Impact Factor ISSN: 1449-1907
- Issue 9; 2026
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Volume 23; 2026
- Past Issues
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
1. Introduction
2. PARP-1 inhibitors as...
3. Can PARP-1 inhibitors...
4. Can PARP-1 inhibitors...
5. PARP inhibition remains...
6. Summary
Acknowledgements
Conflicts of interest
References

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2006; 3(4):117-123. doi:10.7150/ijms.3.117 This issue Cite
Review
PARP-1 inhibitors: are they the long-sought genetically specific drugs for BRCA1/2-associated breast cancers?
Joseph A. De Soto, Chu-Xia Deng ![]()
Genetics of Development and Diseases Branch, National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health, 10/9N105, 10 Center Drive, Bethesda, MD 20892, USA.
Received 2006-6-6; Accepted 2006-7-12; Published 2006-7-15
Abstract
Recent studies demonstrated that PARP-1 [poly(ADP-ribose) polymerase-1] inhibitors kill breast cancer associated gene-1 and –2 (BRCA1/2) deficient cells with extremely high efficiency while BRCA+/- and BRCA+/+ cells are relatively non-responsive to the treatment. It was therefore proposed that PARP-1 inhibitors might be the long-sought genetically specific drugs that are both safe and effective for treating BRCA1/2-associated breast cancers. However, a report published in a recent issue of the International Journal of Biological Sciences revealed that PARP-1 inhibitors, although able to kill naïve BRCA1 mutant cells with high specificity both in vitro and in vivo, exhibit minimal specificity in inhibiting the growth of mouse mammary tumor cells irrespective of their BRCA1 status in allograft nude mice. Non-specific inhibition in human BRCA1+/+, BRCA1+/-, and BRCA1-/- breast cancer cells by PARP-1 inhibitors was also observed. Additional mutations occurring during cancer progression may be a culprit, although the exact cause for the resistance of BRCA1-/- breast cancer cells to PARP-1 inhibitors remains elusive. These findings suggest that PARP inhibition may serve as an approach for the prevention of BRCA related breast cancer and may be useful in combination with other chemotherapeutic agents in the treatment of breast cancer.
Keywords: PARP inhibitors, BRCA1, breast cancer, therapeutic treatment, AG14361
1. Introduction
Breast cancer is the leading cause of cancer incidence and the second leading cause of cancer mortality in women. Familial breast cancer is a small subset of this disease contributing to about 5 to 10% of breast cancer cases. Germline mutations of the breast tumor suppressor genes BRCA1 and BRCA2 have been found to contribute to most of the familial breast cancer cases [1-5]. BRCA1 was mapped in 1990 [6] and cloned in 1994 [7]. Numerous studies in the past decade have revealed that BRCA1-deficiency results in genetic instability, primarily due to centrosome amplification, defective cell cycle checkpoint, and impaired DNA damage repair (reviewed in [8-15]). Hence, BRCA1 is essential for maintaining genome integrity while genetic instability associated with BRCA1 deficiency is responsible for breast cancer formation [16, 17].
One of the major causes of the genetic instability associated with BRCA1 deficiency is that BRCA1 mutant cells have an impaired ability to undergo homologous recombination (HR) [12, 14, 18-20], and therefore cannot effectively repair HR-mediated DNA damage, such as DNA double strand breaks (DSBs). In the absence of BRCA1, DSBs can be gradually accumulated, which may result in the activation of oncogenes and inactivation of tumor suppressor genes, eventually leading to tumor formation. On the other hand, defective DNA damage repair may also make BRCA1 deficient cancer cells more sensitive to DNA damaging agents, such as irradiation, Mitomycin C (MMC), and adriamycin [19, 21-23]. Thus, one of the therapeutic approaches to kill BRCA1 deficient cancer cells efficiently is treating these cells with DNA damaging reagents thereby introducing acute DNA damage. However, the majority DNA damage reagents are not specific and can cause the similar extent of damage in both BRCA1 mutant and wild type cells. More recently it was shown that PARP-1 inhibitors could kill BRCA1/2 deficient cells with high specificity bringing great promise to the BRCA1 cancer patient [24, 25]. Here we will review the current progress of PARP-1 inhibitors in the chemoprevention and therapeutic treatment of BRCA1/2-associated breast cancers and discuss possible options for using these drugs efficiently.
2. PARP-1 inhibitors as potential chemotherapeutic drugs
Using a clonogenic survival assay, Bryant et al. (2005) found that a BRCA2-deficient cell line V-C8, compared with the BRCA2 wild type control, exhibited extreme sensitivity to AG14361, a very potent PARP-1 inhibitor (KI=5nM), and NU1025, a moderately potent PARP-1 inhibitor (KI =50nM). NU1025 treatment also profoundly reduced colony formation (up to 100 fold) of MCF-7 and MDA-MB-231 cells when BRCA2 was depleted by RNAi compared with untreated control cells [24]. Studying mouse embryonic stem (ES) cells carrying targeted mutations of BRCA1 (homozygous deletion of exons 22-24, referred to as BRCA1-/-) or BRCA2 (trEx11/ΔEx27, referred to as BRCA2-/-), Farmer et al. (2005) revealed similar sensitivity of these cells to PARP-1 inhibitors in vitro [25].
Will this work in vivo? To investigate this, both groups transplanted BRCA2-deficient ES cells [25] or V-C8 cells [24] into athymic nude mice and treated the xenografted tumors with PARP-1 inhibitors. Their data revealed that PARP-1 inhibition completely blocked the growth of BRCA2-deficient tumors, but it had no effect on BRCA2-wild type tumors. It was subsequently suggested that PARP-1 inhibition might represent a new concept in cancer treatment for BRCA1/2 mutation carriers.
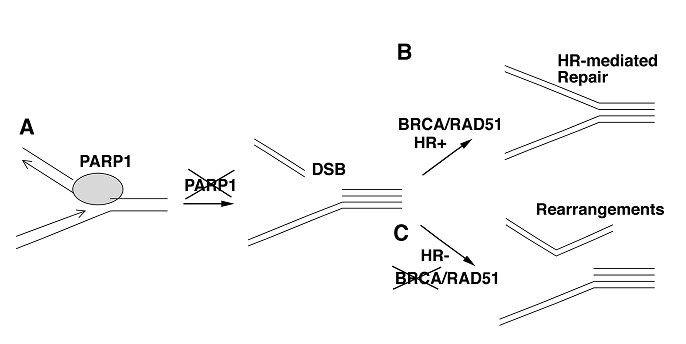
Why are BRCA1- and BRCA2-deficient cells extremely sensitive to PARP-1 inhibition? The products of the genes, BRCA1, BRCA2 and PARP-1, are nuclear proteins and are critical components of the cellular response to DNA damage [12, 14, 18-20, 26-28]. PARP-1 binds to and efficiently repairs DNA single strand breaks (SSBs) formed during base excision repair (BER) of small DNA adducts induced by alkylating agents or ionizing radiation [29]. It has been shown that PARP-1 inhibition leads to persistent single strand breaks (SSBs) in DNA [27], which result in formation of double strand breaks (DSBs) when they meet at replication forks (Fig. 1A). In the presence of wild type BRCA1 and BRCA2, DSBs can be efficiently repaired by RAD51 mediated homologous recombination (Fig. 1B). It has been shown that BRCA1 and BRCA2 interact with RAD51, and play essential role in homologous recombination [28, 30, 31]. Thus, in the event of DNA damage associated with PARP inhibition, BRCA1 or BRCA2 mutant cells cannot repair DNA DSBs properly, leading to the collapse of replication forks, and illegitimated DNA ends joining, consequently leading to growth arrest and apoptosis (Fig. 1C). This model provides a molecular basis for the reason that why BRCA1- and BRCA2-deficient cells are extremely sensitive to PARP-1 inhibition.
A model showing DSB formation and DNA rearrangement in the absence of BRCA and PARP1. (A) PARP-1 binds and repairs single strand breaks (SSBs) in DNA. These SSBs result in formation of double strand breaks (DSBs) when they meet replication forks. (B) In the presence of wild type BRCA1 and BRCA2, DSBs can be efficiently repaired by RAD51 mediated homologous recombination (HR). (C) BRCA1 or BRCA2 mutant cells cannot repair DNA DSBs properly upon the inhibition of PARP, leading to DNA replication arrest and illegitimated DNA ends joining.
3. Can PARP-1 inhibitors specifically kill human BRCA2-deficient pancreatic cancer cells?
Because PARP-1-null mice are viable and healthy [32], and inhibitors that are specific for PARP-1 are relatively non-toxic and do not directly damage DNA, PARP-1 inhibitors might be both safe and effective for BRCA1/2-associated breast cancers. However, it is noticed that these data were obtained by using non-cancerous and genetically naïve BRCA1/2-/- ES cells, BRCA2-/- V-C8 hamster cells, and two BRCA2-proficient breast cancer cell lines, MCF-7 and MDA-MB-231, that carried acute suppression of BRCA2 by siRNA. Cancer cells originated from naturally occurring BRCA1/2 mutations were not tested. To investigate this, Gallmeier and Kern (2005) treated a cell line CAPAN1, which harbors the frequent, naturally occurring BRCA2 6174delT frameshift mutation accompanied by loss of the second allele with NU1025 and 3-aminobenzamide (3AB), a weak PARP-1 inhibitor with the KI=500nM [33]. Their data indicated that PARP inhibition by these drugs did not result in significant cell death of CAPAN1 cells. This observation casts doubt about the uniformity of the specific cell killing of BRCA2-deficient cells upon PARP inhibition. This observation also urged caution as to whether prior findings were fully generalizable for the specific treatment of human cancers harboring naturally occurring, inactivating forms of BRCA2 mutations.
The finding that CAPAN1 cells are relatively insensitive to NU1025 was quickly confirmed [34]. McCabe et al. (2005) compared sensitivity of the CAPAN1 cells to 4 PARP inhibitors, including NU1025, KU0051529 (a weak PARP inhibitor, IC50 730 nM) and two very potent PARP inhibitors, KU0058684 (IC50 3.2 nM) and KU0058948 (IC50 3.4 nM). Their data showed that the CAPAN1 cells were indeed extremely sensitive to the two potent PARP inhibitors although they were much less sensitivity to NU1025 and KU0058948. These results indicate that treatment with potent PARP inhibitors might still be an exciting potential therapy for cancers involving BRCA2 deficiency [34].
However, the puzzle has not been fully solved. NU1025 was previously reported to kill BRCA2-depleted MCF-7 and MDA-MB-231 cells with extremely high efficiency, i.e. 100-fold reduction in colony formation at about 10 μM of NU1025 [24]. In contrast, in CAPAN1 cells, this dose of NU1025 [34] and even at much higher concentrations up to 200 μM [33] failed to exhibit any specific killing. While the reason for this discrepancy remains unclear, it was proposed that genetic alterations during tumor progression due to genetic instability associated with BRCA1- and BRCA2-deficiency might be a culprit [33]. In other words, some specific mutations might block the sensitivity of BRCA1/2-/- cancer cells to PARP-1 inhibition.
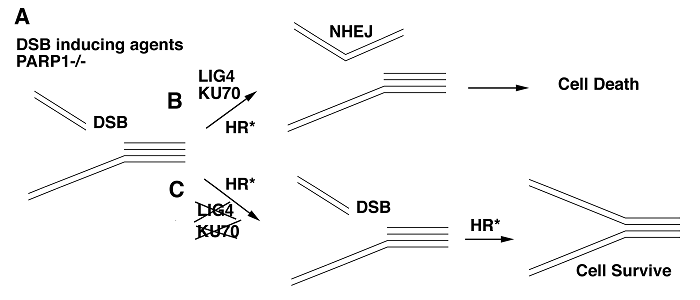
Consistent with this, it was shown recently that the chicken DT40 PARP-1-/- mutant cells displayed reduced levels of homologous recombination (HR) and were sensitive to various double strand break (DSB)-inducing genotoxic agents [35]. Interestingly, when either Ku-70 or Ligase IV was also mutated, the PARP-1-/- cells became resistant to the killing by DSB-inducing drugs. The underlying mechanism is that the absence of Ku70 or ligase IV prevents illegitimated DNA ends ligation of DNA fragments by non-homologous mediated end joining (NHEJ) in PARP-1-/- cells, therefore, preventing their lethality (Fig. 2). This study provides an example that lethality associated with DSBs and PARP-1 inhibition could indeed be rescued by specific types of mutations.
A model showing how Ku70 or ligase IV deficiency represses lethality caused by DSB inducing agents in PARP-1-/- DT40 cells. (A) Treatment of DSB inducing agents in PARP-1-/- DT40 cells decreases ability of HR (HR*) and causes DSBs. (B) Ku-70 protein competes with HR machinery and binds to DSBs, directing the repair toward the non-homologous end joining (NHEJ), where ligase IV (LIG4) also plays an important role. This results in lethality of PARP-1-/- cells. (C) The absence of Ku70 or Ligase IV prevents illegitimated DNA end ligation of DSBs through NHEJ in PARP-1-/- cells. This allows more time for PARP-1 deficient cells to repair DSBs, therefore, preventing their lethality.
4. Can PARP-1 inhibitors specifically kill BRCA1 mutant breast cancer cells?
These observations cast significant doubts on the effectiveness and specificity of PARP inhibition on BRCA associated tumorigenesis. Furthermore, previous in vivo studies only tested two cell lines, BRCA2 mutant ES cells and C-V8 cells while BRCA1 mutant breast cancer cells were not used. To provide a study with more clinical relevance, we compared sensitivity to PARP inhibition of 12 cell lines, including ES cells, mouse mammary tumor cells, and human breast cancer cells using three PARP inhibitors: AG14361, NU1025, and 3-aminobenzamide (3AB) [36].
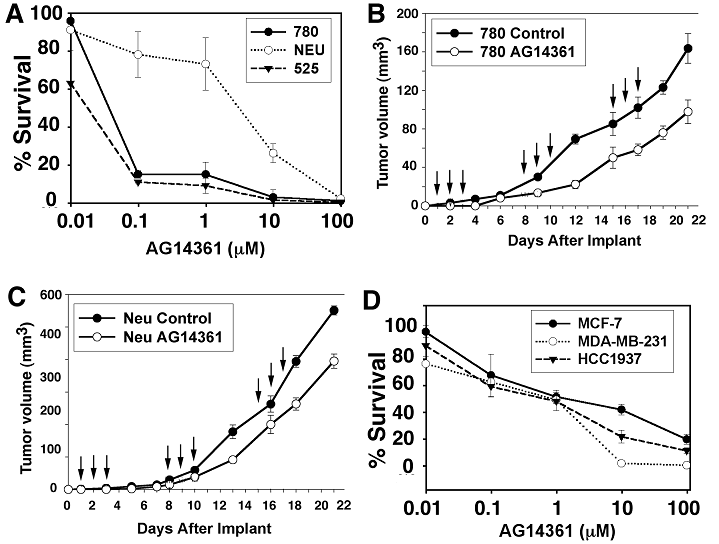
We first tested a BRCA1 mutant ES cell line [BRCA1trEx11/Δ11 that carries one BRCA1 mutant allele truncated at exon 11 [37] and one allele with a deletion of exon 11 by a Cre-LoxP approach [38], referred to as BRCA1-/-]. Our data revealed that the BRCA1-/- cells were significantly more sensitive to AG14361 treatment than BRCA1+/- and BRCA1+/+ ES cells both in vitro and in vivo. Indeed, AG14361 treatment completely blocked tumor growth in 90% nude mice that had received transplantation of BRCA1-/- ES cells. Only one tumor that escaped PARP inhibition grew, though much slower compared with tumors developed in the nude mice that had received transplanted BRCA1+/+ ES cells. Next, we tested sensitivity to PARP inhibition using three mammary tumor cell lines that are either hypomorphic (BRCA1Δ11/Δ11) or null (BRCA1-/-) for BRCA1. In the colonogenic assay, we found that all BRCA1-/- cell lines were much more sensitive to the potent PARP inhibitor AG14361 than BRCA1+/+ tumor cells (Fig. 3A). However, AG14361 treatment exhibited only mild specificity in mouse BRCA1-/- mammary cancer cells (Fig. 3B) in comparison with their controls (Fig. 3C) in the xenografted nude mice. These observations indicated that BRCA1-/- mammary cancer cells are indeed much more resistant to PARP inhibition than BRCA1-/- ES cells in the allograft model. Finally, we studied response to PARP- inhibition of four human breast cancer cell lines: MCF-7 (BRCA1+/+), MDA-MB-231 (BRCA1+/-), HCC1937 (BRCA1-/-) and SUM1315MO2 (BRCA1-/-). Using both clonogenic survival assay and MTT assay, we found that PARP-1 inhibitors seem to have a similar inhibition effects on their growth regardless of the BRCA1 genotypes (Fig. 3D) [36].
Effect of a potent PARP-1 inhibitor, AG14361, on BRCA1 mutant and control cancer cells. (A) Clonogenic survival assay. Five hundred 780 (BRCA1Δ11/Δ11), and 525 (BRCA1-/-), and NEU (BRCA1+/+) cells were exposed to AG14361 for 10 consecutive days, 18 hours after plating. (B,C) Response of in vivo allograft of BRCA1-/- (B), and BRCA1+/+ (C) mammary tumor cells to AG14361. The recipient nude mice were treated intra-peritoneally with AG14361 (30 mg/kg) 24 hours after transplantation of 1 X 106 cells for three consecutive days starting on days 2, 9 and 16 (arrows). Our data indicated that the treatment failed to achieve a more than marginal specific killing of BRCA1 mutant cancer cells as both BRCA1-/- and BRCA1+/+ cells showed about a 30-40% reduction in growth. (D) Clonogenic assay. MCF-7, MDA-MB-231 and HCC1937 cells were treated and exposed to AG14361 continuously for 10 days.
We believe that the non-cancerous cells are generally naïve, whereas tumor cells represent cells that have had multiple genetic mutations and that in a tumor there may be numerous fundamentally different cancer cells when classified by the type and frequency of individual genomic changes. It has been shown that tumorigenesis involves alterations of at least 6 major biological pathways/processes [39]. Indeed, our work in BRCA1 mutant mouse models have also revealed numerous genetic/molecular alterations in BRCA1 mutant tumors, and have demonstrated that BRCA1-associated tumorigenesis has been selected to survive genomic changes [21, 22, 38, 40-43]. This may account for the differential resistance to PARP inhibition observed between BRCA mutant cancer cells and pre-cancerous cells [36].
5. PARP inhibition remains promising in chemoprevention and therapeutic treatment for BRCA related breast cancer
Clearly, a common finding among above studies is that BRCA-/- pre-cancerous cells are very sensitive to PARP inhibition than wild type cells while cancer cells exhibit different response to PARP inhibition under different conditions [24, 25, 33, 34, 36]. Animal studies showed that BRCA1- and BRCA2-deficient embryos die during embryonic development [37, 44-46]. Adults with BRCA1/2 associated hereditary breast cancer inherit one mutated allele through their germline and later suffer loss of heterozygosity of the wild type allele in order for breast cancer to arise. There must be at least one previous mutation in another gene however, that is permissive, for viability of the cell in the event of a second BRCA mutation. Current data suggest that ATM, CHK2 and p53 may be candidate genes for these permissive mutations as impaired functions of these genes, either at heterozygous or homozygous forms, attenuates apoptosis induced by BRCA1 deficiency and allows mutant embryos survive to adulthood [40, 42]. The extended survival of BRCA deficient cells allows for further genomic alterations leading to the eventual development of cancer.
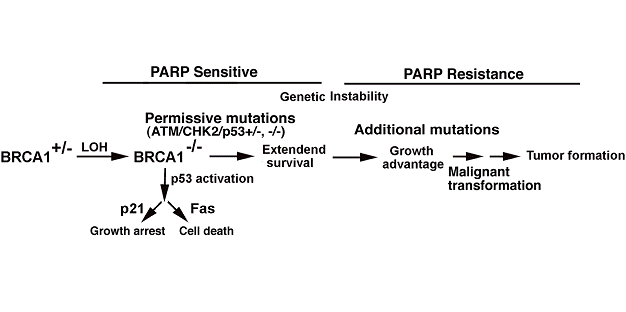
Thus, we propose that the BRCA associated tumorigenesis may be divided into two phases regarding sensitivity to PARP inhibition (Fig. 4). In the early stages of tumorigenesis, BRCA1 mutant cells are sensitive to PARP inhibition. Evidence indicates that treatment of BRCA2-deficient small intestine with a potent PARP-1 inhibitor results in high levels of apoptosis 6-12 hours after treatment [47]. It was shown that p53 deficiency in MDA-MB-231 cells did not render resistance of these cells to PARP inhibition upon siRNA mediated deletion of PARP1 [24]. This suggests that BRCA deficient cells that have recently received a permissive mutation, such as p53, might still be sensitive to PARP-1 inhibitors in this pre-cancerous state.
A model depicting BRCA1 associated tumorigenesis in regarding to the development of PARP resistance. In familial cancer, one mutant BRCA1 allele is inherited from the germline. One additional mutation in the BRCA1 locus (LOH) results in a BRCA1 deficiency, which could result in genetic instability, activation of p53-dependent cell cycle checkpoints and apoptosis pathways, leading to growth arrest and cell death. However, genetic instability triggered by BRCA1 loss could facilitate mutations in general. When permissive mutations, such as ATM, CHK2 or p53 heterozygous or homozygous mutations, occur, BRCA1-/- cells survive. Cells in this phase may be sensitive to PARP inhibition. However, PARP resistance may develop when further genetic alterations occur leading to full growth of breast cancers.
However, in the second stage of tumorigenesis, BRCA deficient tumor cells may become insensitive to PARP inhibition due to additional mutations (Fig. 4). In this regard, we showed that HCC1937 (5832insC) and SUM1315M02 (185delAG), which both produce a truncated and inactive form of BRCA-1 which together account for up to 85% of BRCA-1 pathogenic mutations in humans, were not any more sensitive to PARP-1 inhibitors than wild type or heterozygous mammary tumor cell lines.
Despite a difficulty to identify mutations that render the resistance of BRCA cancer cells to PARP inhibition, there is evidence to suggest that PARP inhibitors may be useful in treating tumors in combination with cytotoxic drugs by increasing the sensitivity of the tumors to these agents. Indeed, it has been shown that PARP-1 inhibitors not only significantly enhance the anti-tumor efficacy of the alkylating agent temozolomide (TMZ) in solid or hematological tumors, but also, restore the sensitivity to TMZ in resistant tumors [48, 49]. In addition, PARP-1 inhibitors have also been shown to similarly sensitize tumors cells to the topoisomerase I inhibitor, irinotecan in colon carcinomas [50, 51]. Furthermore, unpublished data from this lab suggest that PARP-1 inhibitors may also show synergism with some anti-metabolites and anti-mitotic agents. PARP-1 inhibitors also are attractive agents based on what seems to be not only few side effects but also a protective effect in normal tissue. Indeed, reports from clinical trials using PARP-1 inhibitors have successfully completed phase I studies and entered phase II studies for various ischemic disorders [52]. Furthermore, PARP-1 inhibitors seem to protect against the cardiotoxicity of doxorubicin and the nephrotoxicity of cisplatin [53, 54]. The use of PARP-1 inhibitors in combination with standard chemotherapeutic agents also, seems attractive in the sense that by sensitizing tumor cells to cytotoxic agents one might be able to give a lower dose and maintain the same relative efficacy while at the same time reducing the toxic side effects.
6. Summary
We have reviewed the current data suggesting that PARP inhibition may serve as potent approach for prevention of BRCA related breast cancer. However, the use of this strategy for therapeutic treatment for hereditary breast cancers is dependent on the continued susceptibility of BRCA mutant cells to PARP inhibitors, which may be achieved by using a combination with other reagents. The future use of PARP-1 inhibitors as safe and efficacious agents in preventing the hereditary form of breast cancer and as sensitizing agents in the treatment of breast cancer seems bright. Clinical trials are needed to elucidate the best use of PARP inhibitors in combination of other therapeutic drugs in reducing the morbidity and mortality of breast cancers.
Acknowledgements
We thank S. Prindiville and members of Deng lab for critical discussion. This work was supported by the intramural Research Program of National Institute of Diabetes, Digestive and Kidney Diseases, National Institutes of Health, USA.
Conflicts of interest
The authors have declared that no conflict of interest exists.
References
1. Garber JE, Offit K. Hereditary cancer predisposition syndromes. J Clin Oncol. 2005 ;23:276-92
2. Brody LC, Biesecker BB. Breast cancer susceptibility genes. BRCA1 and BRCA2. Medicine (Baltimore). 1998 ;77:208-26
3. Ellisen LW, Haber DA. Hereditary breast cancer. Annu Rev Med. 1998 ;49:425-36
4. Paterson JW. BRCA1: a review of structure and putative functions. Dis Markers. 1998 ;13:261-74
5. Eccles DM, Pichert G. Familial non-BRCA1/BRCA2-associated breast cancer. Lancet Oncol. 2005 ;6:705-11
6. Hall JM, Lee MK, Newman B. et al. Linkage of early-onset familial breast cancer to chromosome 17q21. Science. 1990 ;250:1684-9
7. Miki Y, Swensen J, Shattuck-Eidens D. et al. A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science. 1994 ;266:66-71
8. Venkitaraman AR. Cancer susceptibility and the functions of BRCA1 and BRCA2. Cell. 2002 ;108:171-82
9. Deng CX. Tumor formation in Brca1 conditional mutant mice. Environ Mol Mutagen. 2002 ;39:171-7
10. Brodie SG, Deng CX. BRCA1-associated tumorigenesis: what have we learned from knockout mice? Trends Genet. 2001 ;17:S18-22
11. Deng CX, Brodie SG. Roles of BRCA1 and its interacting proteins. Bioessays. 2000 ;22:728-37
12. Deng CX, Wang RH. Roles of BRCA1 in DNA damage repair: a link between development and cancer. Hum Mol Genet. 2003 ;12:R113-23
13. Deng CX. Roles of BRCA1 in centrosome duplication. Oncogene. 2002 ;21:6222-7
14. Zhang J, Powell SN. The role of the BRCA1 tumor suppressor in DNA double-strand break repair. Mol Cancer Res. 2005 ;3:531-9
15. Cantor SB, Andreassen PR. Assessing the link between BACH1 and BRCA1 in the FA pathway. Cell Cycle. 2006 ;5:164-7
16. Deng CX. Tumorigenesis as a consequence of genetic instability in Brca1 mutant mice. Mutat Res. 2001 ;477:183-9
17. Deng CX. BRCA1: cell cycle checkpoint, genetic instability, DNA damage response, and cancer evolution. Nucleic Acids Res. 2006 ;34:1416-26
18. Snouwaert JN, Gowen LC, Latour AM. et al. BRCA1 deficient embryonic stem cells display a decreased homologous recombination frequency and an increased frequency of non-homologous recombination that is corrected by expression of a brca1 transgene. Oncogene. 1999 ;18:7900-7
19. Moynahan ME, Cui TY, Jasin M. Homology-directed dna repair, mitomycin-c resistance, and chromosome stability is restored with correction of a Brca1 mutation. Cancer Res. 2001 ;61:4842-50
20. Xu X, Aprelikova O, Moens P. et al. Impaired meiotic DNA-damage repair and lack of crossing-over during spermatogenesis in BRCA1 full-length isoform deficient mice. Development. 2003 ;130:2001-12
21. Brodie SG, Xu X, Qiao W. et al. Multiple genetic changes are associated with mammary tumorigenesis in Brca1 conditional knockout mice. Oncogene. 2001 ;20:7514-23
22. Xu X, Weaver Z, Linke SP. et al. Centrosome amplification and a defective G2-M cell cycle checkpoint induce genetic instability in BRCA1 exon 11 isoform-deficient cells. Mol Cell. 1999 ;3:389-95
23. Kim SS, Cao L, Li C. et al. Uterus hyperplasia and increased carcinogen-induced tumorigenesis in mice carrying a targeted mutation of the Chk2 phosphorylation site in Brca1. Mol Cell Biol. 2004 ;24:9498-507
24. Bryant HE, Schultz N, Thomas HD. et al. Specific killing of BRCA2-deficient tumours with inhibitors of poly(ADP-ribose) polymerase. Nature. 2005 ;434:913-7
25. Farmer H, McCabe N, Lord CJ. et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature. 2005 ;434:917-21
26. Malanga M, Althaus FR. The role of poly(ADP-ribose) in the DNA damage signaling network. Biochem Cell Biol. 2005 ;83:354-64
27. Dantzer F, Schreiber V, Niedergang C. et al. Involvement of poly(ADP-ribose) polymerase in base excision repair. Biochimie. 1999 ;81:69-75
28. Wong AK, Pero R, Ormonde PA. et al. RAD51 interacts with the evolutionarily conserved BRC motifs in the human breast cancer susceptibility gene brca2. J Biol Chem. 1997 ;272:31941-4
29. Bryant HE, Helleday T. Poly(ADP-ribose) polymerase inhibitors as potential chemotherapeutic agents. Biochem Soc Trans. 2004 ;32:959-61
30. Scully R, Chen J, Plug A. et al. Association of BRCA1 with Rad51 in mitotic and meiotic cells. Cell. 1997 ;88:265-75
31. Chen J, Silver DP, Walpita D. et al. Stable interaction between the products of the BRCA1 and BRCA2 tumor suppressor genes in mitotic and meiotic cells [In Process Citation]. Mol Cell. 1998 ;2:317-28
32. Wang ZQ, Stingl L, Morrison C. et al. PARP is important for genomic stability but dispensable in apoptosis. Genes Dev. 1997 ;11:2347-58
33. Gallmeier E, Kern SE. Absence of Specific Cell Killing of the BRCA2-Deficient Human Cancer Cell Line CAPAN1 by Poly(ADP-ribose) Polymerase Inhibition. Cancer Biol Ther. 2005 ;4:703-6
34. McCabe N, Lord CJ, Tutt AN. et al. BRCA2-Deficient CAPAN-1 Cells are Extremely Sensitive to the Inhibition of Poly (ADP-Ribose) Polymerase: An Issue of Potency. Cancer Biol Ther. 2005 ;4:934-6
35. Hochegger H, Dejsuphong D, Fukushima T. et al. Parp-1 protects homologous recombination from interference by Ku and Ligase IV in vertebrate cells. EMBO J. 2006 ;25:1305-14
36. De Soto JA, Wang X, Tominaga Y. et al. The Inhibition and Treatment of Breast Cancer with Poly (ADP-ribose) Polymerase (PARP-1) Inhibitors. Int J Biol Sci. 2006 ;2:179-185
37. Shen SX, Weaver Z, Xu X. et al. A targeted disruption of the murine Brca1 gene causes gamma-irradiation hypersensitivity and genetic instability. Oncogene. 1998 ;17:3115-24
38. Xu X, Wagner KU, Larson D. et al. Conditional mutation of Brca1 in mammary epithelial cells results in blunted ductal morphogenesis and tumour formation [see comments]. Nat Genet. 1999 ;22:37-43
39. Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000 ;100:57-70
40. Xu X, Qiao W, Linke SP. et al. Genetic interactions between tumor suppressors Brca1 and p53 in apoptosis, cell cycle and tumorigenesis. Nat Genet. 2001 ;28:266-71
41. Weaver Z, Montagna C, Xu X. et al. Mammary tumors in mice conditionally mutant for Brca1 exhibit gross genomic instability and centrosome amplification yet display a recurring distribution of genomic imbalances that is similar to human breast cancer. Oncogene. 2002 ;21:5097-107
42. Cao L, Kim S, Xiao C. et al. ATM-Chk2-p53 activation prevents tumorigenesis at an expense of organ homeostasis upon Brca1 deficiency. EMBO J. 2006 ;25:2167-77
43. Cao L, Li W, Kim S. et al. Senescence, aging, and malignant transformation mediated by p53 in mice lacking the Brca1 full-length isoform. Genes Dev. 2003 ;17:201-13
44. Hakem R, de la Pompa JL, Sirard C. et al. The tumor suppressor gene Brca1 is required for embryonic cellular proliferation in the mouse. Cell. 1996 ;85:1009-23
45. Sharan SK, Morimatsu M, Albrecht U. et al. Embryonic lethality and radiation hypersensitivity mediated by Rad51 in mice lacking Brca2 [see comments]. Nature. 1997 ;386:804-10
46. Xu X, Li C, Garrett-Beal L. et al. Direct removal in the mouse of a floxed neo gene from a three-loxP conditional knockout allele by two novel approaches. Genesis. 2001 ;30:1-6
47. Hay T, Jenkins H, Sansom OJ. et al. Efficient deletion of normal Brca2-deficient intestinal epithelium by poly(ADP-ribose) polymerase inhibition models potential prophylactic therapy. Cancer Res. 2005 ;65:10145-8
48. Tentori L, Graziani G. Chemopotentiation by PARP inhibitors in cancer therapy. Pharmacol Res. 2005 ;52:25-33
49. Tentori L, Leonetti C, Scarsella M. et al. Systemic administration of GPI 15427, a novel poly(ADP-ribose) polymerase-1 inhibitor, increases the antitumor activity of temozolomide against intracranial melanoma, glioma, lymphoma. Clin Cancer Res. 2003 ;9:5370-9
50. Calabrese CR, Almassy R, Barton S. et al. Anticancer chemosensitization and radiosensitization by the novel poly(ADP-ribose) polymerase-1 inhibitor AG14361. J Natl Cancer Inst. 2004 ;96:56-67
51. Miknyoczki SJ, Jones-Bolin S, Pritchard S. et al. Chemopotentiation of temozolomide, irinotecan, and cisplatin activity by CEP-6800, a poly(ADP-ribose) polymerase inhibitor. Mol Cancer Ther. 2003 ;2:371-82
52. Graziani G, Szabo C. Clinical perspectives of PARP inhibitors. Pharmacol Res. 2005 ;52:109-18
53. Pacher P, Mabley JG, Soriano FG. et al. Activation of poly(ADP-ribose) polymerase contributes to the endothelial dysfunction associated with hypertension and aging. Int J Mol Med. 2002 ;9:659-64
54. Racz I, Tory K, Gallyas FJr. et al. BGP-15 - a novel poly(ADP-ribose) polymerase inhibitor - protects against nephrotoxicity of cisplatin without compromising its antitumor activity. Biochem Pharmacol. 2002 ;63:1099-111
Author contact
![]() Correspondence to: Dr. Chu-Xia Deng, Phone: (301) 402-7225, Fax: (301) 480-1135. Email: chuxiadniddk.nih.gov
Correspondence to: Dr. Chu-Xia Deng, Phone: (301) 402-7225, Fax: (301) 480-1135. Email: chuxiadniddk.nih.gov