apoptotic cell morphological and biochemical damage, and induces changes in translational expression levels of apoptosis-related genes in fetal brain in vivo is unknown.
The present study was therefore designed to test the hypothesis that maternal administration of cocaine during pregnancy caused apoptotic cell death in fetal rat brain. To understand the possible mechanisms underlying cocaine-induced apoptosis in the developing brain, we measured the activities of caspase-3, caspase-8, and caspase-9 and examined the effects of cocaine on Bax and Bcl-2 protein expression in fetal rat brain.
Material and Methods
Materials
Cocaine, Hoechst 33258, ethidium bromide and apoptotic DNA ladder kit were purchased from Sigma (St. Louis, MO). Bax antibody was from PharMingen (San Diego, CA). Bcl-2 antibody was from Santa Cruz Biotechnology (Santa Cruz, CA). Horseradish peroxidase (HRP)-conjugated anti-mouse IgG was from Amersham Life Science (Clearbrook, IL). Proteinase K and DNase-free Rnase were purchased from Boehringer Mannheim (Indianapolis, IN). Colorimetric assay kits for caspase-3, caspase-8, and caspase-9 were from R&D Systems (Minneapolis, MN).
Experimental animals and cocaine administration
Time-dated pregnant Sprague-Dawley rats were purchased from Charles River Laboratories (Portage, MI), and were housed individually in Plexiglas acrylic plastic cages (46 × 24 × 20 cm) in an AAALAC accredited animal facility. Maternal cocaine administration was conducted as described previously [9]. Briefly, eighteen pregnant rats were randomly divided into three groups: 1) control, 2) cocaine 30 mg/kg/day, and 3) cocaine 60 mg/kg/day. Cocaine HCl was dissolved in saline at 10 mg/ml and injected subcutaneously into the pregnant rats at ~10:00 A.M. once a day, starting at day 15 of gestation. Saline-injected pregnant rats served as controls. Food and water were provided as desired. Pregnant dams were sacrificed by cervical dislocation on day 21 of gestation, and the fetal and maternal brains were isolated. For tissue slide preparation, fetal rat brains were fixed in 10% buffered formalin and embedded in paraffin. For the other studies, fresh tissues were used.
All procedures and protocols used in the present study were approved by the Institutional Animal Care and Use Committee of Loma Linda University and followed the guidelines put forward in the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
DNA fragmentation on agarose Gel
The characteristic formation of oligonucleosome-sized fragments of multiples of ~200 bp producing typical DNA ladders on agarose gels is the biochemical hallmark of apoptosis. DNA ladders in fetal rat brains were examined as previously described using the apoptotic DNA ladder kit from Sigma [8, 9]. DNA was extracted from the brain according to the instruction of the kit. DNA (20 μg) was electrophoresed at 70 volts in 1.8% agarose gel in TBE buffer containing 1 μg/ml ethidium bromide, and photographed with ultraviolet illumination. A 100-bp DNA ladder molecular weight marker was added to each gel as a reference for analysis of internucleosomal DNA fragmentation.
Quantitative analysis of apoptotic cells
Fluorescent DNA-binding dyes (Hoechst 33258) were used to define nuclear chromatin morphology as a quantitative index of apoptosis as described previously [9, 26]. Whole fresh fetal brains were isolated from each litter of the experimental groups and immediately fixed in 10% buffered formalin and embedded in paraffin. Then the fetal brain was sectioned (4-μm thick) vertically at the middle of each hemisphere, and six sections from each brain were analyzed for the presence of apoptotic nuclei. The tissue sections were deparaffinized with xylene and rehydrated with graded dilutions of ethanol in water. The tissue sections were then stained with Hoechst 33258 at 8 μg/ml for 10 min at dark room. The slides were rinsing in distill water 3x for 5 minutes each time, and mounted with mounting medium. The nuclear morphology was examined by fluorescence microscopy. Individual nuclei were visualized at ×400, and cells were scored as apoptotic if they exhibited unequivocal nuclear chromatin condensation and/or fragmentation. Sample identity was concealed during scoring. To quantify apoptosis, one section was used to count apoptotic cells from a specific brain region, and counts from each side were averaged together. Adjacent sections were examined to verify the location of specific brain regions on a particular section, in order to obtain consistency in counting. 1,000 nuclei from random microscopic fields were analyzed and the percentage of apoptotic cells was calculated as the number of apoptotic cells/number of total cells × 100%.
Western blot analysis
The fresh fetal brain was homogenized in an ice-cold lysis buffer (20 mM HEPES, pH 7.5, 10 mM KCl, 1.5 mM MgCl2, 1 mM EDTA, 1 mM EGTA, 1 mM DTT, 1 mM PMSF, 2 μg/ml aprotinin, 10 μg/ml leupeptin), followed by centrifugation at 12,000 ×g for 15 min at 4 ºC. The supernatant was collected, and protein concentration was determined using a standard colorimetric assay (Bio-Rad). Total protein was used to determine Bax and Bcl-2 expression as described previously [9, 26]. Equal amount of proteins (50 μg) were loaded in each lane and separated in 10% SDS-PAGE, transferred to nitrocellulose membranes, and incubated with monoclonal antibody against Bax (1:250) or Bcl-2 (1:500) in TBS-T buffer containing 5% nonfat milk for 1 h at room temperature. Bax and Bcl-2 protein expressions were detected from the same membrane. After washing, the membranes were incubated for 1 h with horseradish peroxidase (HRP)-conjugated anti-mouse IgG1 (1:2000) at room temperature, and visualized using an enhanced chemiluminescence detection system. Results were quantified using a scanning densitometer (model 670, Bio-Rad).
Caspase activity assay
Activities of caspase-3, caspase-8 and caspase-9 were determined using the corresponding caspase activity detection kits (R&D Systems) as described previously [9, 26]. The assay is based on spectophotometric detection of the chromophore p-nitroanilide (pNA) after cleavage from the labeled substrates of DEVD-pNA (for caspase-3), IETD-pNA (for caspase-8), and LEHD-pNA (for caspase-9), respectively. The pNA light emission can be quantified using a spectrophotometer or a microtiter plate reader at 405-nm. Comparison of the absorbance of pNA from an apoptotic sample with control allows determination of the fold increase in caspase activity. We followed the assay procedure from the kits with some modification to determine the caspase activities in our samples. Briefly, fresh whole fetal brain and half of maternal forebrain from each litter of each experimental group were isolated and homogenized in a chilled cell lysis buffer, and then incubated on ice for 10 minutes and centrifuge for 1 minute in a microcentrifuge (10,000x g). The supernatant was transferred to a fresh tube and protein concentration was determined using a standard colorimetric assay (Bio-Rad). The protein concentration of each sample was adjusted to 200 μg per 50 μL of cell lysate using chilled cell lysis buffer. Then added 50 μL of 2X Reaction Buffer and 5 μL substrates of DEVD-pNA (for caspase-3), IETD-pNA (for caspase-8), and LEHD-pNA (for caspase-9), respectively. Samples were incubated at 37 ºC for 4 h and the enzyme-catalyzed release of pNA was quantified at 405 nm using a microtiter plate reader. The values of cocaine treated samples were normalized to the untreated controls, allowing determination of the fold increase in caspase activity.
Statistical analysis
Data were presented as the mean ± SEM. In all cases, n refers to the number of dams in each treatment group. Statistical analyses were performed by one-way ANOVA followed by Newman-Keuls post-hoc test. Differences were considered significant when P < 0.05.
Results
Effects of maternal cocaine administration on fetal brain weight
Previously, we reported that the cocaine treatment reduced fetal body weight [9]. In this experiment, maternal cocaine exposure (30 mg/kg/day) significantly decreased fetal brain weight (0.194 ± 0.002 g vs. 0.178 ± 0.002 g, p < 0.05). Cocaine also significantly decreased the ratio of fetal brain/body weight (g/g) (0.0372 ± 0.0006 vs. 0.0352 ± 0.0005, p < 0.05). However, there were no significant differences in fetal brain weight and the ratio of brain/body weight (g/g) between cocaine 30 mg/kg/day and 60 mg /kg/day groups (Table 1).
Effects of maternal cocaine administration on fetal brain weights
| Group | Brain weight (g) | Ratio of Brain/Body weight |
|---|---|---|
| Control | 0.194 ± 0.002 | 0.0372 ± 0.0006 |
| Cocaine 30mg/kg/d | 0.178 ± 0.002* | 0.0352 ± 0.0005* |
| Cocaine 60mg/kg/d | 0.180 ± 0.001* | 0.0356 ± 0.0004* |
Values are means ± SEM
*P < 0.05 vs. control
Effects of cocaine on fetal brain apoptosis
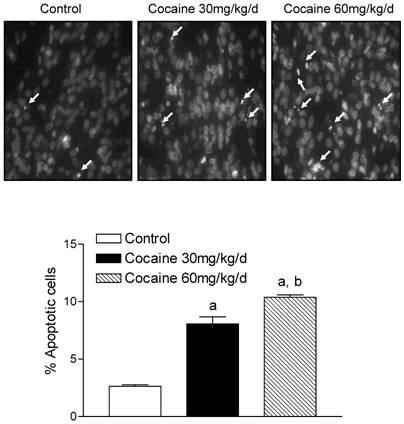
Assessment of nuclear chromatin morphology by Hoechst 33258 staining indicated that cocaine increased condensed and segmented apoptotic nuclei in the fetal brain (Fig. 1, the upper panel). In accordance, cocaine induced formation of oligonucleosome-sized fragments of DNA as ladders of ~200 bp on agarose gels, a hallmark of apoptosis (Fig. 2). Quantification of cocaine-induced apoptotic nuclei defined by the fluorescent DNA-binding dye Hoechst 33258 indicated that cocaine produced a dose-dependent increase in apoptosis in the fetal brain (Fig. 1, the lower panel).
Effect of maternal cocaine administration on apoptosis in fetal rat brain. Cocaine was administered subcutaneously to the pregnant rats for 7 days as described in Methods. Tissue sections of fetal rat brain were stained with DNA-binding fluorescence dye Hoechst 33258, and nuclear morphology was examined by fluorescence microscopy. The top panels show nuclear morphologic changes induced by cocaine (30 mg/kg/day & 60 mg/kg/day). The images were randomly chosen from the fetal brains in each group. The arrows show condensed and fragmented apoptotic nuclei. The bottom panel shows quantitative data obtained from five animals from different mothers of each group. Data are means ± SEM. a P < 0.05 vs. control; b P < 0.05 vs. cocaine 30 mg/kg/day.
Cocaine-induced nucleosomal DNA fragmentation on agarose gels in fetal rat brain. Cocaine was administered subcutaneously to the pregnant rats for 7 days as described in Methods. Cocaine-induced nucleosomal DNA fragmentation in fetal brain was separated in 1.8% agarose gels. The apoptotic DNA ladders shown were in the brain treated with 30 mg/kg/day (lane 3) and 60 mg/kg/day (lane 4), but not in the control brain (lane 2). DNA markers, ØX174 DNA fragments cut by HaeIII as size marker are shown in lane 1. The same results were obtained from five additional separate experiments.

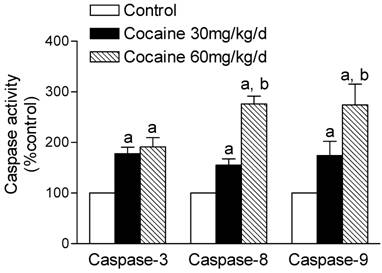
Effects of cocaine on caspase activities
The activation of caspase is a unique feature of apoptotic cell death. We determined cocaine-induced activation of the protease activities of caspase-3, caspase-8, and caspase-9. As shown in Figure 3, cocaine (30, 60 mg/kg/day) produced a dose-dependent increase (% of control) in caspase-3 (177.7 ± 12.9, 191.1 ± 18.5; P < 0.05), caspase-8 (155.2 ± 12.4, 276.3 ± 15.1; P < 0.05) and caspase-9 (174.3 ± 27.9, 274.3 ± 40.9; P < 0.05) activities in the fetal brain. In contrast, cocaine did not significantly affect caspase-3 (140.4 ± 20.0, 148.3 ± 20.4; P > 0.05), caspase-8 (139.9 ± 36.0, 149.3 ± 20.9; P > 0.05) and caspase-9 (127.5 ± 14.6; 145.9 ± 26.6, P > 0.05) activities in the maternal brain.
Effects of maternal cocaine administration on caspase activities in fetal rat brain. Cocaine was administered subcutaneously to the pregnant rats for 7 days as described in Methods. Caspase activities in fetal rat brain were determined using the caspase activity detection kits (see Methods). Data are expressed as % of control, and are means ± SEM for 5 to 6 experiments. a P < 0.05 vs. control, b P < 0.05 vs. cocaine 30 mg/kg/day.
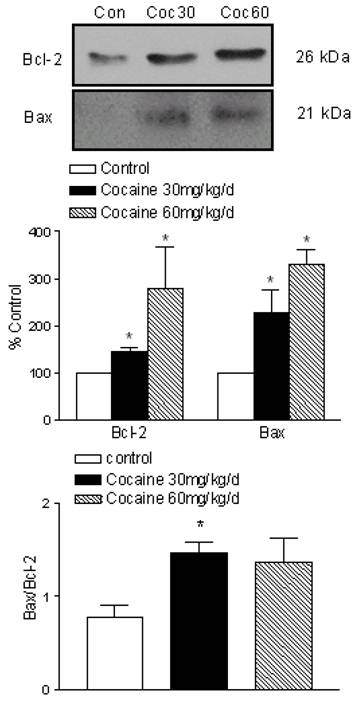
Effects of cocaine on Bax and Bcl-2 protein levels
To determine whether cocaine leads to changes in Bcl-2 family protein levels in fetal brain, we examined the Bcl-2 and Bax protein expression. As shown in Figure 4, Bax protein was minimally detected in control fetal brain. However, it was dose-dependent increased (% of control) in cocaine 30 mg/kg/day (227.1 ± 48.61, P < 0.05) and cocaine 60 mg/kg/day (330.7 ± 31.6, P < 0.05). Bax was minimally present, whereas Bcl-2 was constitutively expressed in normal fetal rat brain. As shown in Figure 4, cocaine also significantly increased Bcl-2 protein levels at the dose of 30 mg/kg/day (145.2 ± 8.16, P < 0.05) and 60 mg/kg/day (278.8 ± 87.65, P < 0.05). The Bax-to-Bcl-2 ratio in fetal rat brain was determined at each point by using value that was normalized to the control protein level within each group (Fig. 4, bottom panel). The Bax-to-Bcl-2 ratio was significantly higher at the dose of cocaine 30 mg/kg per day than the control group (P < 0.05), but no difference at the dose of cocaine 60 mg/kg per day.
Effects of maternal cocaine administration on Bax and Bcl-2 protein expression in fetal rat brain. Cocaine was administered subcutaneously to the pregnant rats for 7 days as described in Methods. Immunoblot analysis of Bax and Bcl-2 proteins was performed in fetal rat brain. The top panel shows the representative immunoblots obtained for Bcl-2 and Bax at the expected size of 26 kDa and 21 kDa, respectively, and shows an increase in Bcl-2 and Bax in the brains treated with cocaine. The middle panel shows quantitative data obtained from five separate experiments. The bottom panel shows the Bax-to-Bcl-2 ratio in fetal rat brain. Data are expressed as % of control, and are means ± SEM. *P<0.05 vs. control.
Discussion
We previously showed that maternal cocaine administration resulted in a decrease in fetal rat body weight [9]. The present study demonstrated that the maternal cocaine treatment caused a significant decrease in fetal brain weight, as compared with the saline control group. This finding is consistent with the previous report in pregnant C57BL/6 mice, in which maternal subcutaneous administration of cocaine from gestation days 12-18 produced significant decreases in fetal body and brain weight [27]. The pair-fed studies demonstrated that maternal undernutrition was not a likely mediator of the effects caused by cocaine [27, 28]. Moreover, our data indicate that cocaine decreases fetal brain/body weight ratio, suggesting that cocaine have higher affinity toxic effects on the fetal brain than the body. Dow-Edwards [29] reported that fetal brain had between 26-42% more concentration of cocaine than fetal plasma after 90 min following either 30 or 60 mg/kg cocaine given via intragastric intubation to Wistar pregnant rats. It was also reported that cocaine affinity for brain tissue is similar in the fetus and dam after subcutaneous injection of cocaine, whereas the cocaine metabolite benzoylecgonine concentrations in fetal brain were greater than those observed in maternal brain [30]. Therefore, fetal brain exposure to cocaine is somewhat prolonged. Our current finding that cocaine had no effect on the activities of the caspases in the maternal brain but only in the fetal brain, further support the idea that these high levels of cocaine or its active cocaine metabolite may contribute to the production of neuronal apoptotic alterations in cocaine-exposed offspring. Nassogne et al [6] reported that cocaine selectively affected embryonic neuronal cells, causing first a dramatic reduction of both number and length of neurites and then extensive neuronal death in co-cultures of neurons and glial cell from mouse embryonic brain. Taken all together, our study with previous reports demonstrated that cocaine exposure in utero causes severe alterations in the fetal brain, they could account for the qualitative or quantitative defects in neuronal pathways that cause a major handicap in brain function following in prenatal exposure to cocaine.
Although prenatal cocaine use during pregnancy appears detrimental to the fetus, a causal effect, mechanism of injury, and the pathway of cocaine induced injury have not well been documented [4]. Previous studies have reported that cocaine does have indirect effect on the developing fetus. Cocaine increases circulating catecholamine levels, which may induce uterine artery vasoconstriction and cause fetal chronic hypoxia, then, could result in altered fetal somatic and fetal brain development [31]. Our finding that cocaine decreased fetal body weight is consistent with our previous studies that chronic hypoxia caused fetal growth restriction [32]. Cocaine induced hypoxia and increased the susceptibility to hypoxia-induced brain damage as the outcomes associated with “crack baby syndrome” represents a common underlying mechanism [33]. On the other hand, cocaine can exert direct effects on both the fetal central and peripheral nervous systems. Studies have demonstrated that prenatal cocaine exposure has direct long-term effects on brain structure and function [23-25, 31]. Our current finding of the asymmetric growth restriction with decreased fetal brain-to-body weight ratio further suggests that direct cytotoxic effects of cocaine on the fetal brain are likely to exist. These data suggests that prenatal cocaine exposure has both indirect and direct effect on the developing fetal brain.
Nassogne et al [22] demonstrated that cocaine induced injury by apoptosis in vitro cultured cortical neuronal cells of fetal mice. Whereas, our present study has demonstrated that cocaine induces apoptosis in the fetal brain when it was administrated to the mother in vivo. In the present study, cocaine-induced apoptosis in fetal brain was clearly demonstrated by morphological changes such as cell shrinkage and rounding, characteristic features of apoptotic death. Moreover, simultaneous assessment of nuclear chromatin morphology further verified that these cells eventually manifested typical apoptotic condensed and fragmented nuclei. Similar finding of cocaine-induced apoptosis has been reported in mice hepatocytes [14]. In addition, we have confirmed that the process of apoptosis defined on the basis of cellular and nuclear chromatin morphology correlates with apoptosis defined on the basis of internucleosomal DNA fragmentation assessed by DNA gel electrophoresis. The discrete ladder of DNA fragments demonstrated by gel electrophoresis indicates the presence of DNA cleavage at linker regions producing double-strand DNA fragments of integral multiples of about 200 bp in the cocaine-induced injury fetal brain. The demonstration of this nucleosomal ladder in the treatment brain strongly suggests that apoptotic DNA degradation with internucleosomal digestion by an endonuclease is involved in the cocaine-induced cell death in the brain. In present study, DNA was extracted from the whole fetal brain, so DNA fragmentation reflected the whole brain region. Future studies will be needed to study the specific brain region and cell type undergoing apoptosis in response to cocaine exposure.
Apoptosis is a process of active cellular self-destruction that requires the expression of specific genes [34, 35]. Despite the diversity of signals that can induce cell death, these pathways share several features in their execution. One mechanism, which is consistently implicated in apoptosis, reflects an orchestrated series of biochemical events that is carried out by a group of cytosolic proteases, termed caspases. The current finding that activities of caspase-8, caspase-9 and caspase-3 were increased after prenatal cocaine exposure provides strong evidence that apoptosis is activated after cocaine exposure in fetus and may contribute to secondary cell injury and cell death. The increased activities of caspae-9 and caspase-3 induced by cocaine suggests that cocaine-induced apoptosis in the fetal brain was likely mediated by mitochondria/cytochrome c pathway [36]. On the other hands, cocaine-induced activation of caspase-8, which releases two active subunits, p18 and p10, into the cytosol, activates additional caspases that cleave other apoptosis-related substrates. Caspase-8 may be involved in death receptor-mediated apoptosis pathway [37].
Bcl-2 gene families are identified as apoptosis regulating genes. Off these genes, bax, bad, bak and bik promote cell death, whereas bcl-2 and bcl-XL inhibit apoptosis and promote cell survival [38, 39]. It has been shown that the Bcl-2 protein physically interacts with several of its homologous proteins, in the form of heterotypic dimers. The most important interactions are considered to lie in Bcl-2/Bax dimerization. Thus, we studied the temporal profile of bcl-2 and bax gene products in terms of protein expression in the fetal brain after cocaine exposure. The current findings that cocaine significantly increased the protein levels of Bcl-2 and Bax in fetal rat brain are consistent to previous reports that gene transcriptional levels of Bcl-2 and Bax are up-regulated in fetal mouse cerebral wall [24]. The current result showed that bax gene expression was markedly induced and dose dependent increased, suggesting that bax was upregulated and played an important role in the induction of apoptotic death in the fetal brain after cocaine exposure. However, in contrast to the aforementioned bcl-2 inhibiting apoptotic cell death, the present study in fetal rat brain found that bcl-2 expression was also increased in a dose dependent manner after cocaine exposure compared to saline control. The increase in anti-apoptotic Bcl-2 protein in the fetal brain may serve as a compensatory protection of the neural cells upon cocaine insult. Previous study [40] found that total Bcl-2 protein is increased in injured brain after traumatic brain injury. Neurons are resistant to ischemic injury when Bcl-2 protein is over-expressed in vivo [41-43]. Whether programmed cell death that occurs after brain injury is maladaptive or beneficial has been addressed in several studies in animal models of stroke, trauma, cerebral ischemia and excitotoxicity [44, 45]. These studies support the hypothesis that Bcl-2 protects neurons from injury. Thus, Bcl-2 expression could be an important factor that promotes survival of neurons injured after cocaine exposure. Although the expressions of Bcl-2 and Bax, both of them, were increased, it was very interesting that the ratio of Bax/Bcl-2 (pro- to anti-apoptotic proteins) was also increased after cocaine exposure in the current study. The findings support the notion that the relative concentrations of pro-apoptotic and anti-apoptotic genes may act as a rheostat for the cell death program [46].
In conclusion, our study has demonstrated that cocaine induces apoptosis in the fetal brain when it is administrated to the mother. As reported previously, our study also demonstrates fetal growth retardation after cocaine use. Moreover, our data indicate that cocaine decreases fetal brain/body weight ratio. The finding of the increased caspases activities re-enforces the conclusion that cocaine induces apoptosis in the fetal brain. The current studies also suggest that multiple mechanisms may be involved in cocaine-induced apoptosis in the fetal brain. One of the apoptotic pathways is regulated by specific genes. Of these genes, Bax is the key gene in upregulation of the cocaine-induced apoptosis in the fetal brain. However, Bcl-2 expression could be an important factor that promotes survival of cocaine-injured neurons. These findings demonstrate that genes can be orchestrated in cocaine-induced apoptosis in fetal brain, and provide a rational for the further development of pharmacological and molecular therapies targeting programmed cell death after cocaine use.
Acknowledgements
The authors thank Dr. Yuhui Xiao for the technical assistance. This work was supported in part by the National Institutes of Health grants HL-82779, HL-83966 (L.Z.) and by the California Tobacco-Related Disease Research Program Award 14FT-0075 (D.X.).
Conflict of Interest
The authors have declared that no conflict of interest exists.
References
1. Chasnoff IJ. Cocaine, pregnancy, and the growing child. Curr. Probl. Pediatr. 1992;22:302-321
2. Chasnoff IJ, Burns KA, Burns KA. Cocaine use in pregnancy: Perinatal morbidity and mortality. Neurotoxicol. Teratol. 1987;9:291-293
3. Chasnoff IJ, Burns WJ, Schnoll SH, Burns KA. Cocaine use in pregnancy. N. Engl. J. Med. 1985;313:666-669
4. Gingras JL, Weese-Mayer DE, Hume RFJr, O Donnell KL. Cocaine and development: mechanism of fetal toxicity and neonatal consequences of prenatal cocaine exposure. Early Hum. Dev. 1992;31:1-24
5. Holzman C, Paneth N. Maternal cocaine use during pregnancy and perinatal outcomes. Epidemiol. Rev. 1994;16:315-34
6. Nassogne MC, Evrard P, Courtoy PJ. Selective neuronal toxicity of cocaine in embryonic mouse brain cocultures. Proc. Natl. Acad. Sci. U. S. A. 1995;92:11029-11033
7. Li G, Xiao Y, Zhang L. Cocaine induces apoptosis in fetal rat myocardial cells through the p38 mitogen-activated protein kinase and mitochondrial/cytochrome c pathways. J. Pharmacol. Exp. Ther. 2005;312:112-119
8. Xiao Y, He J, Gilbert RD, Zhang L. Cocaine induces apoptosis in fetal myocardial cells through a mitochondria-dependent pathway. J. Pharmacol. Exp. Ther. 2000;292:8-14
9. Xiao Y, Xiao D, He J, Zhang L. Maternal cocaine administration during pregnancy induces apoptosis in fetal rat heart. J. Cardiovasc. Pharmacol. 2001;37:639-648
10. He J, Xiao Y, Casiano CA, Zhang L. Role of mitochondrial cytochrome c in cocaine-induced apoptosis in coronary artery endothelial cells. J. Pharmacol. Exp. Ther. 2000;295:896-903
11. He J, Xiao Y, Zhang L. Cocaine induces apoptosis in human coronary artery endothelial cells. J. Cardiovasc. Pharmacol. 2000;35:572-580
12. He J, Xiao Y, Zhang L. Cocaine-mediated apoptosis in bovine coronary artery endothelial cells: role of nitric oxide. J. Pharmacol. Exp. Ther. 2001;298:180-187
13. Wu YB, Shen ML, Gu GG, Anderson KM, Ou DW. The effects of cocaine injections on mouse thymocyte population. Proc. Soc. Exp. Biol. Med. 1997;214:173-219
14. Cascales M, Alvarez A, Gasco P, Fernandez-Simon L, Sanz N, Bosca L. Cocaine-induced liver injury in mice elicits specific changes in DNA ploidy and induces programmed death of hepatocytes. Hepatology. 1994;20:992-1001
15. Li H, Jiang Y, Rajpurkar A, Dunbar JC, Dhabuwala CB. Cocaine induced apoptosis in rat testes. J. Urol. 1999;162:213-216
16. Hutchins JB, Barger SW. Why neurons die: cell death in the nervous system. Anat. Rec. 1998;253:79-90
17. Johnson EM Jr, Deckwerth TL. Molecular mechanisms of developmental neuronal death. Annu. Rev. Neurosci. 1993;16:31-46
18. Wyllie AH, Kerr JF, Currie AR. Cell death: the significance of apoptosis. Int. Rev. Cytol. 1980;68:251-306
19. An SF, Gray F, Scaravilli F. Programmed cell death in brains of HIV-1-positive pre AIDS patients. Lancet. 1995;346:911-912
20. Dickson DW. Apoptosis in the brain. Physiology and pathology. Am J Pathol. 1995;146:1040-1044
21. Linnik MD, Zobrist RH, Hatfield MD. Evidence supporting a role for programmed cell death in focal cerebral ischemia in rats. Stroke. 1993;24:2002-2008
22. Nassogne MC, Louahed J, Evrard P, Courty PJ. Cocaine induces apoptosis in cortical neurons of fetal mice. J. Neurochem. 1997;68:2442-2450
23. Mitchell ES, Snyder-Keller A. c-fos and cleaved caspase-3 expression after perinatal exposure to ethanol, cocaine, or the combination of both drugs. Brain Res Dev Brain Res. 2003;147:107-117
24. Novikova SI, He F, Bai J, Badan I, Lidow IA, Lidow MS. Cocaine-induced changes in the expression of apoptosis-related genes in the fetal mouse cerebral wall. Neurotoxicol Teratol. 2005;27:3-14
25. Novikova SI, He F, Bai J, Cutrufello NJ, Lidow MS, Undieh AS. Maternal cocaine administration in mice alters DNA methylation and gene expression in hippocampal neurons of neonatal and prepubertal offspring. PLoS ONE. 2008;3:e1919
26. Bae S, Xiao Y, Li G, Casiano CA, Zhang L. Effect of maternal chronic hypoxic exposure during gestation on apoptosis in fetal rat heart. Am. J. Physiol. Heart Circ. Physiol. 2003;285:H983-H990
27. Middaugh LD, Boggan WO, Bingel SA, Patrick KS, Xu W. A murine model of prenatal cocaine exposure: effects on the mother and the fetus. Pharmacol. Biochem. Behav. 1996;55:565-574
28. Song J, Guan XW, Ren JQ, He W. Developmental toxicity of cocaine exposure in mid-pregnancy mice. Acta. Pharmacol. Sin. 2002;23:1029-1034
29. Dow-Edwards DL. Fetal and maternal cocaine levels peak rapidly following intragastric administration in the rat. J. Subst. Abuse. 1990;2:427-437
30. Spear LP, Frambes NA, Kirstein CL. Fetal and maternal brain and plasma levels of cocaine and benzoylecgonine following chronic subcutaneous administration of cocaine during gestation in rats. Psychopharmacology (Berl). 1989;97:427-431
31. Slotkin TA. Fetal nicotine or cocaine exposure: which one is worse? J. Pharmacol. Exp. Ther. 1998;285:931-945
32. Xiao D, Ducsay CA, Zhang L. Chronic hypoxia and developmental regulation of cytochrome c expression in rats. J. Soc. Gynecol. Investig. 2000;7:279-283
33. Olsen GD. Potential mechanisms of cocaine-induced developmental neurotoxicity: a minireview. Neurotoxicology. 1995;16:159-167
34. Kerr JF, Wyllie AH, Currie AR. Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics. Br. J. Cancer. 1972;26:239-257
35. Oppenheim RW, Prevette D, Tytell M, Homma S. Naturally occurring and induced neuronal death in the chick embryo in vivo requires protein and RNA synthesis: evidence for the role of cell death genes. Dev. Biol. 1990;138:104-113
36. Green DR, Reed JC. Mitochondria and apoptosis. Science. 1998;281:1309-1312
37. Ashkenazi A, Dixit VM. Death receptors: signaling and modulation. Science. 1998;281:1305-1308
38. Hara A, Hirose Y, Wang A, Yoshimi N, Tanaka T, Mori H. Localization of Bax and Bcl-2 proteins, regulators of programmed cell death, in the human central nervous system. Virchows Arch. 1996;429:249-253
39. Vaux DL, Cory S, Adams JM. Bcl-2 gene promotes haemopoietic cell survival and cooperates with c-myc to immortalize pre-B cells. Nature. 1988;335:440-442
40. Clark RS, Kochanek PM, Chen M, Watkins SC, Marion DW, Chen J, Hamilton RL, Loeffert JE, Graham SH. Increases in Bcl-2 and cleavage of caspase-1 and caspase-3 in human brain after head injury. FASEB. J. 1999;13:813-821
41. Lawrence MS, McLaughlin JR, Sun GH, Ho DY, McIntosh L, Kunis DM, Sapolsky RM, Steinberg GK. Herpes simplex viral vectors expressing Bcl-2 are neuroprotective when delivered after a stroke. J. Cereb. Blood Flow Metab. 1997;17:740-744
42. Linnik MD, Zahos P, Geschwind MD, Federoff HJ. Expression of bcl-2 from a defective herpes simplex virus-1 vector limits neuronal death in focal cerebral ischemia. Stroke. 1995;26:670-1674
43. Martinou JC, Dubois-Dauphin M, Staple JK, Rodriguez I, Frankowski H, Missotten M, Tschopp J. Overexpression of BCL-2 in transgenic mice protects neurons from naturally occurring cell death and experimental ischemia. Neuron. 1994;13:1017-1030
44. Friedlander RM, Gagliardini V, Hara H, Fink KB, Li W, MacDonald G, Fishman MC, Greenberg AH, Moskowitz MA, Yuan J. Expression of a dominant negative mutant of interleukin-1 beta converting enzyme in transgenic mice prevents neuronal cell death induced by trophic factor withdrawal and ischemic brain injury. J. Exp. Med. 1997;185:933-940
45. Hara H, Friedlander RM, Gagliardini V, Ayata C, Fink K, Huang Z, Shimizu-Sasamata M, Yuan J, Moskowitz MA. Inhibition of interleukin 1beta converting enzyme family proteases reduces ischemic and excitotoxic neuronal damage. Proc. Natl. Acad. Sci. U. S. A. 1997;94:2007-2012
46. Adams JM, Cory S. The Bcl-2 protein family: arbiters of cell survival. Science. 1998;281:1322-1326
Author contact
![]() Correspondence to: DaLiao Xiao, PhD, Center for Perinatal Biology, Department of Physiology and Pharmacology, Loma Linda University, School of Medicine, Loma Linda, CA 92350. Tel: 909-558-4325; Fax: 909-558-4029; Email: Dxiaoedu
Correspondence to: DaLiao Xiao, PhD, Center for Perinatal Biology, Department of Physiology and Pharmacology, Loma Linda University, School of Medicine, Loma Linda, CA 92350. Tel: 909-558-4325; Fax: 909-558-4029; Email: Dxiaoedu