Impact Factor ISSN: 1449-1907
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Issue 4; 2026
- Volume 23; 2026
- Past Issues
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
1. Introduction
2. Materials and Methods
3. Results
4. Discussion
Acknowledgements
Conflicts of interest
References

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2006; 3(4):148-151. doi:10.7150/ijms.3.148 This issue Cite
Short Research Communication
Mutation Analysis of hCDC4 in AML Cells Identifies a New Intronic Polymorphism
Daniel Nowak, Maximilian Mossner, Claudia D. Baldus, Olaf Hopfer, Eckhard Thiel, Wolf-Karsten Hofmann ![]()
Department of Hematology, Oncology and Transfusion Medicine, Charité, University Hospital Benjamin Franklin, Berlin, Germany
Received 2006-8-25; Accepted 2006-10-25; Published 2006-10-26
Abstract
hCDC4 (FBW7, FBXW7) is a new potential tumor suppressor gene which provides substrate specificity for SCF (Skp–Cullin–F-box) ubiquitin ligases and thereby regulates the degradation of potent oncogenes such as cyclin E, Myc, c-Jun and Notch. Mutations in the hCDC4 gene have been found in several solid tumors such as pancreas, colorectal or endometrial cancer. We carried out a mutation analysis of the hCDC4 gene in 35 samples of patients with Acute Myeloid Leukemia (AML) to elucidate a possible role of hCDC4 mutations in this disease. By direct DNA sequencing and digestion with Surveyor nuclease one heterozygous mutation in the 5' untranslated region of exon 1, transcript variant 3 was detected. Additionally, we could identify a new intronic SNP downstream of exon 10. The new variation was present in 20% of AML samples and was furthermore confirmed in a panel of 51 healthy individuals where it displayed a frequency of 14%. In conclusion we provide first data that in contrast to several solid tumors, mutations in the hCDC4 gene may not play a pivotal role in the pathogenesis of AML. Furthermore, we describe a new intronic polymorphism with high frequency in the intron sequence of the hCDC4 gene.
Keywords: hCDC4, AML, Mutation Analysis, SNP
1. Introduction
The F-box and WD40 domain protein 7 (hCDC4, FBW7, FBXW7) has recently emerged as a potent new potential tumor suppressor gene [1, 2]. The highly conserved protein consists of an NH2 terminal F-box and seven WD40 repeats in the COOH terminal region and acts as an adaptor protein providing substrate specificity for SCF (Skp–Cullin–F-box) ubiquitin ligases which are involved in tagging proteins for degradation in the proteasome.
hCDC4 has been shown to target specifically cyclin E [3], Myc [4], c-Jun [5] and Notch [6] for proteasomal degradation and therefore negatively regulates several key oncoproteins.
Mutations in the CDC4 gene have been detected in several solid tumors such as colorectal cancer [7, 8], endometrial cancer [9, 10] or cell lines [11]. Furthermore, defective hCDC4 may be involved in cellular pathways leading to chromosomal instability [12].
As the disruption of the above described cellular oncogenic pathways also plays an important role in hematological malignancies we were interested whether mutations of hCDC4 can also be observed in Acute Myeloid Leukemia (AML) or high risk Myelodysplastic Syndrome (MDS). Therefore we carried out a mutational analysis of the hCDC4 gene in 35 samples of AML patients in order to elucidate whether hCDC4 mutations may be relevant for the genesis of this disease.
2. Materials and Methods
Nucleic acid preparation
Heparinized bone marrow (BM) samples from 22 and peripheral blood (PB) samples from 13 Patients with AML were obtained at the time of their initial diagnosis after informed consent. For control, 51 PB samples were obtained from voluntary healthy individuals after informed consent. Mononuclear cells were separated by density gradient centrifugation through Ficoll-Hypaque (Biochrom, Berlin, Germany). Genomic DNA (gDNA) was extracted from mononuclear cells using TRIZOL reagent (Invitrogen, Life Technologies, Grand Island, NY) according to the manufacturer's protocol. The content of gDNA was adjusted to 30 ng/μl for further analyses.
Polymerase chain reaction
The common exons 2 to 11 and the three known variants of exon 1 of the hCDC4 gene (Figure 1A) were amplified by polymerase chain reaction (PCR). Primers were synthesized by Metabion International AG (Martinsried, Germany). Reaction conditions were as follows: An initial denaturation step at 95°C for 15 minutes was followed by 35 cycles consisting of denaturation at 95°C for 30 seconds (s), annealing at 58°C for 30s and elongation at 72°C for 60s followed by a final elongation step at 72°C for 7 min.
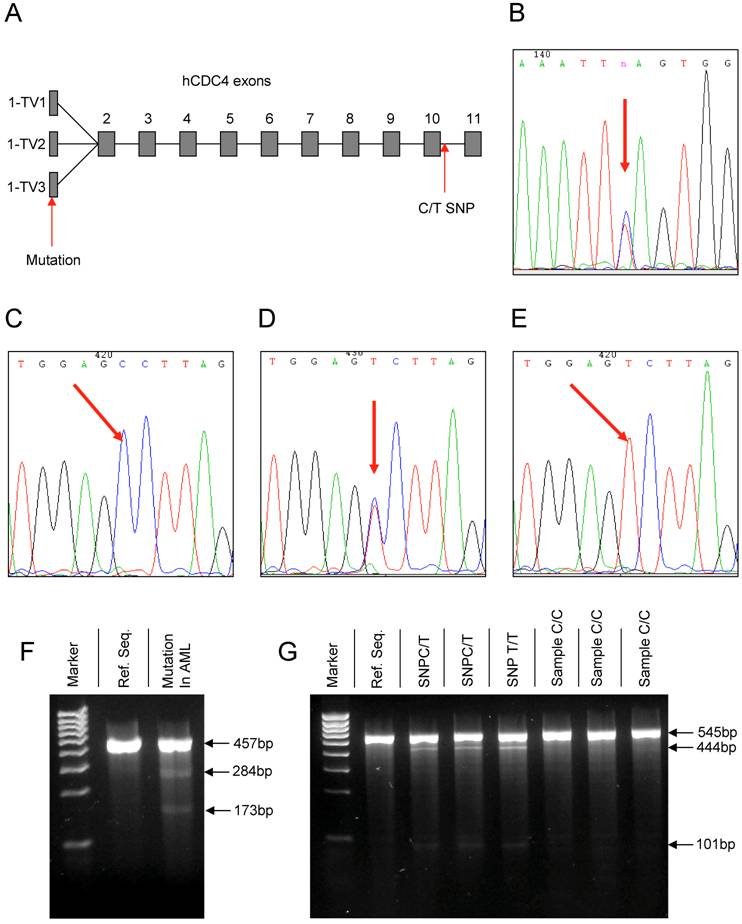
(A) Overview depicting the common exons 2 – 11 and the three transcript variants (TV) of exon 1 of the hCDC4 gene. Arrows mark the position of the detected mutation in the 5' UTR of Exon 1-TV3 and the new intronic SNP downstream of exon 10. (B) Chromatograph of the mutation detected in exon 1. (C-E) Chromatographs of the different variations of the new intronic SNP in the hCDC4 gene on chromosome 4, position 153602874. (F) Gel electrophoresis of a Surveyor nuclease digested PCR product of the AML sample containing the mutation in exon 1 versus digestion of the reference sequence (Ref. Seq.) (negative control). (G) Gel electrophoresis of surveyor nuclease digested PCR products of wild type samples containing the newly identified C/T SNP versus the reference sequence (Ref. Seq.) and samples lacking the SNP.
Primer sequences can be supplied upon request. PCR products were separated by agarose gel electrophoresis on a 2% agarose gel and subsequently purified with the QIAquick PCR purification system (QIAGEN, Hilden, Germany).
Direct DNA sequencing and mutational analysis
Purified PCR products were sequenced at the DLMBC sequencing service (Dr. M. Meixner) in the Department of Biochemistry of the Charité, Berlin, Germany using the ABI PRISM Big Dye Terminator system (Applied Biosystems, Germany). Sequences were analyzed using the Chromas software (Technelysium Pty Ltd, Tewantin, Australia) and GeneDoc software (http://www.psc.edu/biomed/genedoc). Sequences were analyzed in alignment with the NCBI and ENSEMBLE reference sequences of hCDC4: ENST00000281708 / NM_033632, ENST00000263981 / NM_018315, ENST00000296555 / NM_001013415. Sequences with deviations were re-amplified and re-sequenced for confirmation.
Surveyor nuclease digestion
Exon 1 (transcript variant 3) and exon 10 were amplified from selected samples with PCR parameters as described above. After a control electrophoresis on 2% agarose gels, the PCR products were directly subjected to processing with Surveyor nuclease [13] (Transgenomic, Omaha, USA) according to the protocol supplied by the manufacturer: PCR products from a reference sequence were mixed at equimolar amounts with wild type samples, denatured at 95°C for 2 minutes and subsequently re-hybridized. In case of sequence deviations in the wildtype samples from the reference sequence this resulted in the formation of heteroduplexes containing mismatches. In the following incubation with Surveyor nuclease for 20 min. at 42°C heteroduplexes were cleaved at mismatch sites by the enzyme leading to DNA fragments with reduced length. These digestion products were subjected to electrophoresis on 2% agarose gels.
3. Results
Mutational analysis of hCDC4 in AML
In this study the common exons 2 to 11 and the three known transcript variants of exon 1 of hCDC4 (Figure 1A) were analyzed by direct DNA sequencing in 35 samples derived from bone marrow or peripheral blood of AML patients at the time point of initial diagnosis. This resulted in the identification of one heterozygous mutation in the 5' untranslated region (5'UTR) of Exon 1 (transcript variant 3, NM_001013415) comprising a heterozygous T > C exchange of nucleotide (nt) 108 of the exon sequence (Figure 1B). Being located in an untranslated region of the exon, the mutation has no consequence for the hCDC4 protein.
Sequence analysis of hCDC4 exon 10 identifies a new intronic SNP
During the analysis of exon 10 and adjacent intronic sequences we could for the first time identify a new single nucleotide polymorphism (SNP) in the intron sequence flanking the 3' end of exon 10 in 7 out of 35 (20%) patient samples. The SNP consisted of a C > T exchange (Figure 1 C-E) and was positioned at nt 67 of the intron sequence (chromosome 4, position 153602874).
Because this variation had as yet not been classified as a known polymorphism we wanted to determine whether it could possibly be disease specific and therefore carried out a validation in 51 DNA samples of healthy individuals. This led to the confirmation of the SNP as it was also present in 7 out of 51 (13.7%) healthy individuals. The new C > T SNP was submitted to dbSNP (NCBI), identifier: NCBI_ss# P1_1 49855991. Genotype frequencies are summarized in Table 1.
Genotype and frequency of the newly identified SNP in the hCDC4 gene
| SNP C/T, Chromosome 4, Chromosome Pos. 153602874 | |||
|---|---|---|---|
| AML (n=35) | Healthy individuals (n=51) | ||
| Genotype | n / (%) | Genotype | n / (%) |
| C/C | 28 / (80) | C/C | 44 / (86) |
| C/T | 7 / (20) | C/T | 6 / (12) |
| T/T | 0 / (0) | T/T | 1 / (2) |
Confirmation of sequence deviations with surveyor nuclease
In order to confirm the mutation found in exon 1 (transcript variant 3) and the new SNP we subjected selected samples to a mismatch specific digestion procedure employing Surveyor nuclease.
As depicted in Figure 1F the AML sample containing the mutation in exon 1 features cleavage products of approximately 284 bp and 173 bp size as compared to no detectable cleavage products in the reference sequence (negative control). These fragments corresponded to the lengths of the sequence surrounding the detected mutation in exon 1. Similarly, analysis of samples containing the C/T SNP downstream of exon 10 led to the identification of cleavage products of approximately 101 bp and 444 bp in size in samples either containing the heterozygous C/T or homozygous T/T SNP while these fragments could not be detected in samples lacking the SNP and therefore corresponded to the reference sequence (Figure 1 G).
4. Discussion
The elucidation of the molecular pathogenesis of AML has led to the realisation that the emergence of this disease is linked with mutations and aberrations in numerous genes controlling transcription, proliferation and apoptosis [14]. The hCDC4 gene has recently been identified as a new potent tumor suppressor as it has been shown to be responsible for the specific degradation of central oncogenes such as c-Jun, Notch, cyclin E and c-myc [3-6]. As these oncogenes have all been demonstrated to play roles in AML and MDS [15-18] and the hCDC4 gene features mutations in several solid tumors [7-11], we wanted to examine whether hCDC4 might also be mutated in DNA samples of AML.
As demonstrated here, direct sequencing of 35 DNA samples derived from patients with AML revealed one heterozygous mutation in the untranslated region of transcript variant 3 of exon 1 in form of a T > C exchange. A mutation of the hCDC4 gene at this location has not yet been described in previous mutation analyses. In contrast to this silent mutation detected here, mutations discovered previously in solid tumors such as colon or pancreas cancer had the tendency to be accumulated in exons 4, 8 and 9 and were of missense or nonsense type [7-11]. Therefore, the observation that only one heterozygous mutation with no consequence on the translated protein was found while screening 14 exons of 35 patient samples suggests that mutations of hCDC4 do not play a significant role in the pathogenesis of AML.
During sequence analysis of exon 10 in the AML samples we identified a new intronic SNP which we subsequently confirmed by sequencing DNA samples of 51 healthy individuals. Interestingly, this SNP had not yet been registered in any SNP databases despite of its high frequency of 20% in samples of patients with AML and 13,7% in healthy individuals. After its confirmation by re-sequencing and digestion of selected samples with mismatch specific Surveyor nuclease we therefore submitted the new variation to dbSNP (NCBI). The difference of genotype frequency between AML patients and healthy individuals is not significant (p=0.31) and therefore most probably not disease specific.
In conclusion we provide first data that mutations of the hCDC4 gene may not play a pivotal role in the pathogenesis of AML. This supplements studies which have discovered hCDC4 mutations in subsets of solid tumors and implies that mutations of hCDC4 are restricted to certain tumor types. Furthermore, we describe a new SNP with high frequency in the hCDC4 gene on chromosome 4, position 153602874.
Acknowledgements
This work was supported by the "Gutermuth-Foundation".
Conflicts of interest
The authors have declared that no conflict of interest exists.
References
1. Minella AC, Clurman BE. Mechanisms of tumor suppression by the SCF(Fbw7). Cell Cycle. 2005 ;4(10):1356-9
2. Fuchs SY. Tumor suppressor activities of the Fbw7 E3 ubiquitin ligase receptor. Cancer Biol Ther. 2005 ;4(5):506-8
3. Strohmaier H, Spruck CH, Kaiser P, Won KA, Sangfelt O, Reed SI. Human F-box protein hCdc4 targets cyclin E for proteolysis and is mutated in a breast cancer cell line. Nature. 2001 ;413(6853):316-22
4. Yada M, Hatakeyama S, Kamura T, Nishiyama M, Tsunematsu R, Imaki H, Ishida N, Okumura F, Nakayama K, Nakayama KI. Phosphorylation-dependent degradation of c-Myc is mediated by the F-box protein Fbw7. Embo J. 2004 ;23(10):2116-25
5. Nateri AS, Riera-Sans L, Da Costa C, Behrens A. The ubiquitin ligase SCFFbw7 antagonizes apoptotic JNK signaling. Science. 2004 ;303(5662):1374-8
6. Tsunematsu R, Nakayama K, Oike Y, Nishiyama M, Ishida N, Hatakeyama S, Bessho Y, Kageyama R, Suda T, Nakayama KI. Mouse Fbw7/Sel-10/Cdc4 is required for notch degradation during vascular development. J Biol Chem. 2004 ;279(10):9417-23
7. Kemp Z, Rowan A, Chambers W, Wortham N, Halford S, Sieber O, Mortensen N, von Herbay A, Gunther T, Ilyas M, Tomlinson I. CDC4 mutations occur in a subset of colorectal cancers but are not predicted to cause loss of function and are not associated with chromosomal instability. Cancer Res. 2005 ;65(24):11361-6
8. Rajagopalan H, Jallepalli PV, Rago C, Velculescu VE, Kinzler KW, Vogelstein B, Lengauer C. Inactivation of hCDC4 can cause chromosomal instability. Nature. 2004 ;428(6978):77-81
9. Spruck CH, Strohmaier H, Sangfelt O, Muller HM, Hubalek M, Muller-Holzner E, Marth C, Widschwendter M, Reed SI. hCDC4 gene mutations in endometrial cancer. Cancer Res. 2002 ;62(16):4535-9
10. Cassia R, Moreno-Bueno G, Rodriguez-Perales S, Hardisson D, Cigudosa JC, Palacios J. Cyclin E gene (CCNE) amplification and hCDC4 mutations in endometrial carcinoma. J Pathol. 2003 ;201(4):589-95
11. Moberg KH, Bell DW, Wahrer DC, Haber DA, Hariharan IK. Archipelago regulates Cyclin E levels in Drosophila and is mutated in human cancer cell lines. Nature. 2001 ;413(6853):311-6
12. Perez-Losada J, Mao JH, Balmain A. Control of genomic instability and epithelial tumor development by the p53-Fbxw7/Cdc4 pathway. Cancer Res. 2005 ;65(15):6488-92
13. Qiu P, Shandilya H, D'Alessio JM, O'Connor K, Durocher J, Gerard GF. Mutation detection using Surveyor nuclease. Biotechniques. 2004 ;36(4):702-7
14. Steffen B, Muller-Tidow C, Schwable J, Berdel WE, Serve H. The molecular pathogenesis of acute myeloid leukemia. Crit Rev Oncol Hematol. 2005 ;56(2):195-221
15. Iida H, Towatari M, Tanimoto M, Morishita Y, Kodera Y, Saito H. Overexpression of cyclin E in acute myelogenous leukemia. Blood. 1997 ;90(9):3707-13
16. Rangatia J, Vangala RK, Singh SM, Peer Zada AA, Elsasser A, Kohlmann A, Haferlach T, Tenen DG, Hiddemann W, Behre G. Elevated c-Jun expression in acute myeloid leukemias inhibits C/EBPalpha DNA binding via leucine zipper domain interaction. Oncogene. 2003 ;22(30):4760-4
17. Staber PB, Linkesch W, Zauner D, Beham-Schmid C, Guelly C, Schauer S, Sill H, Hoefler G. Common alterations in gene expression and increased proliferation in recurrent acute myeloid leukemia. Oncogene. 2004 ;23(4):894-904
18. Luo H, Li Q, O'Neal J, Kreisel F, Le Beau MM, Tomasson MH. c-Myc rapidly induces acute myeloid leukemia in mice without evidence of lymphoma-associated antiapoptotic mutations. Blood. 2005 ;106(7):2452-61
Author contact
![]() Correspondence to: Daniel Nowak, MD, Charité, Campus Benjamin Franklin, Department of Hematology, Oncology and Transfusion Medicine, Hindenburgdamm 30, 12203 Berlin, Germany. Phone: +49 (0)30 8445 2931 Fax: +49 (0)30 8445 4468 Email: daniel.nowakde
Correspondence to: Daniel Nowak, MD, Charité, Campus Benjamin Franklin, Department of Hematology, Oncology and Transfusion Medicine, Hindenburgdamm 30, 12203 Berlin, Germany. Phone: +49 (0)30 8445 2931 Fax: +49 (0)30 8445 4468 Email: daniel.nowakde