Impact Factor ISSN: 1449-1907

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2008; 5(5):240-243. doi:10.7150/ijms.5.240 This issue Cite
Short Research Communication
IGF-1 regulates cAMP levels in astrocytes through a β2-adrenergic receptor-dependant mechanism
Daniel Chesik, Nadine Wilczak, Jacques De Keyser ![]()
Department of Neurology, University Medical Center Groningen, Hanzeplein 1, 9713 GZ Groningen, the Netherlands
Received 2008-6-10; Accepted 2008-8-4; Published 2008-8-6
Abstract
We have recently demonstrated that neonatal astrocytes derived from mice lacking beta-2 adrenergic receptors (β2AR) possess higher proliferation rates, as compared to wild-type cells, an attribute that was shown to involve insulin-like growth factor (IGF) signaling. In the present study, we demonstrate that basal cAMP levels in β2AR knockout astrocytes were significantly lower than in wild type cells. Furthermore, treatment with IGF-1 reduced intracellular cAMP levels in wild type astrocytes, yet had no effects on cAMP levels in β2AR deficient astrocytes. Our data suggests that IGF-1 treatment influences cAMP production through a β2AR-dependant mechanism in astrocytes. A deficit of β2AR on astrocytes, as previously reported in multiple sclerosis, may influence cell proliferation, an action which could have implications in processes involved in astrogliosis.
Keywords: beta adrenergic receptors, insulin-like growth factor, cyclic adenosine monophosphate, astrocytes, multiple sclerosis
Introduction
Beta-adrenergic receptors (βARs) are members of the superfamily of G-protein coupled receptors (GPCRs) and involved in fundamental processes such as cell growth, differentiation, and metabolism. They are stimulated by catecholamines, epinephrine and norepinephrine (NE) and play important roles in cardiovascular, respiratory, metabolic, reproductive and central nervous system (CNS) functions [1]. One of the major cellular signaling pathways of the βARs is mediated by the G-protein Gsα leading to activation of adenylyl cyclase and increases in the second messenger cyclic adenosine monophosphate (cAMP), an activator of cAMP-dependent protein kinase A (PKA).
The type 1 insulin-like growth factor receptor (IGF-1R) is activated by its ligands, insulin-like growth factor-1 and -2 (IGF-1 and -2), which results in intrinsic tyrosine kinase receptor activity and the transduction of intracellular signaling pathways, including MAPK and PI3K pathways [2]. It is becoming evident that signaling pathways induced by receptor tyrosine kinases (RTK) may interact with GPCR pathways at a variety of intracellular levels, including direct receptor-receptor interactions [3,4]. For example, insulin and IGF-1 have been shown to stimulate insulin and type 1 IGF receptor-catalyzed phosphorylation of the β2AR, which has been shown to result in loss of receptor function and its activation of adenylyl cyclase [5,6].
Multiple sclerosis (MS) is an inflammatory demyelinating disease of the CNS characterized by infiltration of macrophages and T-cells into brain parenchyma. This is accompanied by cytokine and chemokine expression and release. Astrocytes respond to this insult with onset of cellular reactivity, which is particularly prominent in MS and ultimately leads to the formation of chronic lesions [7,8]. IGF is essential for proper CNS development and a potent stimulator of myelin synthesis and, therefore, possesses therapeutic potential for remyelination strategies in MS. However, due to its mitogenic capacity on astrocytes, treatment based on enhancing IGF-1 signaling could augment the process of astrogliosis and further exacerbate astrogliotic scaring, a mechanism which is thought to impede remyelination processes. Investigations in our laboratories have demonstrated a deficiency of the β2AR on astrocytes in lesions and normal appearing white matter of MS patients, whereas these receptors were present on neurons [9]. Astrocytic β2ARs are known to engage in a variety of cellular functions, such as regulation of immune-inflammatory responses, glutamate uptake, and energy metabolism [10-13]. Many of these functions operate via the Gsα - adenylyl cyclase pathway, which enhances cAMP, leading to PKA activation and further phosphorylation of down stream targets. Because of the importance of both the β2 adrenergic and type 1 IGF receptor signaling in CNS and the potential role of a deficit of the β2AR on astrocytes in MS lesions, we investigated the influence of IGF-1, a known modulator of β2AR functions and a potential candidate for therapeutic purposes in MS, on cAMP production in astrocytes derived from mice deficient in β2ARs.
Materials and methods
Materials
Tissue culture plasticware was obtained from Nalge Nunc International (Roskilde, Denmark). All other cell culture materials were purchased from Gibco BRL (UK). For immunohistochemistry, primary antibodies used were rabbit anti-GFAP (Sigma; St Louis, MO, USA) and mouse anti-S-100β (Swant, Bellinzona, Switzerland). Secondary antibodies for immunofluorescent stainings were, Alexa-fluor 488 goat-anti-mouse-IgG (FITC-conjugated) and Alexa-fluor 568-goat-anti-rabbit (TRITC-conjugated) (Sigma). Anti-fading fluorescent mounting medium was from DAKO (Ca., USA). Human recombinant IGF-1 and NE were purchased from Sigma.
Generation of Knockout Mice
Mice lacking functional β2-adrenergic receptors have been generated previously and the strategy for disrupting this gene has been described [14]. The β2AR-deficient mouse (β2AR -/-) was generated by homologous recombination resulting in the insertion of a neomycin resistance gene cassette into the fourth transmembrane domain of the β2-AR gene. These mice are viable, fertile and showed no overt phenotypic abnormalities. All mice were maintained under specified pathogen-free conditions and animal studies were in accordance with the University and government authority's guidelines. Using PCR techniques, mRNA for the β2AR gene could be detected in β2AR +/+, but was absent in β2AR -/- mice astrocytes (data not shown).
Cell cultures
Astrocyte cultures from wild-type (WT) and β2AR knockout (KO) mice were prepared according to a shake-off protocol described previously [15]. Cerebral hemispheres of 1 day-old mouse pups were freed from the meninges and mechanically disrupted using a Pasteur pipette. After centrifugation (10 min, 300*g) single cell suspensions were transferred to culture flasks (1 brain/flask) and cultivated for 5 days in growth medium (DMEM containing; 10% FCS, 5 μg/ml pyruvate, 2 mmol/l glutamine, 50 U/ml penicillin, and 50 μg/ml streptomycin and 1000mg glucose/liter). Growth medium was replaced with fresh medium twice a week. 7 to 12 days after plating, O2A precursor cells and microglia were removed by shaking-off overnight at 250 rpm and 37°C. Two shake-off procedures were performed followed by trypsinisation of the astrocytic monolayer. The suspended cells were filtrated through 100 µm mesh nylon membranes, centrifuged (300*g, 15 min), counted and plated into poly-L-lysine- (PLL)-coated culture 10cm2 dishes (1,000,000 cells/dish) or 12-well multiwell dishes (50,000 cells/well), for mRNA isolation or immunocytochemistry, respectively. 2 h after plating, growth medium was removed and cells rinsed with phosphate buffered saline (PBS) followed by addition of a chemically-defined, insulin-free medium (CDM: DMEM containing 5 μg/ml pyruvate, 2 mmol/l glutamine, 50 U/ml penicillin, 50 μg/ml streptomycin, 5 μg/ml transferrin, and 5 ng/ml selenite) or supplemented with 10% FCS for further cultivation. Purity of cultures were examined by staining with the astrocytic markers S-100β and found to be more than 95% pure (data not shown).
cAMP assay
Astrocytes were cultured in 96-well plates at a density of 20,000 cells / well, incubated in CDM for 48 hrs and treated with IGF-1 (100ng/ml) or NE (10-5M) prior to cell lysis. The cAMP Biotrak competitive enzyme immunoassay system kit from Amersham Bioscience (Buckinghamshire, UK) was utilized for cAMP measurement and applied according to manufacturer's suggestions. Samples were subjected to cAMP extraction and incubated with anti-cAMP antiserum, which was immobilized onto secondary antibody precoated-microplates. Peroxidase-labelled cAMP conjugate was applied for assay competition. Following substrate conversion, absorption was spectrophotometrically measured at 450nm. A cAMP standard was provided for quantitative calculation of cAMP concentrations of astrocyte samples per well.
Statistical analysis
All experiments were performed a minimum of 3 times each, in triplet. For any given experiment, each data bar represents the mean +/- SEM of values obtained in separate experiments. Statistical significance was determined by one-way Anova analysis. Values of P<0.05 were considered significant.
Results
Previous examination of isolated astrocyte cultures by light microscopy revealed contrasting properties regarding proliferation rates of astrocytes derived from WT mice, as compared to those derived from mice with a β2AR deletion. While cell morphology remained unaltered, KO astrocytes grew more rapidly, a characteristic which was visible and quantifiable after 3 days in culture [16].
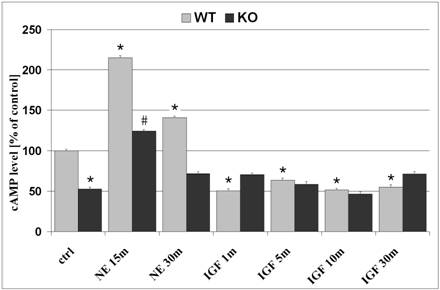
The effects of cAMP on cell proliferation and differentiation are well documented and was our motive for evaluation of cAMP levels in WT and KO astrocytes. In untreated cells, basal cAMP levels demonstrated 47.3% lower concentrations in KO astrocytes as compared to WT cells (p<0.05; figure 1). NE, a non-specific agonist which interacts with both alpha- and beta-adrenergic receptor sites and generally regulates cAMP concentrations. Treatment of astrocytes with NE (10-5M) for 15 minutes, prior to harvesting of cell lysates, resulted in enhanced levels of cAMP in both WT and KO cells by 115% and 135.3%, as compared to untreated cells, respectively. Despite the absence of β2ARs in KO cells, NE increased cAMP levels, which is likely a result of stimulation of other adrenergic receptors that signal via Gs and activate adenylyl cyclase. NE-induced increase in cAMP production reached a maximum after 15-minute treatment and declined after a 30-minute exposure to NE in both WT and KO cells. In response to IGF-1 (50ng/ml; 1 minute treatment), cAMP levels were reduced in WT cells by 50% and remained reduced after a 30-minute exposure to IGF-1 (figure 1; p<0.05, as compared to untreated WT cells). β2AR deficient astrocytes demonstrated no changes in cAMP levels in response to IGF-1 treatment.
Intracellular cAMP levels in astrocytes. Astrocytes were plated in 96-well plates (20,000 cells/well) and cultivated in CDM containing 0% FCS. Basal levels of untreated cells demonstrate 47.3% lower cAMP levels in KO astrocytes, as compared to WT cells. Treatment with NE for 15 minutes increased cAMP levels in both WT and KO astrocytes by 115.1% and 135.3%, respectively. NE-induced cAMP concentration was reduced in both WT and KO after 30 minutes treatment. Treatment with IGF-1 for 1 minute reduced cAMP levels by 50% in WT cells only, an effect that was still observed after 5, 10 and 30 minute treatments. β2AR deficient astrocytes demonstrated no changes in cAMP levels in response to IGF-1 treatment. Data represents mean +SEM. *, p<0.05 compared to untreated WT cells; #, p<0.05 compared to untreated KO cells.
Discussion
cAMP is involved in cellular proliferation-differentiation processes as demonstrated on astrocytes with dibutyryl cAMP (dBcAMP), which initiates a more differentiated status of astrocyte with reduced proliferative capacity [17]. dBcAMP has also been shown to inhibit IGF-1-induced mitogenesis in cells by inhibiting Raf-1 kinase activity, an effect that has been attributed to phosphorylation of Raf-1 by protein kinase A [17-19]. Interestingly, we have shown that cultured β2AR KO astrocytes, which proliferate more rapidly than wild type cells [16], display reduced basal cAMP levels. This reduction in basal cAMP in β2AR deficient cells might be accounted for by the absence of constitutive activity, which is known for the β2AR. In addition to agonist-induced activation, β2AR can undergo spontaneous conformational changes, resulting in ligand-independent activation [20].
In addition to Gs activation and consequent cAMP production, the β2AR can activate Gi proteins, a property that is unique amongst the beta-adrenergic receptors. Activation of Gi results in enhanced survival, an effect which is thought to be mediated by elevated activity of Akt/PKB [21]. An absence of β2AR signaling in astrocytes and consequent reduction of Akt/PKB activation might be compensated by increased IGF signaling as demonstrated previously [16]. This compensatory mechanism might explain enhanced astrocytic proliferation.
We have demonstrated that treatment with IGF-1 reduced cAMP levels in WT astrocytes, yet had no effect on KO cells. Although IGF-1 had a clear effect on cAMP production in WT cells, the mechanism by which IGF receptor signaling is involved in this regulation is uncertain. As mentioned above, IGF-1 can catalyze the phosphorylation of the β2AR, which results in loss of function of this receptor and inhibition of adenylyl cyclase Gs activation [6,22]. Although we demonstrate that IGF-1 has the potential to regulate cAMP levels in astrocytes in a β2AR-dependent manner, we do not know whether this occurs through direct phosphorylation and subsequent desensitization of the β2AR by the IGF-1R, a notion which remains speculative.
We have previously reported a loss of β2AR on astrocytes in cerebral white matter of patients with MS, a deficit which may contribute to pathology of this disease by several possible mechanisms such as impaired astrocytic glycogenolysis. Insufficient glycogenolysis could decrease energy supplies to axons and may contribute to axonal degeneration [23]. In this context, cAMP physiological effects include crucial roles in regulating energy metabolism, such as lipolysis, gluconeogenesis, and glycogenolysis [24]. Loss of β2AR on astrocytes in MS might also contribute to enhanced astrogliosis and cellular reactivity, a hallmark trait in MS lesions. Regulation of cell growth by the β2AR, a receptor which is involved in processes of proliferation and differentiation, has been implicated in astrocytes in vitro [16].
In summary, β2AR deficient astrocytes have lower basal cAMP levels as compared to wild type cells. This reduction of cAMP levels may be involved in the increased cell proliferation as reported previously. We show here that stimulation with IGF-1 reduces cAMP levels in astrocytes in a β2AR-dependent manner, a reduction of which may promote some of the mitogenic properties of IGF signaling.
Acknowledgements
We would like to thank Prof. Brian Kobilka (Stanford University School of Medicine) and Prof. Lutz Hein (University of Wuerzburg) for kindly providing us with β2-AR -/- mice.
Conflict of Interest
The authors have declared that no conflict of interest exists.
References
1. Gutkind JS. Cell growth control by G protein-coupled receptors: from signal transduction to signal integration. Oncogene. 1998;17:1331-1342
2. Hung KS, Tsai SH, Lee TC, Lin JW, Chang CK, Chiu WT. Gene transfer of insulin-like growth factor-1 providing neuroprotection after spinal cord injury in rats. J Neurosurg Spine. 2007;6:35-46
3. Daub H, Weiss FU, Wallasch C, Ullrich A. Role of transactivation of the EGF receptor in signalling by G-protein coupled receptors. Nature. 1996;379:557-560
4. Dalle S, Imamura T, Rose DW, Worrall DS, Ugi S, Hupfeld CJ. et al. Insulin induces heterologous desensitization of G-protein-coupled receptor and insulin-like growth factor I signaling by down-regulating b-arrestin-1. Mol Cell Biol. 2002;22:6272-6285
5. Hadcock JR, Port JD, Gelman MS, Malbon CC. Crosstalk between tyrosine kinase and G-protein-linked receptors. Phosphorylation of β2-adrenergic receptors in response to insulin. J Biol Chem. 1992;267:26017-26022
6. Karoor V, Malbon CC. Insulin-like growth factor receptor-1 stimulates phosphorylation of the b2-adrenergic receptor in vivo on sites distinct from those phosphorylated in response to insulin. J Biol Chem. 1996;271:29347-29352
7. Eng LF, Ghirnikar RS. GFAP and astrogliosis. Brain Pathol. 1994;4:229-237
8. Fawcett JW, Asher RA. The glial scar and central nervous system repair. Brain Res Bull. 1999;49:377-391
9. De Keyser J, Wilczak N, Leta R, Streetland C. Astrocytes in multiple sclerosis lack β2 adrenergic receptors. Neurology. 1999;53:1628-1633
10. Hertz L, Chen Y, Gibbs ME, Zang P, Peng L. Astrocytic adrenoceptors: a major drug target in neurological and psychiatric disorders? Curr Drug Targets CNS Neurol Disord. 2004;3:239-67
11. Fahrig T. Receptor subtype involved and mechanism of norepinephrine-induced stimulation of glutamate uptake into primary cultures of rat brain astrocytes. Glia. 1993;7:212-218
12. Fillenz M, Lowry JP, Boutelle MG, Fray AE. The role of astrocytes and noradrenaline in neuronal glucose metabolism. Acta Physiol Scand. 1999;167:275-284
13. Feinstein DL, Heneka MT, Gavrilyuk V, Dello Russo C, Weinberg G, Galea E. Noradrenergic regulation of inflammatory gene expression in brain. Neurochem Int. 2002;41:357-365
14. Chruscinski AJ, Rohrer DK, Schauble E, Desai KH, Bernstein D, Kobilka BK. Targeted disruption of the b2 adrenergic receptor gene. J Biol Chem. 1999;274:16694-16700
15. McCarthy KD, de Vellis J. Preparation of separate astroglial and oligodendroglial cell cultures from rat cerebral tissue. J Cell Biol. 1980;85:890-902
16. Chesik D, Glazenburg L, De Keyser J, Wilczak N. Enhanced proliferation of astrocytes from beta(2)-adrenergic receptor knockout mice is influenced by the IGF system. J Neurochem. 2007;100:1555-64
17. Kurino M, Fukunaga K, Ushio Y, Miyamoto E. Cyclic AMP inhibits activation of mitogen-activated protein kinase and cell proliferation in response to growth factors in cultured rat cortical astrocytes. J Neurochem. 1996;67:2246-2255
18. Howe LR, Leevers SJ, Gomez N, Nakielny S, Cohen P, Marshall CJ. Activation of the MAP kinase pathway by the protein kinase raf. Cell. 1992;71:335-342
19. Valverde AM, Teruel T, Lorenzo M, Benito M. Involvement of Raf-1 kinase and protein kinase Cf in insulin-like growth factor I-induced brown adipocyte mitogenic signaling cascades: inhibition by cyclic adenosine 3`,5`-monophosphate. Endocrinology. 1996;137:3832-3841
20. Ballesteros JA, Jensen AD, Liapakis G, Rasmussen SG, Shi L, Gether U. et al. Activation of the beta 2-adrenergic receptor involves disruption of an ionic lock between the cytoplasmic ends of transmembrane segments 3 and 6. J Biol Chem. 2001;276:29171-7
21. Zhu WZ, Zheng M, Koch WJ, Lefkowitz RJ, Kobilka BK, Xiao RP. Dual modulation of cell survival and cell death by beta(2)-adrenergic signaling in adult mouse cardiac myocytes. Proc Natl Acad Sci U S A. 2001;98:1607-12
22. Baltensperger K, Karoor V, Paul H, Ruoho A, Czech MP, Malbon CC. The beta-adrenergic receptor is a substrate for the insulin receptor tyrosine kinase. J Biol Chem. 1996;271:1061-4
23. De Keyser J, Zeinstra E, Mostert J, Wilczak N. β2-Adrenoceptor involvement in inflammatory demyelination and axonal degeneration in multiple sclerosis. Trends Pharmacol Sci. 2004;25:67-71
24. Collins S, Surwit RS. The β-adrenergic receptors and the control of adipose tissue metabolism and thermogenesis. Recent Prog Horm Res. 2001;56:309-328
Author contact
![]() Correspondence to: Daniel Chesik, Department of Neurology, University Medical Center Groningen, Hanzeplein 1, 9713 GZ Groningen, The Netherlands. Tel.: 0031-50-3637719; Fax: 0031-50-3611707; e-mail: d.chesikumcg.nl
Correspondence to: Daniel Chesik, Department of Neurology, University Medical Center Groningen, Hanzeplein 1, 9713 GZ Groningen, The Netherlands. Tel.: 0031-50-3637719; Fax: 0031-50-3611707; e-mail: d.chesikumcg.nl