Impact Factor ISSN: 1449-1907
- Issue 9; 2026
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Volume 23; 2026
- Past Issues
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
1. Cancer Vaccines
2. Immune Checkpoint Inhibitors
3. Adoptive Immune Cell Therapy
4. Oncolytic Virus Therapy
5. Common Challenges Shared...
6. Biomarkers: From...
7. Conclusions
Abbreviations
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2026; 23(6):2108-2138. doi:10.7150/ijms.132708 This issue Cite
Review
Current Status and Evolution of Immunotherapy in Glioma Management
Jiaying Liu1, Guangzhao Yang3, Zhengcong Cao2, Haozhe Qin2,3, Maorong Zhu2, Cheng Zou2, Xiao Liu2,3, Yalong He3 ![]() , Jintao Gu2
, Jintao Gu2 ![]()
1. Basic Medical College, The Fourth Military Medical University, Xi'an, 710000, China.
2. State Key Laboratory of Cancer Biology, Biotechnology Center, School of Pharmacy, The Fourth Military Medical University, Xi'an, 710000, China.
3. Department of Neurosurgery, Xijing Hospital, Xi'an, 710000, China.
Received 2026-2-5; Accepted 2026-4-9; Published 2026-5-1
Abstract

Gliomas, especially glioblastoma (GBM), are the most aggressive primary brain tumors, and their treatment faces multiple challenges. These challenges include incomplete surgical resection, the blood-brain barrier (BBB) that limits drug delivery, and an immunosuppressive tumor microenvironment (TME). Despite the "cold tumor" nature of gliomas, significant progress has been made in immunotherapy strategies. In this review, we focus on the following four strategies: 1) Cancer vaccines, especially personalized vaccines based on neoantigens, can activate specific immune responses; 2) Immune checkpoint inhibitors (ICIs) to reverse T cell exhaustion (e.g., anti-PD-1/PD-L1 antibodies); 3) Adoptive immune cell therapies, particularly chimeric antigen receptor T-cell (CAR-T) therapy, have shown encouraging results in preclinical trials; 4) Oncolytic viruses, which have dual roles of directly lysing tumor cells and stimulating immune responses. Immunotherapy offers a new paradigm to overcome the limitations of conventional glioma therapy. However, tumor heterogeneity, BBB limitation, target selection, and immunosuppressive microenvironment remain major obstacles. Future research should focus on developing combination therapies such as immunotherapy combined with radiotherapy, targeted therapy, exploring novel targets, and using biomarkers for patient stratification, which may help improve the survival outcomes of glioma patients. This review comprehensively evaluates the latest clinical progress of glioma immunotherapy, assesses the current limitations, and explores future directions.
Keywords: glioma, immunotherapy, immune checkpoint inhibitors, CAR-T cell therapy, cancer vaccine, oncolytic virus
Introduction
Glioma is a central nervous system tumor arising from glial cells. It is the most common and most aggressive primary brain tumor in adults [1]. The pathogenesis of glioma is complex, involving various genetic and molecular alterations [1]. The 2021 WHO Classification (5th edition) has shifted the classification of adult-type diffuse gliomas from a purely histologic grading system to an integrated paradigm that combines histologic features with molecular biomarkers [2]. These tumors are primarily categorized into three subtypes: 1) IDH-mutant astrocytoma: cases with CDKN2A/B homozygous deletion are directly assigned WHO grade 4, irrespective of classic histologic features such as microvascular proliferation or necrosis [3]; 2) IDH-mutant oligodendroglioma with 1p/19q codeletion [3]; 3) IDH-wildtype glioblastoma, which is strictly defined as IDH-wildtype grade 4 astrocytoma and represents the most malignant and aggressive subtype [2]. Furthermore, distinct genetic alterations are increasingly recognized to shape the tumor immune microenvironment and consequently modulate the response to immunotherapeutic interventions [4]. Although its incidence is relatively low, the median survival for GBM patients is only approximately 15 months, indicating a very poor prognosis [5].
Standard treatment includes surgical resection, radiotherapy, and temozolomide (TMZ) chemotherapy [6]. Additional therapeutic options include DNA damage response (DDR) inhibitors and targeted molecular therapies [7]. Despite these measures, patient outcomes remain poor, with high recurrence rates [7].
The poor prognosis stems from multiple factors. First, the highly invasive nature of glioma cells enables diffuse infiltration into normal brain tissue, precluding complete surgical resection [8]. Second, the BBB severely hinders effective drug delivery, limiting systemic therapy efficacy [5]. Third, the glioma microenvironment is highly immunosuppressive. It features abundant M2-polarized tumor-associated macrophages (TAMs), increased infiltration of regulatory T cells (Tregs) and myeloid-derived suppressor cells (MDSCs), exhausted cytotoxic T lymphocytes, and upregulation of multiple immune checkpoints [1]. These factors, together with metabolic suppression (e.g., tryptophan catabolism via the IDO pathway) and hypoxia-driven immunosuppression, collectively shape the "cold tumor" phenotype of gliomas [9]. Furthermore, glioma has a relatively low tumor mutational burden and high intra- and inter-tumoral heterogeneity. This further weakens the immune system's ability to recognize and eliminate the tumor [1]. Together, these factors present unique challenges, prompting the exploration of immunotherapy as a new strategy.
Despite inherent immunosuppression, immunotherapy for glioma is feasible [10]. Recent re-evaluation of central nervous system (CNS) immune privilege and the discovery of skull-meningeal channels and intracranial lymphatic vessels provide new perspectives on brain tumor immunology [10]. Studies show that inducing immunogenic cell death (ICD) can transform the glioma microenvironment from "cold" to "hot", promote Th17 cell migration to the tumor, and enhance immunotherapy efficacy [11]. Additionally, immune cell infiltration patterns in the TME correlate with patient prognosis. Stronger immune cell infiltration often predicts better survival [11]. These findings provide a theoretical basis for developing immunotherapies for glioma.
Current glioma immunotherapy mainly includes four classic strategies. Cancer vaccines introduce tumor-specific dendritic cells (DCs) or tumor-associated antigens (TAAs) to activate anti-tumor immune responses [12]. Personalized vaccines, especially those targeting neoantigens, show great potential [1]. ICIs like PD-1, PD-L1, and CTLA-4 inhibitors aim to reverse T cell exhaustion and reactivate anti-tumor immunity [1]. Based on the available literature, adoptive immune cell therapy has not yet demonstrated satisfactory clinical efficacy in glioma studies, and its safety profile requires further investigation [13-15]. However, breakthroughs in new therapies such as universal CAR-T, fourth-generation CAR-T, and novel-target CAR-T in preclinical research have brought hope for improving the prognosis of glioma patients. Oncolytic viruses have a dual mechanism: directly killing tumor cells and activating immune responses. They have made breakthrough progress in clinical trials [1]. This review comprehensively evaluates recent clinical progress in these four main immunotherapy strategies for glioma. It critically analyzes limitations and provides insights into future directions.
1. Cancer Vaccines
Cancer vaccines aim to induce the immune system to recognize and attack TAAs or tumor-specific antigens (TSAs) [12] (Figure 1). In early exploration (around the 1990s), the main type of tumor vaccine was the whole tumor cell vaccine [16]. They had manageable safety but limited clinical efficacy [16]; In the early 2000s, DC vaccines emerged. They are made by isolating monocytes from patients' blood, differentiating them into DCs in vitro, loading them with tumor antigens (e.g., cell lysates, synthetic peptides), and reinfusing them to activate specific T cells [17]. In a large Phase 3 trial of a dendritic cell vaccine, the median overall survival (OS) among 331 patients was 23.1 months. Among the 131 patients with MGMT promoter methylation, the 3-year survival rate reached 46.4%. These findings provide strong evidence supporting the survival benefit of the DC vaccine [16]; Around the 2010s, molecularly targeted peptide vaccines appeared. They use defined immunodominant epitope peptides to induce precise immune responses [18]. IMA950 is the first multi-peptide vaccine targeting glioblastoma to enter clinical trials [19]. In its first-in-human Phase I study, it successfully met the safety and immunogenicity endpoints, marking a transition in glioma vaccine development from cell-based approaches to precision molecular targeting [19]. Since 2015, personalized/neoantigen vaccines have developed rapidly. These are peptide-based personalized cancer vaccines composed of immunogenic mutant epitopes derived from the patient's specific glioma [20].
The figure illustrates the mechanisms of action of four types of vaccines: dendritic cell vaccines, peptide vaccines, DNA vaccines, and RNA vaccines. These vaccines promote the activation and maturation of immune cells by presenting tumor-associated antigens. Subsequently, the activated immune cells induce immunogenic cell death of tumor cells, which in turn leads to the release of a broader repertoire of tumor-associated antigens and neoantigens. The newly released antigens further recruit and activate additional immune cells and also promote the reversion of suppressive immune cells, thereby effectively reversing the immunosuppressive state within the TME.
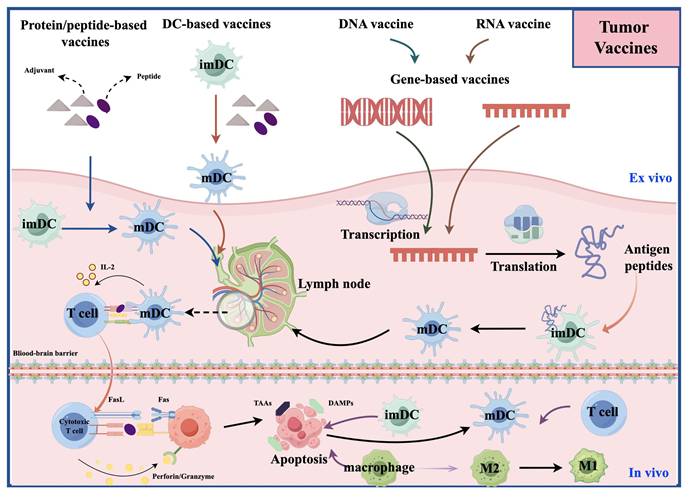
1.1 Dendritic Cell Vaccines
Based on literature from 2015-2025, 14 DC vaccine studies entered clinical trials. Comparative analysis preliminarily confirmed the safety and tolerability of DC vaccines [21-25], demonstrating good prospects in the field of glioma immunotherapy. Representative studies include 1) Wilms Tumor 1 (WT1) mRNA vaccine [21]; 2) autologous tumor antigen (ATA) vaccine [23, 24]; 3) ICT-107 [25]; 4) DC vaccines loaded with glioma stem cell (GSC) (A2B5+) antigen [26]; 5) DC vaccines targeting CMVpp65 [27-29]. First, in a single-arm Phase I/II study of advanced solid tumors, WT1 mRNA-electroporated autologous DCs (WT1-mRNA/DC) demonstrated promising therapeutic activity. The induced type 1 T-cell responses were associated with improved OS [21]. Second, the ATA vaccine's antigens come from self-renewing irradiated autologous tumor cells in culture. It contains personal neoantigens and any GBM TAAs, but not normal cell antigens, offering potential advantages over autologous tumor lysate [23]. However, its production requires sufficient tumor tissue, limiting use in advanced patients [23]. Finally, the efficacy and safety of ICT-107 and GSC antigen-loaded DC vaccines need further validation in larger trials [25, 26].
Specific pretreatment of DC vaccines (e.g., using tetanus-diphtheria (Td) toxoid [28, 29]) combined with adjuvants (e.g., keyhole limpet hemocyanin, KLH) [21] or cytokines (e.g., GM-CSF) [25], or using activators (e.g., IFNγ or lipopolysaccharide [25]), can enhance vaccine potency and patient immune responses to TSAs. This may improve patient outcomes [21, 25]. Many studies indicate that patients with O-6-methylguanine-DNA methyltransferase (MGMT) promoter methylation, combined Telomerase Reverse Transcriptase (TERT) promoter and Isocitrate Dehydrogenase 1/2 (IDH1/2) mutations, low B7-H4 expression, Human Leukocyte Antigen A2 (HLA-A2) positivity, and high immune responses are more sensitive to DC vaccines. This may be associated with better prognosis [25, 26, 30].
1.2 Molecularly Targeted Peptide Vaccines
Representative studies from 2015 to 2025 include 1) the Epidermal Growth Factor Receptor variant III (EGFRvIII) targeted vaccine (Rindopepimut/CDX-110) [31, 32]; 2) IDH mutation-targeted vaccine (IDH1-vac) [33]; 3) Survivin-targeting peptide vaccine (SurVaxM) [34, 35]; 4) and WT1 peptide vaccine [36] as single-target vaccines. Multi-target peptide vaccines include 1) IMA950 [19, 37]; 2) WT1 peptide cocktail vaccine [38]; 3) VEGFR1-VEGFR2 peptide vaccine [39]; 4) glioma-associated antigen (GAA) synthetic peptide vaccine [40].
1.2.1 Single-Target Vaccines
First, Rindopepimut is a single-target peptide vaccine against EGFRvIII. It consists of an EGFRvIII-specific peptide conjugated to KLH. In the ACT III trial, Rindopepimut successfully induced specific immune responses, achieving a 5.5-month PFS rate of 66%, significantly exceeding the preset null hypothesis of 53% (P=0.0168) [32]. In terms of safety, vaccine-related adverse events were common but generally tolerable [32]. However, the subsequent Phase III ACT IV trial showed that Rindopepimut did not improve survival in patients with newly diagnosed glioblastoma (nGBM) [31]. The ACT III trial demonstrated promising efficacy; however, the subsequent Phase III ACT IV trial failed to confirm these findings. Several factors may account for this discrepancy: 1) Patient selection bias. ACT III enrolled a highly selected population with favorable responses to initial therapy [32]. Comparing their survival outcomes with unselected historical controls likely overestimated the therapeutic benefit of the vaccine [32]. 2) Improvements in standard of care. In ACT IV, the median OS in the Rindopepimut arm (20.1 months) was consistent with that observed in ACT III, indicating stable vaccine activity [31]. However, advances in standard therapy likely prolonged survival in the control group relative to historical data, thereby diluting the relative benefit of Rindopepimut and rendering the difference statistically nonsignificant [31]. 3) Instability of target expression. In a small subset of ACT IV patients, EGFRvIII expression was lost at recurrence in approximately 60% of cases, irrespective of treatment received [31]. This finding suggests that single-target strategies against EGFRvIII may be limited and supports the exploration of multi-target approaches in future studies [31]. Second, the IDH1(R132H) peptide vaccine (IDH1-vac) showed good safety in its first trial, but the pseudoprogression rate was high, correlating with increased peripheral T cell responses [33]. Third, SurVaxM has good safety and tolerability [34, 41]. Clinically, benefit was observed in both methylated and unmethylated patients, with longer survival in the former. Notably, this clinical benefit was associated with upregulation of tumor-associated B cells, which predicted better outcomes [35]. However, it is essential to interpret such early-phase findings with caution. A well-documented challenge in GBM research is the frequent discrepancy between encouraging Phase I/II results and the outcomes of subsequent Phase III trials. Initial efficacy signals often fail to be reproduced in larger, more rigorous, and controlled settings, as exemplified by the Rindopepimut trials and other candidate immunotherapies in neuro-oncology [31, 32, 42, 43]. A large randomized trial of SurVaxM for nGBM (NCT02455557) was completed in March 2026, but the results have not yet been reported. Finally, the WT1 peptide (WT2725) vaccine is safe and tolerable but its efficacy needs confirmation [36].
1.2.2 Multi-Target Vaccines
First, IMA950 is a novel multi-target vaccine containing 11 tumor-associated peptides identified from HLA surface receptors in GBM tissue. It was used to induce antigen-specific peripheral CD8+ T cell responses in HLA-A*02-positive patients. In terms of safety, drug-related toxicity was relatively benign [19]. Second, combining TMZ with VEGFR1-VEGFR2 peptide vaccine showed synergistic activity [39]. Third, the GAA synthetic peptide vaccine targets IL-13Rα2, EphA2, WT1, and Survivin. The adjuvant poly-ICLC promotes type 1 polarization of T cell responses against these antigens, enabling effective T cell infiltration into brain tumors and therapeutic benefit [40]. Furthermore, patients with WHO grade II astrocytoma or oligoastrocytoma without prior non-surgical treatment may be particularly suitable for this vaccine [40]. Finally, the WT1 HLA class I/II peptide cocktail vaccine is safe in patients [38].
1.3 Personalized/Neoantigen Vaccines
Representative studies from 2015 to 2025 include 1) IDH1 mutation individualized peptide vaccine [20]; 2) personalized peptide vaccine (PPV) [44]; 3) Glioma Active Personalized Vaccine Consortium (GAPVAC) [45]; 4) autologous formalin-fixed tumor vaccine (AFTV) [46]. First, a clinical trial of a personalized neoantigen-targeting vaccine for IDH1-mutant glioma used whole-exome sequencing to customize treatment, which can cover tumor heterogeneity [20]. Results showed that the vaccine was well-tolerated and induced persistent T cell responses [20]. Moreover, patients with T cell responses to multiple neoantigens had a trend toward longer OS compared to those with no/few responses [20]. Additionally, detection of CDR3 sequences may help track CD4+ and CD8+ T cell clones in blood and tumor tissue [20]. However, this study was retrospective and non-randomized, with variability in the number of peptides administered and the timing of vaccinations. Therefore, the findings warrant further validation in prospective randomized controlled trials. Second, a PPV trial for HLA-A24-positive recurrent GBM (rGBM) found that SART2-93 positivity was a major factor limiting clinical benefit for all patients [44]. In contrast, biomarkers associated with better OS included lower proportions of CD11b+CD14+HLA-DRlow immunosuppressive monocytes, higher proportions of CD4+CD45RA- activated T cells, and medium levels of CCL2, VEGF, IL-6, IL-17, and haptoglobin [44]. Third, AFTV is a personalized vaccine derived from formalin-fixed autologous glioblastoma specimens [46]. Theoretically, it preserves all tumor antigens, including neoantigens, obviating the need for HLA-based patient selection [46]. However, this strategy has two major limitations: the inability to monitor therapeutic response via specific antibody titers, and the dependence of vaccine yield on the amount of resected tumor tissue, precluding flexible dose escalation or prolonged administration. A Phase IIb double-blind randomized trial showed no significant difference in efficacy between the vaccine and placebo groups in the overall population, likely due to the small sample size and the high quality of surgery and TMZ therapy received by the placebo group (median OS 31.5 months) [46]. Subgroup analyses suggested that patients with gross total resection or p53-negative tumors may be more sensitive to AFTV. This hypothesis is being evaluated in a completed Phase III trial (UMIN000010602), the results of which have not yet been reported [46]. Finally, the GAPVAC vaccine consists of APVAC1 (a pre-manufactured unmutated antigen repertoire) and APVAC2 (targeting neoepitopes) for HLA-A*02:01+ or HLA-A*24:02+ nGBM patients [45]. Results showed that APVAC1 induced sustained central memory CD8+ T cell responses, while APVAC2 induced Th1-type CD4+ T cell responses against predicted neoepitopes [45]. Furthermore, both vaccines, administered with poly-ICLC and GM-CSF adjuvants, were considered safe and highly immunogenic [45].
1.4 New Preclinical Advances
Recent advances focus on new delivery systems/pathways, optimized antigen selection, immune microenvironment modulation, and personalized vaccine design.
1.4.1 New Delivery Systems
Nanotechnology offers promising solutions. Optimizing particle size, surface modification, and targeting ligands significantly improves brain enrichment and immune activation [47-50]. Representative platforms include lipid nanoparticles (LNPs), polymer nanoparticles, and inorganic nanomaterials. A key LNP-based preclinical study is the CVGBM vaccine. CVGBM is a novel multi-targeted mRNA-LNP vaccine encoding a fusion protein comprising eight tumor-associated antigen-derived epitopes [48]. Its platform efficacy was validated in a surrogate melanoma mouse model, and antigen presentation specificity was confirmed in vitro [48]. However, immunopeptidomics analysis detected the presentation of only four of the eight encoded epitopes, while the remaining four were not identified [48]. Currently, a Phase I trial for HLA-A*02:01+ MGMT unmethylated GBM is ongoing (NCT05938387) [48]. Although several mRNA-LNP vaccines have entered clinical trials for infectious diseases and cancer, their clinical translation remains limited by poor targeting precision, potential immunogenicity and toxicity, and the difficulty of developing universal delivery systems [51]. Biomimetic nano-DC vaccines show improved biocompatibility, targeting, and immune activation [52]. For example, conjugating biomimetic DC membranes with tumor-associated costimulatory molecules or paclitaxel nanoparticles, combined with ICIs, alleviated T cell exhaustion in mouse models, significantly extended survival, potentially overcoming GBM immunotherapy resistance [53, 54]. However, the clinical translation of cell membrane-coated nanoparticle technology is hindered by challenges in scalable production and purification, safety assurance, quality control, and analytical method development [52].
1.4.2 Delivery Route Selection and Optimization
Vaccine administration route, frequency, and timing significantly impact efficacy and require systematic optimization. 1) The intranasal route is an innovative strategy [49]. It bypasses the BBB, delivering vaccine directly to the brain and cervical lymph nodes [49]. It reduces systemic exposure and has good patient compliance [49]. Sequential targeted sonodynamic nanovaccine (Stars NV) [55], Survivin peptide-CpG nanovaccines (SPOD-NV) [49], and enhanced nanovaccines using biomineralizing virus-like particles [50] are delivered through the nasal cavity, with drug enrichment observed in the brain and lymph nodes [49, 50, 55]. 2) Intravenous delivery is an established technique, allows high dosing, and activates immune cells in systemic lymphoid organs [49]. Serum IgG titers were higher after intravenous delivery (IV) than after intranasal delivery [49]. However, challenges include poor brain/tumor penetration, rapid clearance by liver macrophages, potential cytokine storm, and systemic toxicity risk [49]. A sequential dosing strategy for SPOD-NV vaccine used 5 full IV, 5 full intranasal, or 3 intranasal + 2 IV injections, all combined with anti-CTLA-4 antibody [49]. Mice with full IV dosing had significantly longer median survival than full intranasal [49]. The 3 intranasal + 2 IV group extended survival to 41 days, with a 43% complete response rate [49]. This suggests combining intranasal and IV routes may synergize, producing local and systemic immunity, improving efficacy [49]. 3) Subcutaneous delivery allows antigen uptake by subcutaneous DCs, which migrate to regional lymph nodes [49]. In the SPOD-NV study, subcutaneous + IV sequential dosing was significantly less effective than intranasal + IV, highlighting the key role of intranasal delivery in GBM treatment [49]. Intratumoral delivery directly delivers drugs to the tumor, bypassing the BBB [56]. It enables local immune activation, remodels the TME, and reduces systemic exposure risk [56]. For example, intratumoral pIpC-Dox LNPs significantly increased infiltration and activation of TAMs, DCs, and T cells. Drawbacks include invasive procedure, difficulty covering infiltrative cells, and increased risk with multiple injections [56].
1.4.3 Optimized Antigen Selection
Neoantigens are antigens produced by specific mutations in tumor cells [20]. They are specific and immunogenic, and do not exist in normal tissues, thus becoming ideal vaccine targets [20]. Representative studies include the IDH1-R132H 20-mer peptide vaccine [20] and nanoshield vaccines [57]. The (multi-cationic protein (MCP))-muIDH1 nanoshield vaccine (NaV) uses a MCP scaffold, genetically fuses the muIDH1 neoantigen to MCP, and assembles carboxylated PEG into 100 nm spherical nanoparticles [57]. This enables lysosomal escape, enhanced cross-presentation, NLRP3 inflammasome activation, and prolonged in vivo retention [57]. In tumor-bearing mice, combining NaV with anti-PD1 delayed postoperative recurrence and increased median survival by over 40% [57].
TAAs are normal proteins overexpressed or over-presented on tumor cells [48, 49, 58, 59]. While less immunogenic than neoantigens, they are shared, potentially benefiting more GBM patients [48, 49, 58, 59]. Representative TAAs include 1) Survivin Baculoviral IAP Repeat Containing 5 [49]; 2) Ephrin type-A receptor 2 [49]; 3) Plasminogen activator urokinase receptor [48]; 4) Transforming growth factor beta 2 (TGFB2) [58]. Representative vaccines include 1) SurVaxM [49]; 2) CVGBM mRNA vaccine [48]; 3) and GVac [58]. GVac is a multi-epitope vaccine targeting TGFB2, and its antigen selection and screening have been strongly assisted by computer-aided design [58]. Allergenicity, antigenicity, toxicity, epitope selection, molecular docking, dynamics simulation, and immune simulation can be predicted computationally, theoretically enabling rapid screening of low-toxicity, highly immunogenic candidate vaccines with strong target affinity, though experimental validation is needed [58].
1.4.4 Immune Microenvironment Modulation
GBM's immunosuppressive TME limits vaccine efficacy [7]. New vaccine strategies remodel the TME by reprogramming TAMs, reducing Tregs, and enhancing T cell infiltration [47, 49, 55, 57, 60]. However, the observed TAM reprogramming is mediated not by the vaccine itself, but by secondary components such as sonosensitizers [55] or ICIs [61] incorporated into these strategies. The vaccine Star NV contains a sonosensitizer, the Toll-like receptor agonist R848 (which promotes M1 polarization), and CM-β-CD (for macrophage targeting) [55]. Mechanically, it uses ultrasound to activate sonosensitizers, generate reactive oxygen species (ROS) in tumor tissues to directly destroy GBM cells, and release R848 to achieve TAM transformation to M1 phenotype [55]. In vivo experiments in the glioma mouse model confirmed that this method could kill tumor cells and help to reshape the immunosuppressive microenvironment [55]. Clofazimine (CFZ), an anti-leprosy drug, has potent Wnt/β-catenin inhibitory activity [60]. Combining DC vaccine with CFZ inhibited the Wnt/β-catenin pathway in cancer stem cells, blocking glioma phenotypic heterogeneity/plasticity and β-catenin-mediated immune escape, thereby reducing immunosuppression [60]. Immune checkpoint synergy can also improve the TME. For example, nanoshield vaccine combined with anti-PD-1 improved postoperative relapse-free survival by over 40% in mouse models [57].
2. Immune Checkpoint Inhibitors
Immune checkpoint molecules limit autoimmunity by regulating T cell responses to self-antigens [62]. However, they also limit attacks on cancer cells [62]. ICIs block these molecules, enhancing T cell recognition and killing of cancer cells [62] (Figure 2). Between 1980 and 2000, key immune checkpoints, including CTLA-4 and PD-1 were identified, establishing the foundation for ICIs [63, 64]. In 1996, Allison and colleagues first demonstrated that CTLA-4 blockade enhances antitumor immunity in preclinical models [65]. PD-1 was cloned by Honjo's team in 1992, and its role as an immune checkpoint was elucidated by 2000 [63]. Following regulatory approval of the PD-1 inhibitors Nivolumab and Pembrolizumab in 2014, clinical trials in glioma were initiated. The first-in-human Phase I study of Nivolumab in recurrent glioblastoma (CheckMate 143) established its safety and tolerability [66]. However, the subsequent Phase III CheckMate 143 trial failed to demonstrate an OS benefit for Nivolumab over Bevacizumab in recurrent glioblastoma, underscoring the limitations of ICI monotherapy in this disease [67]. After 2015, ICI trials and combination therapies for glioma increased. According to the literature, ICIs used in trials in the past 10 years include: 1) anti-CTLA-4 (Ipilimumab) [42, 43, 68]; 2) anti-PD-1 (Nivolumab, Pembrolizumab) [42, 69]; 3) anti-PD-L1 (Atezolizumab) [70]. The efficacy of ICI monotherapy is limited, and clinical studies often combine multiple ICIs or other drugs or as adjuvant therapy [71].
As shown in the figure, immune checkpoint molecules such as CTLA-4, PD-1, PD-L1, and TIM-3 negatively regulate T cell activation and T cell-mediated tumor cell killing. Immune checkpoint inhibitors block these inhibitory signaling pathways, relieve T cell exhaustion, and subsequently restore the capacity of T cells to recognize and clear tumor cells.
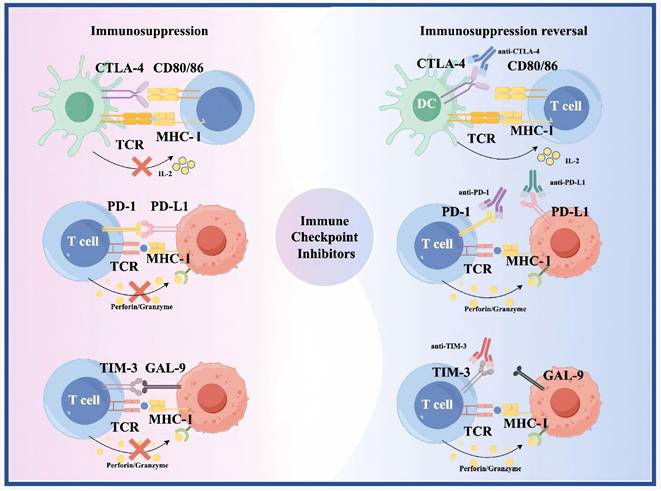
2.1 Clinical Trials and Combination Therapies
Representative clinical studies from the past decade include: 1) Ipilimumab + Nivolumab for nGBM (two studies) [42, 43]; 2) Atezolizumab + Radiotherapy + TMZ Phase I/II trial (NCT03174197) [70]; 3) Evaluation of neoadjuvant Pembrolizumab in rGBM [69]; 4) Nivolumab + Ipilimumab before radiotherapy for nGBM Phase I trial [68]; 5) Bevacizumab + anti-PD-1 therapy (NCT05502991, NCT05540275) [72, 73].
The safety and efficacy of Ipilimumab + Nivolumab for nGBM were demonstrated in the NRG Oncology BN002 (Phase I) trial [42]. However, the randomized Phase II/III NRG Oncology BN007 trial for MGMT-unmethylated nGBM found that Ipilimumab + Nivolumab did not improve Progression-Free Survival (PFS) compared to TMZ [43]. Subsequently, multiple clinical trials have been completed, and despite the use of various immune checkpoint inhibitor regimens, none have demonstrated superiority of ICI monotherapy or combination therapy over standard treatment [43, 74, 75]. Thus, without molecular selection or biomarker guidance, ICIs confer extremely limited survival benefit [43]. Nonetheless, the identification of patients with specific genetic features or immune phenotypes sensitive to ICIs remains of great value, and relevant biomarker analyses are currently ongoing [43]. Atezolizumab + Radiotherapy + TMZ was well-tolerated in a Phase I/II trial (NCT03174197). Unlike DC vaccine sensitivity, this therapy was more effective in PTEN-mutant, mesenchymal subtype GBM patients, less effective in EGFR-mutant, proneural subtype, and significantly better in MGMT-methylated vs. unmethylated patients [70]. Additionally, GBM immune enrichment correlated highly with pre-treatment tumor mesenchymal subtype and patient gut microbiota [70]. Moreover, longer-surviving patients had enriched Bacillota phylum bacteria in stool samples [70]. A trial of neoadjuvant Pembrolizumab in rGBM showed that treatment activity correlated with shortened/reduced cell cycle and proliferation genes and increased T cell/interferon-related genes [69]. This suggests that combined transcription factors that reduce cell cycle/proliferation gene expression or increase T cell/interferon gene expression may improve efficacy [69]. Also, ICIs may have inherent resistance due to chronic inflammation in the TME, suggesting combining anti-inflammatory drugs might restore neoadjuvant ICI activity [69]. Phase I clinical trials of Nivolumab + Ipilimumab in the treatment of nGBM before radiotherapy found that advanced tumors have high LAG-3 levels, downregulation of PPAR signaling and up-regulation of TGF-β and ERBB signaling, and dysregulation of these pathways is associated with ICI resistance [68]. This suggests combining LAG-3 inhibitors, PPAR agonists, and TGF-β/ERBB pathway inhibitors might overcome resistance and improve efficacy [68]. Two studies (NCT05502991, NCT05540275) found that Bevacizumab plus anti-PD-1 significantly improved OS in patients with rGBM. Furthermore, tumor in situ fluid circulating tumor DNA technology can monitor therapeutic response and guide therapy [72, 73]. Collectively, these findings are promising, suggesting pre-treatment screening for patients likely to respond to Bevacizumab + anti-PD-1, refining the target population and reducing medical risk and cost.
2.2 New Preclinical Advances
ICIs show efficacy in many solid tumors but face challenges in glioma. Core challenges include the BBB limiting brain delivery, highly immunosuppressive TME, heterogeneity, scarce tumor-infiltrating lymphocytes (TILs), and dominant TAMs [11]. Recent research focuses on novel delivery systems, combination therapies, TME remodeling, and biomarker identification/patient stratification to improve ICI efficacy.
2.2.1 Novel Delivery Strategies
The BBB is a major obstacle that limits the access of ICIs to GBM tumor sites [5]. To overcome this, researchers utilized low-intensity pulsed ultrasound combined with intravenous microbubbles to achieve transient, reversible, and localized opening of the BBB, thereby enhancing the concentration of aPD-1 in the sonicated regions [76]. Additionally, various delivery systems have been developed, including extracellular vesicles (EV) [77], redox-responsive micelles [78], lipid nanocarriers [79], and nanovesicles [61]. These systems successfully crossed the BBB, achieving efficient brain delivery and tumor-specific accumulation of ICIs, improving treatment safety [61, 78, 79]. However, these delivery systems are still in the research phase and face translational challenges. For instance, although extracellular vesicles possess good biocompatibility and hold promise as therapeutic carriers, they remain limited by heterogeneity, low yields, inefficient drug loading, and rapid clearance by the mononuclear phagocyte system [80]. No EV-based therapy has yet received regulatory approval. Redox-responsive micelles face complex preparation and clinical translation challenges, including amphiphile amount [81], ligand density [82], linkage type [83], polymerization degree [84], reaction conditions [85], and branching degree. These factors collectively influence micelle formation, stability, and scalability. The clinical translation of nanocarriers is hindered by complex physiological barriers, off-target effects of targeting ligands, inherent issues of stability and toxicity, as well as challenges in scalable manufacturing and batch-to-batch consistency [86].
2.2.2 Combination Therapy Strategies
Single ICI efficacy is limited [71]. Researchers explored combinations with chemotherapy [79], radiotherapy [87], photodynamic therapy [88], CAR-NK therapy [89], etc., showing synergy. Moreover, dual/multiple immune checkpoint blockade (ICB) (e.g., PD-1/CTLA-4 [87], αVβ8/PD-1 [90]) enhances immune responses [87]. Furthermore, combining ICIs with epigenetic regulators such as Zinc Finger Protein 638 (ZNF638) inhibition [91] and CpG-STAT3ASO [81] has demonstrated potential to restore immune sensitivity in preclinical settings [91].
2.2.3 Tumor Microenvironment Remodeling
The GBM TME is highly immunosuppressive, dominated by TAMs and MDSCs [61]. Improving this state is key to enhancing ICI efficacy. Strategies include 1) TAM reprogramming (M2 to M1) via inhibiting difluoromethylornithine (DFMO) [61] or Actin Related Protein 2/3 Complex Subunit 1B (ARPC1B) [92]; 2) inhibiting immunosuppressive molecules (Nitric Oxide (NO), ROS, IL-10) to protect T cell function [93]; 3) modulating B cell function to overcome Transforming Growth Factor β (TGFβ)-mediated immunosuppression [90].
2.2.4 Biomarker Identification
Identifying predictive biomarkers is crucial for patient stratification and personalized therapy. Potential biomarkers explored include 1) molecular markers (PD-L1, ZNF638, ARPC1B) [91, 92]; 2) cellular markers (TAMs, B cells, NKT cells) [89, 90, 92]; 3) immune microenvironment features (double-stranded RNA (dsRNA), IFN signaling) [91]; 4) spatial transcriptomics revealing cell interaction networks [90].
3. Adoptive Immune Cell Therapy
Adoptive immune cell therapy is a treatment method in which immune cells with anti-tumor activity are screened, expanded, activated, or genetically engineered in vitro, and then infused back into the patient's body to help the patient's immune system recognize and attack cancer cells [94]. During the 1980s to 2000s, lymphokine-activated killer (LAK) cells and tumor-infiltrating lymphocytes (TILs) were explored clinically for glioma treatment [95]. Early studies by Jacobs et al. demonstrated the feasibility of intratumoral LAK cell injection [96]; however, clinical efficacy was limited and responses were transient, with subsequent trials confirming these therapeutic constraints [97, 98]. TIL-based approaches faced challenges including low TIL abundance in glioma and functional exhaustion within the immunosuppressive TME [99]. However, a recent study using IL-2/IL-15/IL-21-expanded TILs reported complete tumor regression in a glioblastoma patient, suggesting renewed potential for this strategy [100]. Around 2000s-mid 2010s, engineered T cells emerged. CAR-T therapy genetically modifies patient T cells to express CARs targeting specific TAAs, enabling tumor cell recognition and killing [101]. T cell receptor-engineered T cells (TCR-T) differ by introducing optimized TCR genes to recognize intracellular antigens presented by major histocompatibility complex (MHC) molecules [102]. From late 2010s-present, new generation therapies arose, including multi-target CAR-T, logic-gated CAR, armored CAR-T, CAR-NK, CAR-M, neoantigen-targeting CAR-T/TCR-T, combination therapies, and gene-edited universal products [103].
According to the data collected, most of the literature considers adoptive cell therapy to have a good safety profile, with the most serious adverse event usually grade 3 fever [104, 105]. However, recent literature has reported that this therapy is associated with the occurrence of cognitive impairment in patients, suggesting that its safety needs to be further reviewed. In glioma patients specifically, neurological events occur in approximately 41% of cases across early-phase trials [106]. Mechanistic studies in mouse models have demonstrated that CAR-T therapy can disrupt hippocampal neurogenesis and oligodendroglial homeostasis, leading to cognitive deficits [107], and similar reactive microglial states have been confirmed in human brain tissue from patients who received CAR-T therapy for brain tumors [107]. Furthermore, authoritative guidelines from the EBMT and reviews in Nature Reviews Neurology have identified movement and neurocognitive toxicity as emerging concerns across both hematologic and solid malignancies, including CNS tumors [108, 109]. These converging lines of evidence underscore the need for further scrutiny of the long-term safety of adoptive cell therapy.
3.1 Clinical Trials
Since 2015, CAR-T cell therapies for glioma have entered clinical trials. Targets in CAR-T trials primarily include 1) TAAs: EGFRvIII [13, 14, 110]; 2) Interleukin-13 Receptor subunit alpha-2 (IL13Rα2) [111, 112]; 3) Disialoganglioside GD2 (GD2) [113]; 4) Matrix Metalloproteinase-2 (MMP-2) [114].
3.1.1 EGFRvIII Target
EGFRvIII results from an in-frame deletion of exons 2-7 and mutation at the exon 1/8 junction [14]. It is a tumor-specific, oncogenic, immunogenic epitope [14]. Expressed in ~25-30% of nGBM cases, its expression is spatiotemporally heterogeneous, can re-emerge, and varies greatly within a tumor [14]. The efficacy of anti-EGFRvIII CAR-T cell therapy alone has proven to be limited [14]. After treatment, the upregulation of inhibitory molecules, especially PD-L1, in the TME is accompanied by increased infiltration of Tregs, suggesting that the combination of ICIs may improve the efficacy [14]. However, a Phase I trial of repeated anti-EGFRvIII CAR-T infusions + Pembrolizumab was ineffective [13]. While safe and tolerable, disease control was limited, with most patients progressing within months [13]. Reasons included limited peripheral CAR-T cell expansion/persistence, difficulty infiltrating the tumor, EGFRvIII heterogeneity/antigen loss, and increased Tregs/TAMs post-treatment, indicating Pembrolizumab alone couldn't reverse the highly suppressive TME [13]. A Phase I trial of bivalent CAR-T cells targeting EGFR epitope 806 and IL-13Rα2 via intracerebroventricular delivery for EGFR-amplified rGBM showed safety and feasibility [110]. 62% had tumor shrinkage, a few had durable control [110]. The trial enrolled EGFR-amplified patients but not based on IL-13Rα2 expression, potentially affecting efficacy [110]. In five re-infused patients, CAR-T expansion/persistence was worse than the first infusion, with short PFS, suggesting immune clearance or resistance mechanisms [110].
3.1.2 IL13Rα2 Target
IL13Rα2 is a high-affinity IL-13 decoy receptor, negatively regulating IL-13 signaling [111]. It is expressed in ≥ 75% of GBM patients, overexpressed in > 50%, correlating with shorter survival [111]. It is expressed on GSCs, differentiated tumor cells, and TAMs/MDSCs, but not significantly in normal brain [111]. Its expression correlates with the mesenchymal subtype [111]. This first-in-human trial evaluated an off-the-shelf, steroid-resistant allogeneic CAR-T therapy targeting IL13Rα2 in rGBM [112]. By using healthy donor T cells, it overcomes the manufacturing time, cost, and feasibility limitations of autologous CAR-T [112]. This therapy uses zinc-finger nucleases to knock out the glucocorticoid-receptor gene in T cells so that they remain functional in the presence of dexamethasone [112]. In terms of clinical outcomes, treatment was well tolerated, with local tumor necrosis or shrinkage in most patients [112]. However, antitumor activity was transient, and all patients eventually experienced disease progression [112]. The lack of durability of CAR-T cells is a major factor limiting efficacy, in part because of the use of first-generation CAR constructs (Figure 3) containing only the CD3ζ signaling domain, which lack the costimulatory domain and result in limited expansion and survival [112]. In addition, because HLA matching was not performed, one of the six patients developed anti-CAR antibodies, suggesting the presence of host immune rejection [112]. Future improvements include the use of second-generation or higher-generation CAR designs with costimulatory domains, optimized gene editing (e.g., knockout of the TCR to avoid graft-versus-host disease), and the exploration of manufacturing processes that preserve the stem-like properties of T cells and prolong their survival [112].
This figure shows the engineered structure of the first to fourth-generation CAR-T. The core structure of first-generation CAR-T is an extracellular single-chain variable fragment (scFv) plus a single intracellular signaling domain (usually CD3ζ). The core structure of the second-generation CAR-T is extracellular scFv + a costimulatory signal domain + CD3ζ. The core structure of the third-generation CAR-T is extracellular scFv + two or more costimulatory domains + CD3ζ. The core structure of the fourth-generation CAR-T integrates additional genetic elements on the basis of the second-generation CAR, so that it can secrete specific cytokines or express additional receptors. The structures of the fourth-generation CAR-T are diverse, and only the cytokine secreting fourth-generation CAR-T is shown in the figure.
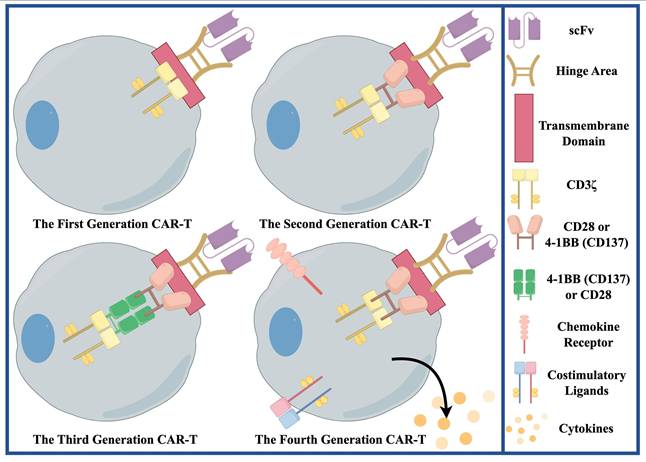
3.1.3 GD2 Target
GD2 is a disialoganglioside, and its expression is highly specific [113]. In normal tissues, it is mainly distributed in the neuronal cell membrane [113]. However, it is highly expressed in a variety of malignant tumors [113]. In GBM, GD2 is enriched on tumor cells and specifically overexpressed on GSCs. ~80% of diffuse intrinsic pontine gliomas express GD2 [113]. A trial of GD2-specific 4SCAR-T cells for GD2-positive GBM used a fourth-generation CAR design with an inducible suicide gene [113]. The results showed that both intravenous and intracavitary injections were safe and tolerable [113]. Median survival from infusion was 10 months [113]. Regarding CAR-T cell kinetics, they expanded in peripheral blood for 1-3 weeks, then persisted at low frequency [113]. Importantly, post-infusion tumor resection showed GD2 antigen loss (mechanism unknown), suggesting need for multi-target therapy [113]. Additionally, after treatment, TAMs increased and tumor-infiltrating NK cells decreased, suggesting that the immunosuppressive microenvironment still needs to be improved [113].
3.1.4 MMP-2 Target
Metalloproteinase MMP-2 is widely expressed in GBM cells, and its main role is to degrade collagen Ⅳ, the main component of the basement membrane of the extracellular matrix [114]. In gliomas, overexpression of MMP-2 is closely related to the malignant behavior of tumors [114]. It accelerates the malignant progression of glioma by destroying the BBB, inhibiting immune cell infiltration, and mediating immune evasion [114]. The CLTX-CAR T-cell study is the first to exploit the ability of CLTX, a peptide derived from scorpion venom, to treat patients with recurrent, MMP-2-expressing GBM through the mechanism of broad MMP-2 binding to glioma cells [114]. Pharmacokinetically, CAR T cells were persistently detected in tumor cavity fluid (TCF) and at low levels peripherally, proving intratumoral delivery allows transport [114]. In terms of safety and efficacy, intratumoral infusion was tolerable with manageable adverse events (AEs), achieving a disease control rate of 75%, although the small sample size precluded assessment of survival benefit [114]. However, several limitations were identified. MMP-2 expression on tumor vasculature, fibroblasts, and myeloid cells poses a potential risk [114]. Furthermore, the inhibitory TME may limit efficacy, as stromal and myeloid components may drive T cell exhaustion, suggesting that combination strategies targeting the TME could improve outcomes [114]. Future directions include modifying peptide ligand/CAR structure for better targeting, enhancing T cell persistence/proliferation, and combining therapies [114].
3.2 New Preclinical Advances
CAR-T therapy has demonstrated significant clinical efficacy in hematologic malignancies, but its application in glioma remains limited. Major challenges: 1) High antigen heterogeneity; 2) Immunosuppressive TME factors (TGF-β, PD-L1); 3) CAR-T cell exhaustion from chronic antigen exposure; 4) BBB limiting migration; 5) Antigen loss/downregulation post-treatment [94]. Recent innovations to overcome these include: 1) Universal targeting platforms (e.g., monomeric streptavidin 2-based Chimeric Antigen Receptor T cells (mSA2 CAR-T)) [115]; 2) Dual/multi-target CAR designs [116]; 3) Armored CAR-T cells [116]; 4) Sonogenetically controlled CAR-T [117]; 5) Combination therapies (e.g., with oncolytic viruses, small molecules) [118, 119]; 6) New targets (e.g., Glycoprotein A Repetitions Predominant, Human Leukocyte Antigen-E) [120, 121]. Conversely, several other CAR-T strategies have demonstrated limited efficacy or encountered significant challenges in preclinical glioma models. These include permanently expressed CAR constructs associated with off-tumor toxicity [122], and B7-H3-targeted CAR-T cells that have yet to overcome intratumoral heterogeneity [123]. Promising preclinical studies are listed below.
3.2.1 Universal CAR-T Platform: mSA2 System
The mSA2 CAR-T platform is a revolutionary universal strategy [115]. It uses mSA2 as the CAR's antigen-binding domain, mediated by biotinylated antibodies [115]. Its advantage is that the targeting specificity of CAR-T cells can be rapidly changed by simple replacement of biotinylated antibodies, without redesign and manufacture of new CAR constructs, so that rapid target switching, multi-target combination, overcoming antigen escape, and reducing manufacturing costs can be achieved [115]. Researchers developed biotinylated antibodies for GBM markers CD276 (B7-H3), EPHA2, CD70, IL13Rα2 [115]. In vitro, mSA2 CAR-T cells specifically recognized primary GBM cell lines antigen-dependently, simultaneously targeting heterogeneous subpopulations, clearing GBM cells within 20 hours via live imaging [115]. In immunodeficient mouse orthotopic GBM models, intracranial injection of mSA2 CAR-T pre-loaded with anti-CD70 or anti-CD276 antibodies significantly suppressed tumor growth, induced apoptosis, and extended survival [115]. This therapy provides a good safety foundation for clinical translation. In a mouse model of GBM, mSA2 CAR-T cells did not interact with brain endogenous biotin, thereby reducing the risk of off-target cross-reactivity [115].
3.2.2 Checkpoint Antibody Receptor-modified (ARMed) CAR-T Cells
The BBB blocks antibody entry into the CNS, a major reason for limited efficacy in anti-PD-1 + CAR-T trials [94]. To overcome this, researchers developed fourth-generation ARMed CAR-T, combining EGFRvIII-targeting CAR with anti-PD-1 minibody secretion [118]. The advantage of this therapy is that ARMed CAR-T is able to cross the BBB and secrete microantibodies at the tumor site, overcoming the BBB limitation of systemic delivery [118]. Mice treated with EGFRvIII ARMed CAR-T had significantly smaller brain tumors versus controls, with a median OS of 34 days and 10% surviving to day 50 (study end) [118]. Additionally, no dose-limiting toxicities were observed, and human T cells remained detectable in peripheral blood at day 29 post-infusion, comparable to CAR T cells alone [118].
3.2.3 NR4A3 Deficiency + FOS Overexpression Synergy Overcomes CAR-T Exhaustion
NR4A family genes (NR4A1, NR4A2, NR4A3) are upregulated in TILs, especially terminally exhausted T cells [124]. FOS expression is highest in CD8⁺ naive populations and is co-expressed with stemness genes including TCF7, CCR7, IL7R, and LEF1 [124]. Based on this, researchers knocked down NR4A3 and overexpressed FOS (FOS OE/NR4A3 KD) in CAR-T cells [124]. Modified CAR-T cells showed long-term tumor suppression, significantly extended mouse survival, and widespread tumor infiltration [124]. This provides a novel genetic modification strategy for solid tumor CAR-T, enhancing stemness and anti-exhaustion capacity by targeting NR4A3 and FOS, offering a preclinical basis for delaying progression [124].
3.2.4 CD44/CD133 Dual-Target IL7Rα Armored CAR-T Cells
IL7Rα is key for T cell survival/proliferation [116]. Armoring CAR-T with ΔIL7Rα intracellular domain enhances tumor-killing [116]. Targeting both CD44 and CD133, classic GSC biomarkers, aims to clear GSCs comprehensively, preventing antigen escape [116]. Since CD44 is also on T cells, to prevent CAR-T fratricide, researchers modified the CAR structure; Tanζ-T28 became the ideal base, successfully constructing Tanζ-T28 CAR-T cells [116]. Evaluating Tanζ-T28 CAR-T in glioblastoma organoid (GBO) models showed decreasing GBO volume, successful CAR-T infiltration, memory T cell enrichment in brain tissue, reduced checkpoint marker expression, and upregulated effector function genes [116]. In addition, a clinical trial (NCT05577091) is ongoing [116].
In recent years, with the continuous advancement of innovative research on CAR-T therapy, the field has encountered challenges in clinical translation. Overall, the clinical translation of CAR-T therapy is hindered by high costs, complex regulatory compliance, substantial quality variability in manufacturing processes, and a widespread shortage of infrastructure and specialized personnel [125]. However, universal platforms such as mSA2 CAR-T may help reduce costs and accelerate the clinical translation process [115].
4. Oncolytic Virus Therapy
Oncolytic virus therapy uses genetically engineered, weakly pathogenic viruses to selectively induce ICD in cancer and stromal cells [7] (Figure 4). It works via multiple mechanisms: direct oncolysis, immune activation, TME remodeling, and vascular disruption [126]. In the early 21st century, clinical trials initially verified the safety and feasibility of herpes simplex virus type 1 (HSV-1) G207 [127], reovirus adenovirus [128], NDV-HUJ oncolytic virus [129] and other modified viruses. A Phase I trial of G207 combined with radiotherapy demonstrated acceptable safety and radiographic responses in rGBM patients, with six of nine patients achieving stable disease or partial response [127]. By 2010, the safety and feasibility of DNX-2401 (Delta-24-RGD) and recombinant poliovirus (PVSRIPO) were established [130, 131]. A pivotal Phase I study of intratumoral DNX-2401 in rGBM reported a 20% 3-year survival rate, including one patient who achieved complete remission lasting three years [130]. Notably, post-treatment tumor analysis revealed CD8+ T-cell infiltration, providing clinical evidence of "cold"-to-"hot" tumor conversion [130]. Concurrently, a Phase I trial of PVSRIPO in rGBM demonstrated a 21% 3-year survival rate versus 4% in historical controls, highlighting its potential to induce durable antitumor immunity [131]. Since the late 2010s, emerging strategies have focused on combination approaches with ICIs, chemoradiotherapy, and the design of next-generation oncolytic viruses [126, 132-135].
The figure illustrates the oncolytic effect mediated by oncolytic viruses following intratumoral injection. This effect promotes the release of a large amount of antigens from the tumor, including tumor-associated antigens (TAAs), damage-associated molecular patterns (DAMPs), pathogen-associated molecular patterns (PAMPs), and viral antigens, thereby facilitating the activation of immune cells and their capacity to kill tumor cells.
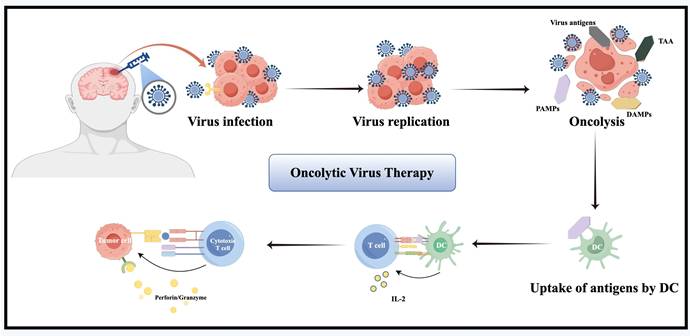
4.1 Clinical Trials
Oncolytic virus platforms in trials over the past decade include: 1) measles virus [136]; 2) adenovirus [132, 137, 138]; 3) herpes virus [139, 140].
4.1.1 Oncolytic Measles Virus
Measles virus is a single-stranded, negative-sense RNA virus. It naturally targets cells via receptors (CD46, Nectin-4), which are often overexpressed on cancer cells [136]. Harnessing this property, oncolytic measles viruses are engineered for enhanced efficacy/safety, overcoming natural limits, and allowing replication monitoring [136]. A prime example is the Measles Virus-based Carcinoembryonic Antigen-expressing Oncolytic Virus (MV-CEA), derived from the safe Edmonston vaccine strain [136]. It causes minimal normal tissue damage, uses CD46 for selective infection, has a strong bystander effect, and expresses soluble CEA for monitoring replication [136]. In clinical testing, this therapy was well tolerated and safe, with a median OS of 11.6 months and a 12-month OS rate of 45.5% in patients with refractory rGBM, which was superior to historical controls [136]. Notably, pre-existing immunity did not prevent virus replication within the tumor [136]. Instead, tumor-intrinsic interferon-stimulated gene (ISG) levels emerged as the key determinant of viral replication [136]. Specifically, high ISG signaling inhibited viral replication, thereby limiting therapeutic benefit [136]. Therefore, this therapy may be best suited for low ISG patients; for high ISG, combining with JAK and STAT inhibitors could block interferon response and enhance replication [136]. Additionally, CD8+ T cell infiltration observed post-treatment waned in subsequent samples from some patients, suggesting single/limited treatments may not sustain long-term immune activation [136]. Future directions include developing measles strains expressing H. pylori neutrophil-activating protein for enhanced immunostimulation or combining measles virus with ICIs [136].
4.1.2 Oncolytic Adenovirus
Adenovirus is a non-enveloped dsDNA virus. Genetic modification enables tumor-selective replication [141]. Several adenoviral platforms have been clinically assessed for GBM, including Ad-TD-nsIL12 [137], DNX-2401 [132], and NSC-CRAd-S-pk7 [138]. Among these, Ad-TD-nsIL12 is a third-generation adenovirus with multiple genetic modifications [137]. Specifically, deletion of E1ACR2 and E3gp19K enabled tumor-specific replication, whereas deletion of E1B19K enhanced antitumor immunity, whereas E1B55K and E3B overcame previous limitations of impaired viral replication capacity and M2-type macrophage infiltration [137]. Furthermore, the IL-12 signal peptide was modified to create a non-secreting form (nsIL12), enabling sustained, low-level IL-12 expression within the TME while avoiding systemic IL-12 toxicity such as lethal inflammatory syndrome [137]. Clinically, a Phase I trial demonstrated that Ad-TD-nsIL12 was tolerable at 1×10¹⁰ vp, with dose-limiting toxicity occurring at 5×10¹⁰ vp. Most adverse events were grade 1-2 and resolved within 48 hours [137]. Median OS was 5.1 months (range: 3.1-21.2 months) [137]. Notably, one patient achieved complete response and one achieved partial response; both responders had IDH1 mutation and MGMT expression, suggesting that exploring IDH mutant sensitivity to oncolytic viruses warrants further investigation [137]. Mechanistically, late TME analysis revealed dominance by M2 macrophages (CD163⁺), which may promote tumor progression [137]. Therefore, combining Ad-TD-nsIL12 with M2 inhibitors or reprogrammers to promote M1 polarization could enhance therapeutic efficacy [137].
Another platform, DNX-2401 (tasadenoturev/Delta-24-RGD), is a conditionally replicating adenovirus featuring a 24-bp deletion in E1A for selective replication and an RGD peptide insertion in the fiber knob to improve GBM cell infectivity via integrin binding [132]. In a multicenter Phase I/II trial (NCT02798406), DNX-2401 combined with Pembrolizumab was safe and tolerable [132]. Median survival in GBM was 12.5 months, with an objective response rate (ORR) of 10.4%, which was not statistically different from the preset 5% control rate [132]. Importantly, post-treatment analysis revealed upregulation of several immune checkpoints (TIGIT, LAG3, B7-H3), suggesting that combining multiple ICIs post-treatment might improve efficacy [132]. Furthermore, an exploratory analysis suggested that patients with a TMEmedium subtype characterized by moderate immune infiltration, medium PD-1 expression, and absence of multiple immunosuppressive factors may be more likely to derive clinical benefit from this approach [132].
4.1.3 Oncolytic Herpes Virus
HSV is a neurotropic dsDNA virus with a large genome (~150kb) [142]. Over the past decade, G47Δ has emerged as a key oncolytic HSV-1 variant [139, 140]. It is a third-generation, triple-mutated virus constructed by deleting the α47 gene and overlapping the US11 promoter from its parental virus G207, which enhances tumor-specific replication and immunogenicity [139]. In clinical studies, previous Phase I/II trials confirmed the safety of G47Δ in progressive GBM, with patients achieving a median survival of 7.3 months (range: 1-14 months) [140]. A subsequent Phase II trial for residual or recurrent GBM showed more impressive results: median OS from G47Δ start was 20.2 months, with a 1-year survival rate of 84.2%—far exceeding the 15% historical control [139]. Mechanistically, biopsy after treatment revealed a sustained increase in CD4⁺ and CD8⁺ T cells without an increase in Foxp3⁺ Tregs, suggesting that G47Δ can effectively improve the immunosuppressive microenvironment and promote immune infiltration [139]. Based on these findings, G47Δ was approved in Japan in June 2021, becoming the country's first oncolytic virus product [139]. However, distant new lesions, CSF dissemination, and local recurrence around the target were common, indicating that local therapy struggles to control systemic or diffuse progression [139]. Additionally, imaging proved inadequate for accurately evaluating efficacy, underscoring the critical need for novel assessment methods [139].
4.2 New Preclinical Advances
Oncolytic virus therapy shows great potential but faces challenges: poor targeting, high toxicity, poor drug control, systemic delivery limited by BBB, weak virus infiltration, host immune clearance, and immunosuppressive TME limiting immune activation. Recent strategies in preclinical research have made important progress. Innovations in virus platforms, delivery strategies, combination therapies, and metabolic regulation are summarized below.
4.2.1 New Oncolytic Virus Platforms
New platforms focus on improving targeting, reducing toxicity, and enabling drug control. 1) Zika virus (ZIKV), 2) infectious bursal disease virus (IBDV), 3) human cytomegalovirus (HCMV), and 4) myxoma virus (MYXV) platforms provide strong support [126, 133, 143-145]. 1) ZIKV shows selective targeting of GSCs, with natural neuroprogenitor tropism and SOX2⁺ cell preference [126, 143]. 2) IBDV is a dsRNA non-human virus, avoiding pre-existing immunity; it infects tumor and myeloid cells [133]. 3) HCMV based on AD169 strain: deleted UL1-UL20/UL/b regions to reduce virulence; restored pentamer complex (gH/gL/pUL128-131) function to enhance tropism; integrated paramyxovirus glycoprotein H/F complex for EGFR targeting; added tet-off system for drug-controlled IL-12 expression [144]. 4) MYXV is an enveloped dsDNA poxvirus. A MYXV construct lacking the M011L gene (vMyx-M011L-KO/EGFP) induced apoptosis in infected brain tumor-initiating cells [145].
4.2.2 Innovative Delivery Strategies
Cell carrier-mediated delivery, surface modification for BBB penetration, and combination with photodynamic therapy (PDT) improve virus delivery to tumors [145-148]. 1) Macrophage carriers protect virus from neutralizing antibodies, effectively infiltrate glioma tissue, and have strong tumor tropism [146]. 2) Adipose-derived stem cell carriers have low immunogenicity (allowing allogeneic use), strong tumor tropism, protect virus from antibodies/T cells, and maintain migration ability post-infection [145]. 3) Cholesterol-modified adenovirus (OA@Cho) uses LDL receptor-mediated transcytosis across BBB for precise glioma targeting/delivery [147]. 4) PDT-generated ROS damage cancer cells, increase tumor vascular permeability, induce ICD, thereby increasing virus susceptibility, promoting virus entry, and enhancing anti-tumor immunity [148].
4.2.3 Combination Therapy Strategies
Combining oncolytic viruses with chemotherapy, ICIs, metabolic inhibitors, PDT, etc., shows synergistic potential for improved efficacy [133-135]. For example, when IBDV is combined with TMZ, IBDV can enhance the cytotoxic activity of TMZ, and TMZ not only does not impair the susceptibility of GBM cells to IBDV, but can enhance the ability of viral replication and promote viral release [133]. Triple therapy (a bromodomain-containing protein 9 (BRD9) inhibitor + oHSV + anti-PD-1/CTLA-4) showed potent anti-tumor effect, nearly eradicating tumors; long-term survivors showed protective immunity upon re-challenge [134].
4.2.4 Metabolic Regulation Intervention
Metabolic reprogramming and targeting metabolic pathways can reduce virus-induced lipid peroxidation [135]. For example, combining oHSV with an IDH R132H inhibitor restored the sensitivity of IDH-mutant glioma to oHSV, inducing ferroptosis and anti-tumor immunity [135]. Mechanistically, this combination blocked glycolysis, cleared ROS, inhibited glutamine metabolism, and suppressed Protein Kinase C in immunocompetent mouse IDH-mutant tumor models, significantly improving virus therapy efficacy [135]. However, the use of metabolic reprogramming inhibitors must ensure selective toxicity to tumor cells, thereby preventing metabolic disruption in normal cells that could render them susceptible to oncolytic virus infection [149].
4.3 Biomarkers and Patient Stratification
Analysis of GBM trial (NCT03152318) data revealed several predictive biomarkers: 1) Low BRD9 mRNA expression correlated with significantly longer PFS [134]. 2) METTL14-low gliomas are more sensitive to oHSV-1 [150]. 3) Wild-type IDH gliomas are more sensitive to oHSV-1 [134]. 4) High PD-L1/PD-1 expression suggests potential benefit from combination ICB [135]. 5) High glycolysis patients may be sensitive to metabolic reprogramming. In these cases, consider combining glycolysis inhibitors [135]. 6) Patients with high oxidative phosphorylation may benefit from oHSV monotherapy [135]. 7) Immune "hot" tumors with rich T cell infiltration suit viral immunotherapy and may benefit from ICIs [134]. 8) Immune "cold" tumors with scarce T cell infiltration (especially GBM) need virus-induced immune activation. For such patients, consider higher doses, multiple administrations, or combining immunomodulators [134].
In recent years, advancements in oncolytic virotherapy have provided new strategies for the treatment of glioma. However, the translation of these therapies from research to clinical application remains hindered by multiple challenges, including safety risks and heterogeneity associated with animal-derived components, difficulties in maintaining viral activity and stability, and the urgent need to upgrade purification technologies toward large-scale, automated, and intelligent manufacturing [151].
5. Common Challenges Shared across Different Immunotherapies
Although the immunotherapeutic strategies described above have shown promise in preclinical models, they have largely failed to translate into durable survival benefit in clinical trials (Table 1). This suggests common barriers across different therapeutic modalities. This section systematically analyzes the core mechanisms underlying these failures, including 1) immunosuppression in the TME [152, 153], 2) tumor antigen heterogeneity and escape [13, 154], 3) blood-brain barrier limitations for drug delivery [155, 156], and 4) adaptive resistance under therapeutic pressure [153, 157].
Summary of key clinical trials of immunotherapy for glioma
| Representative Agent/Strategy | Trial Phase | Enrollment (n) | Patient Population | Median OS (months) | Median PFS (months) | Key Efficacy/Survival Endpoint | Key Toxicities/Safety Profile | Reference |
|---|---|---|---|---|---|---|---|---|
| Cancer Vaccines | ||||||||
| WT1-mRNA/DC | Phase I/II | 40 (13 GBM) | Advanced solid tumors (incl. GBM) | GBM: 43.7 | Not reported | • DCR: 69.2% (T1/T2)• ORR: 23.1% (T2)• Type 1 T-cell responses linked to improved OS | • Well tolerated• Local injection site reactions only• No systemic toxicity | [21] |
| AV-GBM-1 | Phase II | 60 (57 treated) | nGBM (pre-RT/TMZ) | 16.0 | 10.7 | • PFS 57% longer vs historical controls• 3-yr PFS: 12.3%• 8 doses correlated with OS (HR 0.09, p <0.0001) | • Well tolerated• Local injection site reactions only• No treatment discontinuations | [23] |
| ICT-107 | Phase II (RCT, DB) | 124 (81 vaccine, 43 control) | nGBM (HLA-A1⁺/A2⁺, post-RT/TMZ) | 17.0 (vaccine) vs 15.0 (control); p=0.58 (NS) | 11.2 (vaccine) vs 9.0 (control); HR 0.57, p=0.011 | • PFS significantly improved• HLA-A2 subgroup: greater immune benefit• Methylated HLA-A1: OS 47.6 vs 25.8 mo (p = 0.049) | • Well tolerated; no difference in AEs vs control• Most common: fatigue, convulsions, nausea | [25] |
| GSC-DCV | Phase II (RCT, DB) | 43 (22 vaccine, 21 placebo) | nGBM/rGBM, post-resection (≥95%) | 13.7 (DCV) vs 10.7 (placebo); p=0.05Multivariate HR 2.5, p=0.02 | 7.7 (DCV) vs 6.9 (placebo); p=0.75 | • IDH1ʷᵗTERTᴹᵀ subgroup: OS (p<0.01), PFS (p=0.03)• Low B7-H4 subgroup: OS (p=0.02) | • Well tolerated• Mild fever (1 pt), injection site erythema (1 pt) | [26] |
| pp65-DC + DI-TMZ + GM-CSF | Phase I | 11 | nGBM (post-resection >90%, post-RT/TMZ) | 41.1 (vs 19.2 historical, p=0.0001) | 25.3 (vs 8.0 historical, p=0.0001) | • 4/11 pts PFS at 59-64 mo• 100% exceeded RPA predicted survival (median gain 30 mo)• pp65 immune response correlated with OS>40 mo (p=0.031) | • Well tolerated• 1 Grade 3 vaccine-related immunologic reaction (GM-CSF sensitization) | [29] |
| CMV-ATCT-DC | Phase I/II (RCT, single-blind) | 22 enrolled, 15 completed (8 DC, 7 saline) | nGBM (CMV-seropositive, KPS ≥80, post-RT/TMZ) | Not reported (underpowered) | Not reported (underpowered) | • Primary safety endpoint met• Polyfunctional T cells: significant increase in CMV-ATCT-DC vs saline (p=0.0078)• Polyfunctional T cells correlated with OS (R=0.74, p=0.037) | • Well tolerated• No SAEs related to treatment• AEs consistent with standard of care | [27] |
| Td toxoid + CMV pp65-DC | Phase I/II (RCT, blinded) | 12 (6 Td, 6 DC) | nGBM (CMV-seropositive, KPS ≥80, post-RT/TMZ) | Td: not reached (>36.6)DC: 18.5 | Td: not reached (>36.6)DC: 10.8 | • DC migration: significant increase in Td vs DC (p=0.049)• Td significantly improved PFS (p=0.013) and OS (p=0.013)• DC migration correlated with survival | • Well tolerated• No vaccine- or Td-related AEs | [28] |
| Rindopepimut (ACT III) | Phase II | 65 | nGBM (EGFRvIII+, post-GTR, post-CRT) | 21.8 (from study entry) | 9.2 (from study entry) | • PFS at 5.5 mo: 66% (primary endpoint met, p=0.017)• 36-mo OS: 26%• 85% ≥4x Ab titer• 67% target loss in evaluable samples | • Grades 1-2 injection site reactions (nearly all)• Grade 3/4 events rare; no fatal AEs | [32] |
| Rindopepimut (ACT IV) | Phase III | 745 (405 MRD) | nGBM (EGFRvIII+, MRD <2 cm² post-CRT) | 20.1 (vaccine) vs 20.0 (control); HR 1.01, p=0.93 | Not reported | • Primary endpoint not met (futility stopped)• 2-yr OS (SRD): 30% vs 19% (exploratory, p=0.029) | • Grade 3-4: thrombocytopenia (9% vs 6%), fatigue (2% vs 5%), brain edema (2% vs 3%)• Injection site reactions (80% vs 41%, mostly grade 1-2) | [31] |
| IDH1-vac | Phase I | 33 enrolled, 32 vaccinated | nGBM WHO grade 3/4 IDH1(R132H)+, post-RT/TMZ | 3-yr OS rate: 0.84 | 3-yr PFS rate: 0.63 | • Immune response rate: 93.3% (across multiple HLA alleles)• PsPD rate: 37.5% (linked to immune response)• MSS correlated with antigen presentation | • No RLT/DLT; AEs grade 1 only• Most common: injection site induration (65.6%), erythema (46.9%) | [33] |
| SurVaxM (Phase I) | Phase I | 9 enrolled, 8 evaluable | rGBM/AA (survivin⁺, HLA-A*02/03/24) | 86.6 weeks (~21.7) (from study entry) | 17.6 weeks (~4.4) (from study entry) | • Immune response: 75% (cellular & humoral)• 3 pts PR/SD >6 mo• 7/9 pts survived >12 mo• 1 pt CR, alive NED at 174 weeks | • No SAEs related to vaccine• Mostly grade 1: injection site reactions, fatigue, myalgia• Grade 3 seizure (unrelated) | [41] |
| SurVaxM + adj TMZ (Phase IIa) | Phase IIa | 64 enrolled, 63 evaluable | nGBM (survivin⁺, HLA-A*02/03/11/24, post-RT/TMZ) | 25.9 (from first vaccine)28.4 (from diagnosis) | 11.4 (from first vaccine)14.4 (from diagnosis) | • PFS-6 (from diagnosis): 95.2% (primary endpoint met, p<0.0001 vs 54% historical)• 36-mo OS: 41.4%• Ab titer >30,000 correlated with OS (HR 0.41, p = 0.015)• Methylated MGMT: mOS 41.4 mo; Unmethylated: 16.5 mo | • No SAEs related to SurVaxM• Most common: grade 1 injection site reactions (37.5%)• AEs attributable to TMZ (leukopenia, etc.) | [34] |
| WT2725 | Phase I | 62 (20 GBM) | Advanced WT1+ malignancies (HLA-A*0201+/0206+) | Solid tumors: 13.0GBM: 10.2 | Solid tumors: ~2GBM: ~2 | • irCR (GBM): 13.3% (2/15); irPR: 6.7%• CTL response: dose-dependent (0 mg → 27 mg: 0% → 88.9%)• 2 GBM pts irCR, PFS ≥30.6 and ≥37.0 mo | • No DLT; MTD not reached• Most common: grade 1-2 injection site reactions• No discontinuations | [36] |
| IMA950 | Phase I | 45 | nGBM (HLA-A*02+, post-resection, receiving CRT+adj TMZ) | 15.3 (overall) | Not reported (PFS-6: 74%, PFS-9: 31%) | • Immune response: 90% TUMAP responders• ISR correlated with OS (ISR+ mOS 26.7 mo vs ISR- 13.2 mo; HR 0.33, p=0.0001)• MGMT methylated: mOS 28.3 mo vs unmethylated 14.8 mo (p=0.025) | • Well tolerated• Most common: grade 1-2 injection site reactions (26/45)• 2 DLTs (grade 3 fatigue, anaphylaxis) | [19] |
| IMA950 + poly-ICLC | Phase I/II | 19 (16 GBM, 3 AA) | nGBM/AA (HLA-A2+, post-RT/TMZ) | GBM: 19 (from surgery)Whole cohort: 21 | GBM: 9.5 (from surgery)PFS-6/9 (GBM): 81%/63% | • Primary immunogenicity endpoint met• CD8 multi-peptide responses: 46.2%• CD4 responses: 84.6% | • Well tolerated• Most common: injection site reactions, fatigue, headache• 1 Grade 4 interstitial pneumonia (unrelated) | [37] |
| WT1 cocktail vaccine | Phase I | 14 (12 GBM, 2 AA) | rGBM/AA (HLA-A*24:02+, WT1+, refractory to TMZ) | 24.7 weeks (~5.7) | Not reported | • SD at 6 weeks: 43%• 1-year OS rate: 36%• Immune responses: WT1-specific CD8+ (64%), CD4+ (91%) | • No DLT; No grade 3/4 AEs• Grade 1 injection site reactions (all patients) | [38] |
| VEGFRs peptide vaccine + TMZ | Phase I/II | 4 | nGBM (HLA-A*24:02+, post-surgery, receiving RT/TMZ) | Individual: 31.6, 41.8, >46.8, >31.6 | Individual: 18.2, 41.8, >46.8, >31.6 | • 2/4 patients achieved CR (incl. 1 MGMT unmethylated)• Both CR patients alive at analysis• Histopathological evidence of CTL-mediated killing of tumor vessels, cells, and Tregs | • No grade 3/4 AEs• Grade 1 local skin reaction in 1 patient | [39] |
| GAA peptide vaccine + poly-ICLC | Phase I | 23 | LGG (high-risk), HLA-A2⁺ | Not reported | Cohort 1 (no prior RT): 17Cohort 3 (recurrent): 12 | • Cohort 1: 91% responded to ≥1 GAA, 82% to ≥3 GAAs• Cohort 3: 56% responded to ≥1 GAA, 33% to ≥3 GAAs• 3 pts in Cohort 1 remain PFS (37, 42, 47+ mo) | • Grade 1-2 injection site reactions (100%)• One case of Grade 3 fever/fatigue (RLT) | [40] |
| Personalized mutIDH1 multi-peptide vaccine | Phase I/II | 52 | IDH1-mutant glioma (grade 2-4), newly diagnosed or recurrent | Not reached (median f/u 36 mo) | Not reported | • mutIDH1 immune response: 89%• Immunological responders had significantly longer OS (HR 0.09; p=0.0057)• mutIDH1-specific CD4+ T cells were polyfunctional and polyclonal• Identical TCRβ sequences found in patients with shared HLA-DQ alleles | • Well tolerated• Grade 1 injection site reactions only• No grade ≥3 AEs | [20] |
| Personalized peptide vaccination (PPV) based on pre-existing IgG levels | Phase III (RCT, DB) | 88 (58 PPV, 30 placebo) | HLA-A24+ recurrent GBM, refractory to TMZ/RT | PPV: 8.4Placebo: 8.0 (p=0.62) | Not shown (no significant difference) | • Primary endpoint not met• PPV detrimental with SART2-93 peptide (HR 15.9), age ≥70 yr (HR 7.9), weight >70 kg (HR 4.1), PS3 (HR 2.8)• PPV beneficial without SART2-93 + age <70 yr (HR 0.49, p=0.03)• Lower baseline CD11b+CD14+HLA-DRlow monocytes and higher CD3+CD4+CD45RA- T cells correlated with improved OS | • Well tolerated• Most common: injection site reactions (PPV 71%, placebo 53%)• Grade ≥3 AEs: PPV 40%, placebo 37%• One grade 3 pulmonary embolism (PPV-related) | [44] |
| Autologous formalin-fixed tumor vaccine (AFTV) | Phase IIb (RCT, DB, placebo-controlled) | 57 (30 AFTV, 27 placebo) | Newly diagnosed supratentorial GBM, age 16-75 yr, KPS ≥60% | AFTV: 25.6Placebo: 31.5 (p=0.64) | AFTV: 13.3Placebo: 13.3 (p=0.98) | • Primary endpoint not met• Total resection subgroup: 3-yr OS 80% vs 54% (p = 0.16); 3-yr PFS 81% vs 46% (p = 0.067)• p53-negative subgroup: 3-yr OS 79% vs 43% (p = 0.072)• DTH-2 response not associated with survival• Met predefined criteria for Phase III trial | • Well tolerated; no significant difference in AEs• Grade ≥3 AEs: AFTV 23%, placebo 30%• No serious AEs attributed to AFTV | [46] |
| APVAC1 + APVAC2 + poly-ICLC + GM-CSF | Phase I | 16 enrolled, 15 vaccinated | nGBM (HLA-A*02:01⁺/24:02⁺, post-GTR, receiving CRT) | 29.0 (n=15 evaluable) | 14.2 | • APVAC1 immune response: 92.3%• APVAC2 immune response: 80% (Th1-type CD4⁺)• Low baseline Tregs correlated with higher memory cell induction | • Well tolerated• Most common: grade 1-2 injection site reactions• Two cases of anaphylaxis (potentially GM-CSF-related)• One grade 3 cerebral edema | [45] |
| Immune Checkpoint Inhibitors | ||||||||
| Ipilimumab + Nivolumab + adj TMZ | Phase I | 32 enrolled, 31 evaluable | nGBM (unifocal, supratentorial, post-GTR/NTR, post-CRT) | Combination cohort (n=19): 20.7 (from treatment start) | Combination cohort (n=19): 16.1 | • Primary safety endpoint met: 1 DLT in Cohort 1, 1 DLT in Cohort 2, 0 DLTs in Cohort 3• 1-year OS rate (combination): 68.4%• 2-year OS rate (combination): 31.6% | • Well tolerated; no Grade 5 AEs• No unexpected toxicity• Most common Grade 3-4: skin disorders (38.5%), lab abnormalities (23.1%) | [42] |
| Ipilimumab + Nivolumab + RT (no TMZ) | Phase II/III (RCT) | 159 (79 immunotherapy, 80 TMZ) | nGBM (MGMT-unmethylated, KPS ≥70, post-resection) | Immature (~13 mo each)HR 0.95, p=0.36 | 7.7 (immunotherapy) vs 8.5 (TMZ)HR 1.47, p=0.96 | • Primary endpoint not met (PFS not improved)• Trial terminated• 2 treatment-related deaths on immunotherapy arm (autoimmune disorder, colitis) | • No significant difference in AEs between arms (p = 0.69)• Grade ≥3 AEs: 37.8% (TMZ) vs 41.0% (immunotherapy)• Grade 5 events: 0 vs 2 | [43] |
| Atezolizumab + RT/TMZ | Phase I/II | 60 | nGBM (WHO 2016 criteria, KPS ≥60) | All pts: 18.0IDH-wt: 16.1MGMT methylated: 25.4MGMT unmethylated: 14.6 | Not reported | • ORR: 23.3% (CR 10% + PR 13.3%)• DoR: 13.91 mo• High ESTIMATE Immune Score tumors: mOS 24.8 vs 14.5 mo (HR 0.45, p = 0.02)• Mesenchymal subtype: mOS 26.5 mo | • Well tolerated; no new safety signals• Grade ≥3 treatment-related AEs: 56.7% (most common: lymphopenia)• 3 patients removed due to hepatitis/pneumonitis (DLTs) | [70] |
| Nivolumab + Ipilimumab (pre-radiation) | Phase I | 15 | nGBM (Grade 3/4, post-surgery, pre-RT) | 19.3 (95% CI, 12.9-NA) | 1.3 (95% CI, 0.92-2.99) | • Feasibility demonstrated (treatment started median 38 days post-op)• MGMT methylated: mOS 35.7 mo vs unmethylated: 12.6 mo (p = 0.004)• Paired tumor analysis (n = 5): post-progression tumors showed ↑ TGF-β/ERBB, ↓ PPAR signaling, ↑ monocytes, ↓ B cells.• Growing tumors trended toward higher baseline LAG-3. | • Well tolerated; no DLTs• Most common: rash, pruritus, fatigue, nausea, anorexia• Grade 3: lipase ↑ (2), anorexia (1), pruritus (1), rash (3)• One Grade 4 cerebral edema• No Grade 5 events | [68] |
| Neoadjuvant + Adjuvant Pembrolizumab | Phase II (single-arm expansion) | 25 (expansion) + 32 (initial) = 57 pooled | rGBM (surgically accessible, 1st/2nd relapse, KPS ≥70) | Expansion: 6.8Initial neoadjuvant: 13.7Initial adjuvant: 7.5 | Expansion: 2.5Initial neoadjuvant: 3.3Initial adjuvant: 2.5 | • Primary pharmacodynamic endpoint met: Cell cycle gene signature decreased in neoadjuvant vs adjuvant tumors (p=0.009)• IFN-high/CC-low subgroup (n=13): mOS 13.8 mo• No confirmed OS benefit for neoadjuvant regimen | • Well tolerated; no new safety signals• Grade ≥3 AEs in 6 pts: cerebral edema (2), headache, fatigue, adrenal insufficiency, hyperthyroidism | [69] |
| Tislelizumab + Low-Dose Bevacizumab | Phase II | 32 | rGBM | 14.3 | 8.2 | • ORR: 56.3%• 12-month OS: 43.8%• >20% reduction in MAF/TMB post-treatment correlated with improved PFS and OS (p = 0.0005-0.008)• Baseline MUC16 mut, H3F3B amp, SRSF2 amp associated with worse OS | • Any grade AEs: 90.6% (most common: anemia, fatigue, increased ALT)• Grade ≥3: 6.2% (incl. one grade 4 acute pancreatitis)• No grade 5 events | [73] |
| Adoptive Immune Cell Therapy | ||||||||
| CART-EGFRvIII cells + Pembrolizumab | Phase I | 7 | nGBM (EGFRvIII+, MGMT-unmethylated) | 11.8 (90% CI, 9.2-14.2) | 5.2 (90% CI, 2.9-6.0) | • Primary safety endpoint met: no DLTs• Antigen reduction: EGFRvIII decreased post-treatment in 6/7 pts• ↑ IFN-stimulated T cells in post-treatment tumors correlated with time from relapse to death (Spearman p=0.03) | • Safe and well-tolerated• No CRS, no ICANS• One patient experienced severe (grade 3-4) immune-related AE (liver/kidney injury) likely related to Pembrolizumab | [13] |
| CART-EGFRvIII cells (single infusion) | Phase I | 10 | rGBM (EGFRvIII+, MGMT-unmethylated) | 251 days (~8) | Not evaluable | • Primary safety & feasibility endpoint met• On-target activity: EGFRvIII expression decreased in 5/7 assessed pts (p = 0.03)• Trafficking: Higher CAR T concentration in brain vs blood in 2 pts• TME changes: ↑ immunosuppressive molecules (IDO1, PD-L1, FoxP3, IL-10) | • No EGFR-directed toxicity, no systemic CRS, no neurotoxicity typical of CD19-CARs• 3 subjects with clinically significant neurologic events (possibly related to disease or local inflammation)• No DLTs | [14] |
| CART-EGFR-IL13Rα2 (bivalent, ICV) | Phase I | 18 | rGBM (EGFR-amplified, IDH-wt) | Not reached (f/u 8.1 mo) | 1.9 (90% CI, 1.1-3.4) | • ORR: 8% (1/13)• Tumor regression: 62% (8/13)• Durable disease: 2 pts PFS >6 mo (7.7, 16.6)• MTD/RDE: 2.5 × 10⁷ cells | • CRS: 100% (all grades 1-2)• Grade 3 neurotoxicity: 56% (no grade 4-5)• One DLT (grade 3 anorexia, weakness, fatigue) at MTD | [110] |
| IL-13(E13Y)-zetakine CD8+ CTLs | Phase I | 3 | rGBM | Not reported (mean ~11) | Not reported | • Feasibility demonstrated• Antigen loss: In one patient, IL13Rα2 expression decreased post-treatment• Radiographic response: In one patient, ↑ necrotic volume on MRI post-retreatment | • Well tolerated at 10⁷ and 5×10⁷ cell doses• At 10⁸ cell dose: Grade 3 headache (1 pt), Grade 3 neurological event (shuffling gait, tongue deviation; 1 pt); both transient and manageable | [111] |
| GRm13Z40-2 cells (allogeneic, GR-knockout) | Phase I | 6 | rGBM (unresectable, IL13Rα2+, on dexamethasone) | 2.9 (post-treatment) | Not reported | • Proof-of-Concept: First-in-human allogeneic, ZFN-modified, steroid-resistant CAR T product• Anti-tumor activity: Transient tumor reduction/necrosis at infusion site in 4/6 pts | • Well tolerated• All treatment-related AEs ≤ grade 3, transient, manageable• No DLTs, GVHD, or severe treatment-related neuroinflammation | [112] |
| GD2-specific 4SCAR-T (IV ± intracavitary) | Phase I | 8 | rGBM (GD2+, IDH-wt; 4 adults, 4 children) | 10 (range, 3-24) from infusion | Not reported | • ORR: 50% (4/8 PR, 3-24 mo duration)• Disease control (PR+SD): 62.5%• Antigen loss & TME modulation: ↓ GD2, ↑ CD8+, ↓ CD163+ M2 macrophages (1 pt) | • Safe and well-tolerated• No severe AEs (grade ≥4) or CAR-T-related neurotoxicity• One patient: grade 2 seizures and grade 3 headache probably related to infusion | [113] |
| CLTX-CAR T cells (intracavitary/intratumoral) | Phase I (Interim) | 4 | rGBM (MMP-2+, heavily pretreated) | <6 (3 pts)1 pt >20 | Not reported | • Disease Control Rate: 75% (3/4 SD)• Regional control: No recurrence at infusion site for >9 weeks (1 pt)• Proof-of-Concept: First-in-human toxin-based CAR platform | • Well tolerated; no DLTs or CRS• No grade ≥3 CNS events attributed as probable/definite• Grade 3 cerebral edema (possibly related) in 2 pts• No humoral immunogenicity detected | [114] |
| Oncolytic Virotherapy | ||||||||
| MV-CEA | Phase I | 22 | rGBM (anti-MV immune at baseline) | 11.6 | 3.4 | • 12-month OS: 45.5% (favorable vs historical controls)• Disease control rate (SD): 88.9% (Group A), 92.3% (Group B)• Biomarker (ISG signature): DLDA score inversely correlated with viral replication (R=-0.6, p=0.04)• TME remodeling: ↑ CD8+ and CD68+ infiltration (day 5) | • Well tolerated; no DLTs up to max dose (2×10⁷ TCID50)• Only grade 1-2 AEs• No viremia, viral shedding, or treatment-induced immunosuppression | [136] |
| DNX-2401 + Pembrolizumab | Phase I/II | 49 | rGBM/rGS (1st/2nd recurrence, unresected) | 12.5 | 3.4 | • Primary ORR endpoint (10.4%) not met (p=NS vs 5%)• Secondary 12-mo OS endpoint (52.7%) met (vs 20% control)• Clinical benefit rate (SD or better): 56.2%• 3 patients alive at 45, 48, and 60 mo• Biomarker: Responses only in the "TMEmedium" subtype (exploratory) | • Well tolerated; no DLTs• Most common: brain edema (37%), headache (31%), fatigue (29%)• Grade 3 AEs: 22%; one grade 4 event• No treatment-related deaths | [132] |
| Ad-TD-nsIL12 | Phase I | 8 | rHGG (with ventricular system connection) | 5.1 (range, 3.1-21.2) from first injection | Not reported | • Primary MTD endpoint: 1×10¹⁰ vp (grade 3 seizures as DLT at 5×10¹⁰ vp)• Antitumor activity: 1 CR, 1 PR (both in Cohort 2)• TME remodeling: ↑ CD4+/CD8+ T cell infiltration, confirmed viral replication | • Well tolerated at ≤1×10¹⁰ vp• Most common: fever, cognitive disturbance, nausea/vomiting• Grade 3 seizures (DLTs) at highest dose in 2/2 pts• No viral DNA in blood/CSF | [137] |
| NSC-CRAd-S-pk7 (neural stem cell-loaded) | Phase I | 12 | nGBM (WHO grade III/IV) | 18.4 | 9.05 | • Primary safety endpoint met: No DLT up to highest dose• MGMT unmethylated subgroup (exploratory): mOS 18.0 mo, mPFS 8.8 mo (favorable vs historical)• TME remodeling: ↑ CD8+ T cell infiltration, ↑ PD-1 expression, ↓ survivin/syndecan-1 | • Safe and well-tolerated; no DLT• One patient: grade 3 viral meningitis (probably related, due to inadvertent intraventricular injection)• No treatment-related deaths• RP2D: 1.50×10⁸ NSCs / 1.875×10¹¹ vp | [138] |
| G47Δ (triple-mutated HSV-1) | Phase II | 19 | Residual/rGBM (post-RT/TMZ) | 20.2 (from 1st injection)28.8 (from initial surgery) | 4.7 (from 1st injection) | • Primary endpoint (1-yr OS): 84.2% (met, trial terminated early)• TME remodeling: ↑ CD4+/CD8+ TILs with repeated dosing, low Foxp3+ Tregs• Regulatory outcome: First approved oncolytic virus for malignant glioma (Japan, 2021) | • Safe and well-tolerated; no DLT• Most common: fever (89.5%), vomiting (57.9%), nausea (52.6%)• Grade ≥3 lymphocytopenia: 26.3% (resolved without treatment) | [139] |
| G47Δ (triple-mutated HSV-1) | Phase I/II | 13 | rGBM (post-RT/TMZ) | 7.3 (from last dose)30.5 (from initial surgery) | 8 days (from last dose) | • 1-year OS rate (from last dose): 38.5%• Long-term survival: 3 patients >46 mo• Best overall response (2-yr, exploratory): CR 7.7%, PR 7.7%• TME remodeling: ↑ CD4+/CD8+ T cell infiltration post-treatment | • Safe and well-tolerated• Most common: fever (61.5%), headache (61.5%), vomiting (46.2%)• Grade 3 events: leukopenia, headache, vomiting, neurological disorder (few pts)• No DLT | [140] |
Abbreviations: AA, anaplastic astrocytoma; Ab, antibody; adj, adjuvant; AE, adverse event; AFTV, autologous formalin-fixed tumor vaccine; ALT, alanine aminotransferase; amp, amplification; APVAC, actively personalized vaccination; CAR, chimeric antigen receptor; CBR, clinical benefit rate; CI, confidence interval; CMV, cytomegalovirus; CNS, central nervous system; CR, complete response; CRT, chemoradiotherapy; CRS, cytokine release syndrome; CSF, cerebrospinal fluid; CTL, cytotoxic T lymphocyte; DB, double-blind; DC, dendritic cell; DCR, disease control rate; DI-TMZ, dose-intensified temozolomide; DLT, dose-limiting toxicity; DLDA, diagonal linear discriminant analysis; DoR, duration of response; DTH, delayed-type hypersensitivity; dx, diagnosis; ECOG, Eastern Cooperative Oncology Group; EGFR, epidermal growth factor receptor; EGFRvIII, epidermal growth factor receptor variant III; ESTIMATE, Estimation of Stromal and Immune cells in Malignant Tumors using Expression data; f/u, follow-up; FACS, fluorescence-activated cell sorting; FoxP3, forkhead box P3; GAA, glioma-associated antigen; GBM, glioblastoma; GM-CSF, granulocyte-macrophage colony-stimulating factor; GR, glucocorticoid receptor; GSC-DCV, glioma stem cell-derived dendritic cell vaccine; GTR, gross total resection; GVHD, graft-versus-host disease; H3F3B, H3.3 histone B; HLA, human leukocyte antigen; HR, hazard ratio; HSV, herpes simplex virus; ICANS, immune effector cell-associated neurotoxicity syndrome; ICB, immune checkpoint blockade; ICI, immune checkpoint inhibitor; ICV, intracerebroventricular; IDH, isocitrate dehydrogenase; IDO1, indoleamine 2,3-dioxygenase 1; IFN, interferon; Ig, immunoglobulin; IHC, immunohistochemistry; IL, interleukin; incl., including; IQR, interquartile range; irCR, immune-related complete response; irPR, immune-related partial response; iRANO, immunotherapy Response Assessment in Neuro-Oncology; ISG, interferon-stimulated gene; ISR, IMA950-specific response; ITT, intent-to-treat; IV, intravenous; KPS, Karnofsky Performance Status; LAG-3, lymphocyte activation gene 3; LGG, low-grade glioma; MAF, mutant allele fraction; MGMT, O⁶-methylguanine-DNA methyltransferase; mo, months; MRS, magnetic resonance spectroscopy; MRD, measurable residual disease; MSS, mutation-specificity score; MTD, maximum tolerated dose; MUC16, mucin 16; mut, mutation; MV, measles virus; MVAF, maximal somatic variant allelic frequency; NA, not available; NED, no evidence of disease; nGBM, newly diagnosed glioblastoma; NR, not reached; NS, not significant; NTR, near total resection; ORR, objective response rate; OS, overall survival; PD, progressive disease; PD-1, programmed cell death protein 1; PD-L1, programmed death-ligand 1; PFS, progression-free survival; pfu, plaque-forming units; PLR, platelet-to-lymphocyte ratio; poly-ICLC, polyinosinic-polycytidylic acid stabilized with poly-L-lysine and carboxymethylcellulose; post-, post-treatment; PPV, personalized peptide vaccination; pre-, pre-treatment; PR, partial response; PsPD, pseudoprogression; PS, performance status; pt, patient; RANO, Response Assessment in Neuro-Oncology; RCT, randomized controlled trial; RDE, recommended dose for expansion; rGBM, recurrent glioblastoma; rGS, recurrent gliosarcoma; rHGG, recurrent high-grade glioma; RLT, restricted limiting toxicity; RPA, recursive partitioning analysis; RP2D, recommended Phase 2 dose; RT, radiotherapy; SAE, serious adverse event; SD, stable disease; SRD, significant residual disease; SRSF2, serine and arginine rich splicing factor 2; T1, time point 1; T2, time point 2; TCID50, 50% tissue culture infectious dose; Td, tetanus-diphtheria; TERT, telomerase reverse transcriptase; TGF-β, transforming growth factor beta; TIL, tumor-infiltrating lymphocyte; TISF, tumor in situ fluid; TMB, tumor mutational burden; TME, tumor microenvironment; TMZ, temozolomide; TNF, tumor necrosis factor; Treg, regulatory T cell; TUMAP, tumor-associated peptide; VEGF, vascular endothelial growth factor; VEGFR, vascular endothelial growth factor receptor; vp, viral particles; vs, versus; wk, week; WT1, Wilms tumor 1; yr, year; ZFN, zinc finger nuclease.
5.1 Immunosuppression in the Tumor Microenvironment
Regardless of the immunotherapeutic strategy employed, its ultimate antitumor efficacy depends on the effective activation of a functional immune system [158] (Figure 5). However, the TME of glioma, particularly glioblastoma, is characterized by profound immunosuppression, rendering immune cells—whether vaccine-induced T cells [159] or CAR-T cells [13, 14]—prone to inactivation or exhaustion upon entry into the tumor site.
Schematic overview of four major immunotherapeutic strategies for glioma. 1) Peptide vaccines and dendritic cell (DC) vaccines: Peptide vaccines deliver tumor-associated antigens (TAAs) in combination with adjuvants, thereby inducing DC maturation in vivo. DC vaccines are generated by differentiating patient-derived CD14⁺ monocytes into immature DCs, followed by loading with tumor antigens. Mature DCs promote the activation and differentiation of T cells. Activated T cells cross the blood-brain barrier (BBB) and exert anti-glioma effects through Fas/FasL-mediated apoptosis and the perforin/granzyme release pathway. 2) Oncolytic virus therapy: Oncolytic viruses selectively infect and lyse glioma cells, releasing TAAs and damage-associated molecular patterns (DAMPs), which in turn activate DCs and initiate T cell responses, ultimately leading to tumor cell killing. 3) Immune checkpoint inhibitors: These inhibitors block immune checkpoint signaling pathways, reverse T cell exhaustion, and thereby eliminate glioma cells. 4) Chimeric antigen receptor T (CAR-T) cell therapy: Patient-derived peripheral blood mononuclear cells (PBMCs) are isolated, genetically modified to express chimeric antigen receptors (CARs), expanded ex vivo, and then infused back into the patient. CAR-T cells cross the BBB and specifically target and kill glioma cells.
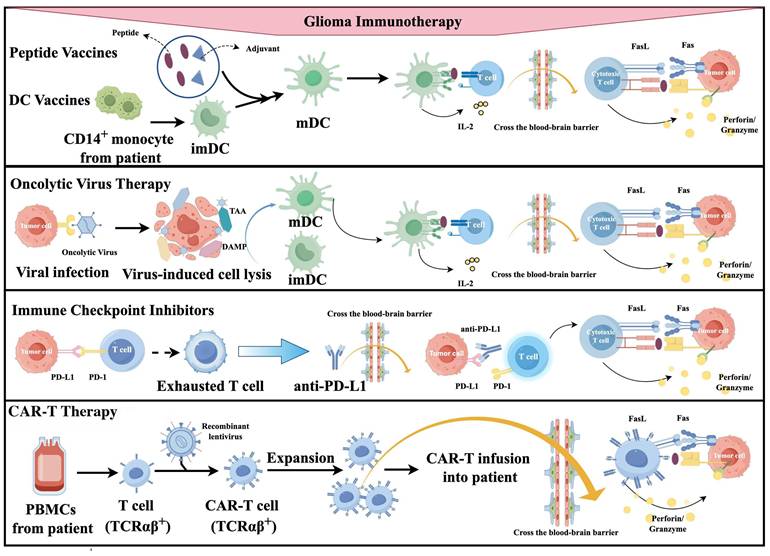
5.1.1 Impact on Vaccines
Vaccines require the presentation of antigens to DCs to subsequently activate T cells [160]. However, in the immunosuppressive TME, dendritic cell function is impaired, and activated T cells are also unable to effectively infiltrate and proliferate [160].
5.1.2 Impact on ICIs
The PD-1/PD-L1 immune checkpoint pathway is just one of many immunosuppressive mechanisms [161]. The TME also contains abundant Tregs, MDSCs, and inhibitory cytokines such as TGF-β and IL-10 [161]. Even upon PD-1/PD-L1 blockade, these additional mechanisms continue to effectively suppress T cell function [162].
5.1.3 Impact on CAR-T Cells
Even if CAR-T cells successfully reach the tumor site, they encounter the same immunosuppressive microenvironment, leading to poor persistence, rapid exhaustion, and an inability to sustain long-term antitumor effects [103].
5.1.4 Impact on Oncolytic Viruses
The primary mechanism underlying the antitumor effect of oncolytic viruses is a T cell-mediated, tumor-specific immune response [163]. However, the TME can inhibit T cell function through various mechanisms, thereby compromising the efficacy of this therapeutic approach [163].
5.2 Tumor Antigen Heterogeneity and Escape
Glioblastoma exhibits significant spatial and interpatient heterogeneity, which is a key factor contributing to immune evasion [164]. The expression of recognizable antigens on tumor cells is a prerequisite for vaccine-induced immune responses and for ICIs to relieve T cell suppression [165]. For oncolytic viruses, however, direct oncolysis depends on the expression of surface receptors on target cells, yet it does not require that all tumor cells express such receptors [166]. As long as a sufficient number of cells express the receptors and become infected, releasing ample damage-associated molecular patterns and tumor-associated antigens, an immune response can be activated to eliminate tumor cells that do not express the receptors [167].
Strategies such as multi-target vaccines [19], dual-target CAR-T cells [116], and "plug-and-play" CAR-T platforms (e.g., mSA2 CAR-T) [115] hold promise for overcoming immune evasion driven by tumor heterogeneity by enabling simultaneous targeting of multiple tumor-associated antigens or flexibly adapting to changes in antigen expression, positioning them as a key direction for future research.
5.3 Blood-Brain Barrier Constraints
Glioblastoma is located within the central nervous system, where the unique BBB not only acts as a physical obstacle to drug delivery but also restricts the entry of immune cells from the periphery into the tumor site [168].
5.3.1 Impact on Macromolecular Therapeutics and Oncolytic Viruses
Monoclonal antibodies (such as PD-1/PD-L1 antibodies), CAR-T cells, and oncolytic viruses are all macromolecules or nanoscale particles that are largely incapable of crossing an intact BBB under normal conditions [145-148]. Even though tumor growth can disrupt the local barrier, such disruption is heterogeneous and insufficient, resulting in suboptimal drug accumulation [156].
5.3.2 The Impact on Vaccines
Vaccines activate T cells in peripheral lymph nodes, and these activated T cells must successfully cross the BBB to exert their antitumor effects [169]. The presence of the BBB significantly limits the infiltration efficiency of these T cells [169]. Various delivery strategies, such as nanocarriers [61, 79] and focused ultrasound [76], offer potential approaches to overcome this physical barrier.
5.4 Adaptive Resistance and Tumor Evolution
Most studies suggest that tumors undergo dynamic evolution under therapeutic pressure. Even when an initial treatment is effective, tumors can acquire adaptive resistance through multiple mechanisms, including upregulation of alternative immune checkpoint molecules, metabolic reprogramming, or activation of bypass growth signaling pathways [68, 132]. However, a few studies have also demonstrated that tumors can evolve even in the absence of pharmacological intervention, under natural conditions [31].
For instance, following treatment with PD-L1 inhibitors, tumors may upregulate the expression of other checkpoint molecules (such as LAG-3, TIM-3, TIGIT, and CTLA-4), thereby achieving "bypass escape" [170]. For CAR-T therapy, in addition to direct antigen loss, tumors can also evade immune recognition by downregulating antigen processing and presentation machinery components, such as MHC class I molecules, thereby impairing the antitumor response of endogenous T cells and indirectly promoting immune evasion [171]. For vaccines, under the immune pressure induced by treatment, tumor cells may escape immune recognition by directly downregulating target antigen expression or by downregulating antigen processing and presentation machinery components, such as MHC class I molecules, thereby hindering the killing of tumor cells by CD8+ T cells [172]. Oncolytic virus infection of tumor cells depends on specific cell surface receptors [173]. Under therapeutic pressure, tumor cells can resist viral infection by downregulating or losing these receptors [173].
This mechanism reveals the underlying reasons for the transient efficacy of monotherapy and highlights the importance of combination therapy. The advantages of combination therapy can be summarized as follows: 1) Broader target coverage. Whether through the combination of ICIs [70], multi-target vaccines [19], or universal CAR-T platforms [115], the range of target coverage exceeds that of monotherapy, enabling more effective elimination of heterogeneous tumor cell populations. 2) Broader coverage of mechanisms of action. Under therapeutic pressure, tumors can acquire adaptive resistance through multiple mechanisms [68, 132]. To circumvent this, combining therapies with distinct mechanisms—such as ICIs with epigenetic modulators [91] or with oncolytic viruses [134]—makes it more difficult for tumors to escape. 3) Synergistic effects. The combination of certain therapies, such as chemotherapy with immunotherapy, can produce synergistic effects that achieve "1+1 > 2" therapeutic outcomes, surpassing the simple additive effects of individual agents [39]. 4) Improved safety. In a combination therapy setting, the dosage of each therapeutic agent can be reduced accordingly, thereby alleviating target organ toxicity and enhancing the safety and tolerability of treatment [174]. In line with the growing recognition of combination strategies, several ongoing clinical trials are evaluating the efficacy of immunotherapy combined with other modalities (Table 2).
Ongoing clinical trials of immunotherapy combination strategies in glioma
| Combination Strategy | Specific Regimen | Trial Phase | Target Population | Primary Endpoint / Objective | NCT Number |
|---|---|---|---|---|---|
| Vaccine + Chemoradiotherapy | Tamavaq™ personalized neoantigen vaccine + Radiotherapy + Temozolomide | Early Phase I | Newly diagnosed glioma | Primary: Safety, efficacy | NCT07077616 |
| Immuno-immuno combination | Autologous TIL therapy + Pembrolizumab (anti-PD-1) | Phase I/II | Advanced brain cancer (gliomas, meningiomas, etc.) | Primary: Efficacy, safety | NCT06640582 |
| Immunotherapy + Chemotherapy | Tarlatamab (DLL3-targeted BiTE) + Metronomic Temozolomide | Phase I/II | Adolescents and adults with recurrent high-grade CNS tumors (including IDH-mutant glioma, ependymoma, medulloblastoma) | Primary: Safety; Secondary: Clinical activity | NCT07243470 |
| Immunotherapy + Anti-angiogenic | CTX-009 (DLL4/VEGF-A bispecific antibody) + CTX-471 (anti-CD137 agonist antibody) | Phase Ib/II | Recurrent glioblastoma (CNS WHO grade 4, IDH-wildtype) | Primary: Safety; Secondary: Efficacy (preliminary) | NCT07392957 |
| Dendritic cell vaccine + Standard of care | DOC1021 (autologous dendritic cell immunotherapy) + Radiotherapy + Temozolomide + Tumor Treating Fields (TTFields) | Phase II | Newly diagnosed glioblastoma (WHO grade 4) | Primary: Safety, efficacy | NCT06805305 |
| Peptide vaccine + Chemotherapy | PEP-CMV vaccine + Adjuvant temozolomide (one cycle) | Phase I | Newly diagnosed MGMT-unmethylated glioblastoma | Primary: Safety, tolerability | NCT06132438 |
| ICI + Anti-angiogenic + Radiotherapy | Retifanlimab (anti-PD-1) + Bevacizumab + Hypofractionated Radiotherapy | Phase II | Recurrent glioblastoma | Primary: Efficacy | NCT06160206 |
| Gene therapy + Immunotherapy + Surgery | TGX-007 (AAV-HSV-tk + IL-12) intratumoral injection + oral valacyclovir + standard of care surgery | Phase I/II | Newly diagnosed or recurrent glioblastoma | Primary: Safety, OBD; Secondary: Efficacy | NCT07346144 |
| CAR-T+Chemotherapy | Autologous GD2-C7R CAR-T cells (GD2 CAR + constitutively active IL-7 receptor) + Cyclophosphamide/Fludarabine lymphodepletion | Phase I | GD2-expressing Brain Tumors (DMG, HGG, DIPG, medulloblastoma) | Primary: MTD; Secondary: CAR-T persistence, anti-tumor activity | NCT04099797 |
| Dual ICI + Surgery | Ipilimumab (anti-CTLA-4) + Nivolumab (anti-PD-1) administered IV (neo-adjuvant) and intracranial (adjuvant via Ommaya reservoir) + maximal safe resection | Phase I | Resectable recurrent glioblastoma | Primary: Safety, feasibility | NCT06097975 |
| Bispecific antibody-armed T cells + Focused ultrasound | EGFR BATs (anti-CD3 × anti-EGFR bispecific antibody-armed autologous T cells) + Low-intensity focused ultrasound (LIFU) with microbubbles for BBB opening | Phase I | Newly diagnosed MGMT unmethylated IDH-wildtype glioblastoma | Primary: Safety, feasibility; Secondary: T cell trafficking | NCT07343986 |
| bispecific antibody-immunotoxin + immunomodulator | D2C7-IT (bispecific antibody) + 2141-V11 (immunomodulator) | Phase I/II | Resected recurrent glioblastoma | Primary: Safety; Secondary: Efficacy | NCT06455605 |
| Radioimmunotherapy + ICI | Actimab-A (anti-CD33 monoclonal antibody conjugated with actinium-225) + Cemiplimab (anti-PD-1) | Phase I | Recurrent glioblastoma | Primary: Safety, tolerability; Secondary: Efficacy | NCT07422363 |
| Allogeneic γδ T cell therapy ± Chemo/Radiotherapy | Allogeneic Vγ9Vδ2 T cells (intraventricular via Ommaya reservoir) ± Temozolomide or radiotherapy | Phase I | WHO grade IV malignant glioma (glioblastoma, DIPG, medulloblastoma, ependymoma) | Primary: Safety, feasibility; Secondary: Efficacy | NCT06396481 |
| Bispecific antibody-immunotoxin+ immunomodulator + Radiotherapy | D2C7-IT (bispecific antibody-immunotoxin) + 2141-V11 (immunomodulator) | Phase I/II | Newly diagnosed MGMT unmethylated glioblastoma | Primary: Safety; Secondary: Efficacy | NCT05734560 |
| CAR-T + Dual ICIs + Neoadjuvant/Adjuvant | IL13Rα2-CAR T cells (intracranial via Rickham catheter) + Nivolumab (anti-PD-1) ± Ipilimumab (anti-CTLA-4) | Phase I | Resectable recurrent glioblastoma | Primary: Safety; Secondary: Efficacy | NCT04003649 |
| IL-7 agonist + PD-1 inhibitor + Surgery | Neoadjuvant Efineptakin Alfa (NT-I7) and Pembrolizumab | Phase II | Recurrent glioblastoma | Primary: Safety; Secondary: Efficacy | NCT05465954 |
| PD-L1 inhibitor + Multi-kinase inhibitor | Atezolizumab (anti-PD-L1) + Cabozantinib (VEGFR/MET/AXL inhibitor) | Phase I/II | Recurrent glioblastoma | Primary: Safety; Secondary: Efficacy | NCT05039281 |
| Dendritic cell vaccine + PD-1 inhibitor | Hybrid dendritic cell vaccine + Pembrolizumab (anti-PD-1) | Phase III | Adult glioblastoma (post standard-of-care surgery, chemotherapy, radiotherapy) | Primary: Efficacy, safety | NCT06749925 |
| Dual ICIs + Chemotherapy + Ultrasound BBB opening | Balstilimab (anti-PD-1) + Botensilimab (anti-CTLA-4) + Liposomal doxorubicin + Soncloud-9 ultrasound device for BBB opening | Phase IIa | Newly diagnosed glioblastoma (post-radiotherapy) | Primary: Safety, feasibility; Secondary: Efficacy | NCT05864534 |
| PD-L1/VEGF bispecific antibody + Chemotherapy | BNT327 (Pumitamig, anti-PD-L1 x VEGF-A bispecific antibody) ± Temozolomide | Phase II | Recurrent glioblastoma (WHO grade IV, post-radiotherapy + Temozolomide) | Primary: Safety; Secondary: Efficacy | NCT07297212 |
| CAR-NK + IL-15 agonist + Anti-VEGF + TTFields | Nogapendekin alfa inbakicept (IL-15 agonist) + PD-L1 t-haNK (CAR-NK cells) + Bevacizumab + Tumor Treating Fields (TTFields) | Phase II/IIB | Recurrent or progressive glioblastoma | Primary: Safety; Secondary: Efficacy | NCT06061809 |
| Allogeneic virus-specific T cells + PD-1 inhibitor | Allogeneic CMV-specific T cells + Pembrolizumab (anti-PD-1) | Phase I/II | Newly diagnosed and recurrent glioblastoma/grade 4 astrocytoma | Primary: MTD, RP2D; Secondary: Anti-tumor activity | NCT06157541 |
Abbreviations: AAV, adeno-associated virus; anti-CD137, anti-cluster of differentiation 137; anti-CTLA-4, anti-cytotoxic T-lymphocyte-associated protein 4; anti-PD-1, anti-programmed cell death protein 1; anti-PD-L1, anti-programmed death-ligand 1; AXL, AXL receptor tyrosine kinase; BBB, blood-brain barrier; BiTE, bispecific T-cell engager; CAR, chimeric antigen receptor; CAR-NK, chimeric antigen receptor natural killer; CAR-T, chimeric antigen receptor T-cell; CMV, cytomegalovirus; CNS, central nervous system; DIPG, diffuse intrinsic pontine glioma; DLL3, delta-like ligand 3; DLL4, delta-like ligand 4; DMG, diffuse midline glioma; EGFR, epidermal growth factor receptor; GD2, disialoganglioside 2; HGG, high-grade glioma; HSV-tk, herpes simplex virus thymidine kinase; ICI, immune checkpoint inhibitor; IDH, isocitrate dehydrogenase; IL, interleukin; IL13Rα2, interleukin-13 receptor alpha 2; LIFU, low-intensity focused ultrasound; MET, mesenchymal-epithelial transition factor; MGMT, O⁶-methylguanine-DNA methyltransferase; MTD, maximum tolerated dose; OBD, optimal biological dose; PD-1, programmed cell death protein 1; PD-L1, programmed death-ligand 1; RP2D, recommended Phase II dose; TIL, tumor-infiltrating lymphocyte; TTFields, tumor treating fields; VEGF, vascular endothelial growth factor; VEGF-A, vascular endothelial growth factor A; VEGFR, vascular endothelial growth factor receptor.
In summary, glioma immunotherapy faces a series of interrelated and overlapping challenges, including a complex immunosuppressive TME, high tumor heterogeneity, the restrictive nature of the BBB, and tumor adaptive resistance and evolution. In the future, strategies such as multi-target approaches, optimization of delivery systems, remodeling of the TME, and the development of rational combination therapies hold promise for overcoming these obstacles one by one, ultimately achieving breakthroughs in glioma immunotherapy.
6. Biomarkers: From Classification to Combination Strategies
The biomarkers discussed in this manuscript have distinct functional roles in clinical practice. To establish a unified classification framework, this section categorizes them into three groups: predictive biomarkers, prognostic biomarkers, and biomarkers guiding combination therapy selection.
6.1 Predictive Biomarkers
Predictive biomarkers are defined as biological characteristics that can be objectively measured and indicate the likelihood of response to a specific therapeutic intervention [175]. These markers are critical for patient selection in targeted therapy and immunotherapy, enabling the identification of patients most likely to derive clinical benefit while minimizing unnecessary toxicity [175]. Table 3 summarizes key predictive biomarkers discussed in this review, along with their associated therapies and current evidence.
Predictive biomarkers for therapeutic response
| Therapeutic Strategy | Biomarker | Predictive Role | Corresponding Study |
|---|---|---|---|
| Cancer Vaccine | HLA-A2, HLA-A1 | HLA typing for patient selection | ICT-107 [25] |
| IDH1, TERT, B7-H4 | IDH1ʷᵗTERTᴹᵀ subgroup showed OS/PFS benefit; low B7-H4 subgroup showed OS benefit | GSC-DCV [26] | |
| MGMT methylation | Methylated patients: mOS 41.4 months vs 16.5 months in unmethylated | SurVaxM + adj TMZ [34] | |
| MGMT methylation | Among ISR⁺ patients, methylated: mOS 28.3 months vs 14.8 months in unmethylated | IMA950 [19] | |
| HLA-A*24:02, WT1 | Patient selection based on HLA typing and WT1 expression | WT1 cocktail vaccine [38] | |
| HLA-A*24:02 | Patient selection based on HLA typing | VEGFRs peptide vaccine + TMZ [39] | |
| IDH1 mutation | Targeting IDH1-mutant glioma patients | Personalized mutIDH1 multi-peptide vaccine [20] | |
| Immune Checkpoint Inhibitor | MGMT methylation | Methylated patients: mOS 35.7 months vs 12.6 months in unmethylated | Nivolumab + Ipilimumab (pre-radiation) [68] |
| CAR T-Cell Therapy | EGFRvIII, MGMT unmethylated | Patient selection based on EGFRvIII expression; MGMT unmethylated status for patient selection | CART-EGFRvIII + Pembrolizumab [13]; CART-EGFRvIII cells [14] |
| EGFR amplification, IDH wild-type | Patient selection based on EGFR amplification and IDH wild-type status | CART-EGFR-IL13Rα2 [110] | |
| IL13Rα2 | Patient selection based on IL13Rα2 expression | GRm13Z40-2 cells [112] | |
| GD2, IDH wild-type | Patient selection based on GD2 expression; IDH wild-type | GD2-specific 4SCAR-T [113] | |
| MMP-2 | Patient selection based on MMP-2 expression | CLTX-CAR T cells [114] | |
| Oncolytic Virotherapy | ISG signature | ISG signature score inversely correlated with viral replication, predicting treatment response | MV-CEA [136] |
| TMEmedium subtype | Treatment response observed only in the "TMEmedium" subtype (exploratory) | DNX-2401 + Pembrolizumab [132] |
Abbreviations: adj TMZ, adjuvant temozolomide; B7-H4, B7 homolog 4; CAR-T, chimeric antigen receptor T-cell; EGFR, epidermal growth factor receptor; EGFRvIII, epidermal growth factor receptor variant III; GD2, disialoganglioside 2; GSC-DCV, glioma stem cell-derived dendritic cell vaccine; HLA, human leukocyte antigen; IDH, isocitrate dehydrogenase; IL13Rα2, interleukin-13 receptor alpha 2; ISG, interferon-stimulated gene; ISR, immune response; MGMT, O⁶-methylguanine-DNA methyltransferase; MMP-2, matrix metalloproteinase-2; mOS, median overall survival; OS, overall survival; PFS, progression-free survival; TERT, telomerase reverse transcriptase; TME, tumor microenvironment; TMZ, temozolomide; VEGFR, vascular endothelial growth factor receptor; WT1, Wilms tumor 1.
6.2 Prognostic Biomarkers
6.2.1 IDH-wildtype glioblastoma
MGMT promoter methylation is one of the most well-established prognostic biomarkers in IDH-wildtype glioblastoma, with its methylation status associated with significant improvements in OS and PFS [176], and it retains prognostic value even in the context of adjunctive therapies such as tumor-treating fields [177]. TERT promoter mutations occur in approximately 80% of IDH-wildtype glioblastomas [178]; however, their prognostic significance remains controversial. Some studies have associated TERT promoter mutations with poorer survival [179], whereas others suggest a synergistic interaction between TERT promoter mutations and MGMT methylation status, with patients harboring both alterations exhibiting the longest survival [180]. In addition, chromosome 7 gain and chromosome 10 loss, along with EGFR amplification, are characteristic genomic alterations in IDH-wildtype glioblastoma [181] and are closely associated with worse OS and PFS [182]. PTEN mutations, observed in approximately 40% of cases, are likewise associated with poor prognosis and treatment resistance [183].
6.2.2 IDH-mutant astrocytomas
In IDH-mutant diffuse astrocytomas, tumor grade is a key prognostic factor, with grade 3 tumors exhibiting a median OS of 8.1 years, whereas grade 4 tumors show a significantly shorter median OS of 4.7 years (p < 0.05) [184]. Homozygous CDKN2A/B deletion is strongly associated with worse survival outcomes; patients with this deletion have a median OS of only 1.8 years compared with 5.5 years in those without the deletion (p < 0.001) [2]. The presence of IDH mutation has now been established as a favorable prognostic factor in diffuse astrocytomas and oligodendrogliomas [185]. MGMT promoter methylation is more prevalent in IDH-mutant gliomas and is independently associated with improved OS and PFS when treated with TMZ; however, its predictive value appears to be largely limited to grade 4 tumors [186]. In addition, TP53 mutations and ATRX loss frequently co-occur, indicating genomic instability, and are generally associated with favorable prognosis [187]; patients with ATRX mutations are younger and have longer OS [188].
6.2.3 IDH-mutant and 1p/19q-codeleted oligodendroglioma
Oligodendrogliomas have long been recognized as having a favorable prognosis, with their defining feature being the concurrent presence of IDH1/2 mutations and 1p/19q codeletion [2]. The molecular landscape of oligodendrogliomas includes frequent TERT promoter mutations [189], retained ATRX expression, and absence of p53 accumulation, which is consistent with the mutual exclusivity between 1p/19q codeletion and TP53 or ATRX alterations [190]. In a subset of cases, CDKN2A/B deletions may be detected, an alteration associated with more aggressive tumor behavior and poorer prognosis [191].
6.3 Biomarkers for Combination Therapy Selection
The rational design of combination therapies increasingly relies on biomarkers that identify synergistic vulnerabilities. Table 4 outlines representative biomarkers and their associated combination approaches.
Biomarker guiding combination therapy selection
| Therapeutic Strategy | Biomarker | Predictive Role | Corresponding Study |
|---|---|---|---|
| Cancer Vaccine | MGMT methylation | Methylated patients: mOS 41.4 months vs 16.5 months in unmethylated | SurVaxM + adj TMZ [34] |
| MGMT methylation | Among ISR⁺ patients, methylated: mOS 28.3 months vs 14.8 months in unmethylated | IMA950 [19] | |
| Immune Checkpoint Inhibitor | MGMT methylation | Methylated patients: mOS 35.7 months vs 12.6 months in unmethylated | Nivolumab + Ipilimumab (pre-radiation) [68] |
| ESTIMATE Immune Score, molecular subtype | High immune score tumors: mOS 24.8 vs 14.5 months; mesenchymal subtype: mOS 26.5 months | Atezolizumab + RT/TMZ [70] | |
| PLR, MUC16, H3F3B, SRSF2 | Low PLR correlated with improved PFS/OS; MUC16 mutation, H3F3B amplification, SRSF2 amplification associated with worse OS | Tislelizumab + Low-Dose Bevacizumab [72, 73] | |
| CAR T-Cell Therapy | EGFRvIII, MGMT unmethylated | Patient selection based on EGFRvIII expression; MGMT unmethylated status for patient selection | CART-EGFRvIII + Pembrolizumab [13]; CART-EGFRvIII cells [14] |
| EGFR amplification, IDH wild-type | Patient selection based on EGFR amplification and IDH wild-type status | CART-EGFR-IL13Rα2 [110] | |
| IL13Rα2 | Patient selection based on IL13Rα2 expression | GRm13Z40-2 cells [112] | |
| Oncolytic Virotherapy | ISG signature | ISG signature score inversely correlated with viral replication, predicting treatment response | MV-CEA [136] |
| TMEmedium subtype | Treatment response observed only in the "TMEmedium" subtype (exploratory) | DNX-2401 + Pembrolizumab [132] |
Abbreviations: adj TMZ, adjuvant temozolomide; CAR-T, chimeric antigen receptor T-cell; EGFR, epidermal growth factor receptor; EGFRvIII, epidermal growth factor receptor variant III; ESTIMATE, Estimation of Stromal and Immune cells in Malignant Tumors using Expression data; H3F3B, H3.3 histone B; IDH, isocitrate dehydrogenase; IL13Rα2, interleukin-13 receptor alpha 2; ISG, interferon-stimulated gene; ISR, immune response; MGMT, O⁶-methylguanine-DNA methyltransferase; MMP-2, matrix metalloproteinase-2; mOS, median overall survival; MUC16, mucin 16; OS, overall survival; PFS, progression-free survival; PLR, platelet-to-lymphocyte ratio; RT, radiotherapy; SRSF2, serine and arginine rich splicing factor 2; TME, tumor microenvironment; TMZ, temozolomide.
7. Conclusions
Recent progress in glioma immunotherapy has been substantial, particularly in efficacy [136, 139], BBB penetration [47-50, 76], TME remodeling [47, 49, 55, 57, 60], overcoming heterogeneity [46, 115], and controlled release with reduced toxicity [144]. However, several critical research gaps remain to be addressed. 1) Target selection. Many targets, such as EGFRvIII, exhibit unstable expression and are prone to loss following treatment, leading to resistance [31]. An ideal target should be closely associated with tumor cell growth, proliferation, and invasion, while maintaining stable expression that is less likely to be lost [192]. Therefore, the identification and screening of optimal targets constitute a critical priority, as this will help mitigate off-target effects and reduce the incidence of therapeutic resistance. 2) Biomarker identification and patient stratification. The identification of reliable biomarkers in glioma faces unique challenges compared with many other solid tumors. (a) The presence of the blood-brain barrier (BBB) severely restricts the release of tumor-derived biomolecules into the peripheral circulation, resulting in extremely low abundance of circulating biomarkers, including circulating tumor DNA (ctDNA) and circulating tumor cells (CTCs) [193, 194]. Studies have shown that the detection rate of plasma ctDNA in patients with glioma is currently below 20%, which is considerably lower than that reported for other malignancies [194]. (b) Glioma exhibits high tumor heterogeneity, making it difficult for a single biopsy or a single biomarker to fully capture the overall tumor status [195]. This characteristic often manifests in clinical trials as a lack of statistically significant overall outcomes or statistically significant efficacy only in specific subgroups, suggesting that current patient stratification strategies remain insufficiently refined [49]. (c) The TME of glioma is highly immunosuppressive, characterized by a low tumor mutational burden (TMB) and a paucity of high-quality neoantigens, which further limits the identification and application of immune-related biomarkers [196]. (d) The expression of certain biomarkers in glioma is dynamic, whereas clinical detection still relies primarily on pathological biopsy, making it difficult to capture real-time changes during treatment [197]. (e) Owing to the infiltrative growth pattern of glioma, obtaining tissue samples carries significant risks; although liquid biopsy holds promise, standardized methods for its application have not yet been established [197]. 3) Preclinical model selection. The representativeness of certain models is limited. In immunotherapy studies in particular, a fully competent immune system is essential, yet the establishment of humanized models often relies on immunodeficient mice, which compromises the representativeness of efficacy assessments [48]. Selecting models that are reproducible, amenable to visualization, biomimetic, and easy to establish is crucial for facilitating clinical translation [198]. 4) Drug design. Drug design should take into account patient compliance, prioritizing convenient routes of administration. From the early stages of design, the involvement of pharmaceutical analysis experts is essential to develop analytical protocols that support preparation, purification, and scalable manufacturing. Additionally, aligning drug development with real-world manufacturing processes and patient needs is critical. The establishment of versatile platforms, such as the mSA2 system, can help shorten the research and development cycle and reduce costs [115]. 5) Drug development. The safety of CAR-T therapy requires further validation [106-109]. Enhancing the persistence of CAR-T cells in vivo [112, 114] and preventing the rapid clearance of oncolytic viruses by the host [145] remain key challenges. Currently, glioma immunotherapy continues to be constrained by the BBB, high tumor heterogeneity, the immunosuppressive microenvironment, and adaptive resistance accompanied by tumor evolution [13, 152-157]. Future progress is expected through improved delivery routes, combination strategies, the identification of optimal targets, and remodeling of the immune microenvironment. 6) Clinical trial considerations. (a) Patient selection bias. Enrolling patients without disease progression following standard therapy as trial subjects, compared with general patient populations, may lead to an overestimation of treatment efficacy [32]. (b) Control arm selection. With the advancement of standard-of-care treatments, OS in historical control groups has improved, making it difficult to reproduce promising results from early-phase trials in subsequent randomized studies [31]. (c) Control arm design. Immunotherapy trials should use standard therapy—at minimum TMZ—as the comparator to determine whether the investigational treatment offers improvements over existing therapies [66]. (d) Insufficient sample size. The low incidence and high mortality rate of gliomas make patient recruitment challenging, limiting the feasibility of many trials and resulting in underpowered analyses [26, 46, 114, 132]. Multicenter international collaboration is urgently needed, along with efforts to standardize treatment protocols. (e) Contradictory findings. Some studies suggest that immunotherapy requires minimal residual disease [46], whereas others have observed long-term survival benefits in patients without complete resection [31]. The rindopepimut trial found no correlation between antibody titers and survival benefit, suggesting that T-cell responses may serve as a more relevant selection criterion [31]; conversely, most trials, including AFTV, have reported that higher antibody levels correlate with better outcomes [46]. 7) Efficacy evaluation. Current imaging modalities are insufficient for assessing treatment response, highlighting the need for novel techniques [139]. Certain therapies, such as AFTV, contain theoretically all tumor antigens, making it impossible to evaluate efficacy based on specific antibody titers, necessitating the establishment of new evaluation criteria [46]. 8) Clinical translation. The production, quality control, and management of novel biological products require standardization [52]. Furthermore, biomarker-guided stratification or individualized treatment approaches are costly and require policy support [7]. Although each of the four major immunotherapy strategies faces distinct translational challenges [51, 52, 86, 125, 151], oncolytic virus therapy currently demonstrates the most promising translational potential. This approach has garnered significant attention due to its unique dual mechanism of "oncolysis plus immune activation" [199, 200]. G47Δ has been approved in Japan for the treatment of malignant glioma, with a Phase II trial reporting a one-year survival rate of 84.2% and a median OS of 20.2 months, marking a milestone in the field [139]. Furthermore, the combination of oncolytic viruses with ICIs has shown synergistic effects in preclinical and early-phase clinical studies, with the potential to convert "cold" tumors into "hot" tumors [134]. In contrast, ICIs are unlikely to confer additional survival benefits in the absence of molecular screening or specific biomarker guidance [43, 74, 75]. Tumor vaccines are limited in monotherapy due to tumor heterogeneity and antigen loss; however, their value as components of combination regimens should not be overlooked [31]. Adoptive cell therapies similarly face challenges such as antigen loss and limited in vivo persistence, resulting in modest efficacy as monotherapies [13, 112, 114]. Nonetheless, the recently developed mSA2 universal platform offers a promising strategy to address antigen loss by enabling simple exchange of biotinylated antibodies, providing new opportunities for this therapeutic modality [115].
Abbreviations
AEs: Adverse Events; AFTV: Autologous Formalin-fixed Tumor Vaccine; ARMed CAR-T: Checkpoint Antibody Receptor-modified Chimeric Antigen Receptor T-cell; ARPC1B: Actin Related Protein 2/3 Complex Subunit 1B; ATA: Autologous Tumor Antigen; BBB: Blood-Brain Barrier; BRD9: Bromodomain-containing Protein 9; CAR-T: Chimeric Antigen Receptor T-cell; CFZ: Clofazimine; CNS: Central Nervous System; DAMPs: Damage-associated molecular patterns; DDR: DNA Damage Response; DFMO: Difluoromethylornithine; DCs: Dendritic Cells; dsRNA: Double-stranded RNA; EGFRvIII: Epidermal Growth Factor Receptor Variant III; EV: Extracellular Vesicles; GAA: Glioma-associated Antigen; GAPVAC: Glioma Active Personalized Vaccine Consortium; GARP: Glycoprotein A Repetitions Predominant; GBM: Glioblastoma; GBO: Glioblastoma Organoid; GSC: Glioma stem cell; HCMV: Human Cytomegalovirus; HLA-A2: Human Leukocyte Antigen A2; HSV-1: Herpes Simplex Virus Type 1; IBDV: Infectious Bursal Disease Virus; ICB: Immune Checkpoint Blockade; ICD: Immunogenic Cell Death; ICIs: Immune Checkpoint Inhibitors; IDH1: Isocitrate Dehydrogenase; IDH1/2: Isocitrate Dehydrogenase 1/2; IDH1-vac: Isocitrate Dehydrogenase 1 Mutation-targeted Vaccine; ISG: Interferon-stimulated Gene; IV: intravenous delivery; JAK: Janus Kinase; KLH: Keyhole Limpet Hemocyanin; LAK: Lymphokine-activated Killer; LNPs: Lipid Nanoparticles; MCP: Multi-cationic Protein; MDSCs: Myeloid-derived Suppressor Cells; METTL14: Methyltransferase-like 14; MGMT: O-6-Methylguanine-DNA Methyltransferase; MHC: Major histocompatibility complex; mSA2 CAR-T: Monomeric Streptavidin 2-based Chimeric Antigen Receptor T-cells; MV-CEA: Measles Virus-based Carcinoembryonic Antigen-expressing Oncolytic Virus; MYXV: Myxoma Virus; NaV: Nanoshield Vaccine; nGBM: Newly Diagnosed Glioblastoma; NO: Nitric Oxide; nsIL12: Non-secreting Interleukin-12; OA@Cho: Cholesterol-modified Adenovirus; oHSV-1: Oncolytic Herpes Simplex Virus-1; OS: Overall Survival; PAMPs: Pathogen-associated molecular patterns; PBMCs: Peripheral blood mononuclear cells; PDT: Photodynamic Therapy; PFS: Progression-Free Survival; PPV: Personalized Peptide Vaccine; PVSRIPO: Poliovirus (Recombinant Nonpathogenic Poliovirus: Rhinovirus Recombinant); rGBM: Recurrent Glioblastoma; ROS: Reactive Oxygen Species; scFv: Single-chain variable fragment; SPOD-NV: Survivin Peptide-CpG Nanovaccine; Stars NV: Sonodynamic Nanovaccine; STAT: Signal Transducer and Activator of Transcription; SurVaxM: Survivin-targeting Peptide Vaccine; TAAs: Tumor-associated Antigens; TAMs: Tumor-associated Macrophages; TCF: Tumor Cavity Fluid; TCR-T: T Cell Receptor-engineered T Cells; TERT: Telomerase Reverse Transcriptase; TGFB2: Transforming Growth Factor Beta 2; TGFβ: Transforming Growth Factor Beta; TILs: Tumor-infiltrating Lymphocytes; TME: Tumor microenvironment; TMZ: Temozolomide; Tregs: Regulatory T Cells; TSAs: Tumor-specific Antigens; WHO: World Health Organization; WT1: Wilms Tumor 1; ZIKV: Zika Virus; ZNF638: Zinc Finger Protein 638.
Acknowledgements
All figures in this review were created using Figdraw.
Author Contributions
JG: Conceptualization, Writing-review & editing, Supervision, Funding acquisition, Visualization.
YH: Writing-review & editing, Supervision, Funding acquisition, Visualization.
JL: Writing-original draft, Writing-review & editing, Formal analysis, Validation, Project administration.
GY: Writing-review & editing, Formal analysis.
ZC, HQ, and MZ: Writing-review & editing, Software.
CZ and XL: Writing-review & editing, Data curation, Investigation.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Fekrirad Z, Barzegar Behrooz A, Ghaemi S, Khosrojerdi A, Zarepour A, Zarrabi A. et al. Immunology Meets Bioengineering: Improving the Effectiveness of Glioblastoma Immunotherapy. Cancers (Basel). 2022 14
2. Louis DN, Perry A, Wesseling P, Brat DJ, Cree IA, Figarella-Branger D. et al. The 2021 WHO Classification of Tumors of the Central Nervous System: a summary. Neuro Oncol. 2021;23:1231-51
3. Komori T. Grading of adult diffuse gliomas according to the 2021 WHO Classification of Tumors of the Central Nervous System. Lab Invest. 2022;102:126-33
4. Craig H, Tamar B, Roger J P, Patrick Y W. Clinical implications of the 2021 edition of the WHO classification of central nervous system tumours. Nat Rev Neurol. 2022 18
5. Choudhary N, Osorio RC, Oh JY, Aghi MK. Metabolic Barriers to Glioblastoma Immunotherapy. Cancers (Basel). 2023 15
6. Yasinjan F, Xing Y, Geng H, Guo R, Yang L, Liu Z. et al. Immunotherapy: a promising approach for glioma treatment. Front Immunol. 2023;14:1255611
7. Zhou Y, Shi F, Zhu J, Yuan Y. An update on the clinical trial research of immunotherapy for glioblastoma. Front Immunol. 2025;16:1582296
8. Bianconi A, Palmieri G, Aruta G, Monticelli M, Zeppa P, Tartara F. et al. Updates in Glioblastoma Immunotherapy: An Overview of the Current Clinical and Translational Scenario. Biomedicines. 2023 11
9. Gagliardi F, De Domenico P, Snider S, Roncelli F, Comai S, Mortini P. Immunomodulatory mechanisms driving tumor escape in glioblastoma: The central role of IDO and tryptophan metabolism in local and systemic immunotolerance. Crit Rev Oncol Hematol. 2025;209:104657
10. Desland FA, Hormigo A. The CNS and the Brain Tumor Microenvironment: Implications for Glioblastoma Immunotherapy. Int J Mol Sci. 2020 21
11. Mishchenko TA, Turubanova VD, Gorshkova EN, Krysko O, Vedunova MV, Krysko DV. Glioma: bridging the tumor microenvironment, patient immune profiles and novel personalized immunotherapy. Front Immunol. 2023;14:1299064
12. Zhao T, Li C, Ge H, Lin Y, Kang D. Glioblastoma vaccine tumor therapy research progress. Chin Neurosurg J. 2022;8:2
13. Bagley SJ, Binder ZA, Lamrani L, Marinari E, Desai AS, Nasrallah MP. et al. Repeated peripheral infusions of anti-EGFRvIII CAR T cells in combination with pembrolizumab show no efficacy in glioblastoma: a phase 1 trial. Nat Cancer. 2024;5:517-31
14. O'Rourke DM, Nasrallah MP, Desai A, Melenhorst JJ, Mansfield K, Morrissette JJD. et al. A single dose of peripherally infused EGFRvIII-directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci Transl Med. 2017 9
15. Bagley SJ, Logun M, Fraietta JA, Wang X, Desai AS, Bagley LJ. et al. Intrathecal bivalent CAR T cells targeting EGFR and IL13Rα2 in recurrent glioblastoma: phase 1 trial interim results. Nat Med. 2024;30:1320-9
16. Sloan AE, Dansey R, Zamorano L, Barger G, Hamm C, Diaz F. et al. Adoptive immunotherapy in patients with recurrent malignant glioma: preliminary results of using autologous whole-tumor vaccine plus granulocyte-macrophage colony-stimulating factor and adoptive transfer of anti-CD3-activated lymphocytes. Neurosurg Focus. 2000;9:e9
17. Yamanaka R, Abe T, Yajima N, Tsuchiya N, Homma J, Kobayashi T. et al. Vaccination of recurrent glioma patients with tumour lysate-pulsed dendritic cells elicits immune responses: results of a clinical phase I/II trial. Br J Cancer. 2003;89:1172-9
18. Izumoto S. Peptide vaccine. Adv Exp Med Biol. 2012;746:166-77
19. Rampling R, Peoples S, Mulholland PJ, James A, Al-Salihi O, Twelves CJ. et al. A Cancer Research UK First Time in Human Phase I Trial of IMA950 (Novel Multipeptide Therapeutic Vaccine) in Patients with Newly Diagnosed Glioblastoma. Clin Cancer Res. 2016;22:4776-85
20. Zelba H, Shao B, Rabsteyn A, Reinhardt A, Greve C, Oenning L. et al. In-depth characterization of vaccine-induced neoantigen-specific T cells in patients with IDH1-mutant glioma undergoing personalized peptide vaccination. J Immunother Cancer. 2025 13
21. Berneman ZN, De Laere M, Germonpré P, Huizing MT, Willemen Y, Lion E. et al. WT1-mRNA dendritic cell vaccination of patients with glioblastoma multiforme, malignant pleural mesothelioma, metastatic breast cancer, and other solid tumors: type 1 T-lymphocyte responses are associated with clinical outcome. J Hematol Oncol. 2025;18:9
22. Ridolfi L, Gurrieri L, Riva N, Bulgarelli J, De Rosa F, Guidoboni M. et al. First step results from a phase II study of a dendritic cell vaccine in glioblastoma patients (CombiG-vax). Front Immunol. 2024;15:1404861
23. Bota DA, Piccioni DE, Duma CM, Kesari S, Carrillo JA, LaRocca RV. et al. Phase 2 trial of personal dendritic cell vaccines in newly diagnosed glioblastoma: 3-year follow-up and correlations with survival. Hum Vaccin Immunother. 2025;21:2556591
24. Wang QT, Nie Y, Sun SN, Lin T, Han RJ, Jiang J. et al. Tumor-associated antigen-based personalized dendritic cell vaccine in solid tumor patients. Cancer Immunol Immunother. 2020;69:1375-87
25. Wen PY, Reardon DA, Armstrong TS, Phuphanich S, Aiken RD, Landolfi JC. et al. A Randomized Double-Blind Placebo-Controlled Phase II Trial of Dendritic Cell Vaccine ICT-107 in Newly Diagnosed Patients with Glioblastoma. Clin Cancer Res. 2019;25:5799-807
26. Yao Y, Luo F, Tang C, Chen D, Qin Z, Hua W. et al. Molecular subgroups and B7-H4 expression levels predict responses to dendritic cell vaccines in glioblastoma: an exploratory randomized phase II clinical trial. Cancer Immunol Immunother. 2018;67:1777-88
27. Reap EA, Suryadevara CM, Batich KA, Sanchez-Perez L, Archer GE, Schmittling RJ. et al. Dendritic Cells Enhance Polyfunctionality of Adoptively Transferred T Cells That Target Cytomegalovirus in Glioblastoma. Cancer Res. 2018;78:256-64
28. Mitchell DA, Batich KA, Gunn MD, Huang MN, Sanchez-Perez L, Nair SK. et al. Tetanus toxoid and CCL3 improve dendritic cell vaccines in mice and glioblastoma patients. Nature. 2015;519:366-9
29. Batich KA, Reap EA, Archer GE, Sanchez-Perez L, Nair SK, Schmittling RJ. et al. Long-term Survival in Glioblastoma with Cytomegalovirus pp65-Targeted Vaccination. Clin Cancer Res. 2017;23:1898-909
30. Liau LM, Ashkan K, Tran DD, Campian JL, Trusheim JE, Cobbs CS. et al. First results on survival from a large Phase 3 clinical trial of an autologous dendritic cell vaccine in newly diagnosed glioblastoma. J Transl Med. 2018;16:142
31. Weller M, Butowski N, Tran DD, Recht LD, Lim M, Hirte H. et al. Rindopepimut with temozolomide for patients with newly diagnosed, EGFRvIII-expressing glioblastoma (ACT IV): a randomised, double-blind, international phase 3 trial. Lancet Oncol. 2017;18:1373-85
32. Schuster J, Lai RK, Recht LD, Reardon DA, Paleologos NA, Groves MD. et al. A phase II, multicenter trial of rindopepimut (CDX-110) in newly diagnosed glioblastoma: the ACT III study. Neuro Oncol. 2015;17:854-61
33. Platten M, Bunse L, Wick A, Bunse T, Le Cornet L, Harting I. et al. A vaccine targeting mutant IDH1 in newly diagnosed glioma. Nature. 2021;592:463-8
34. Ahluwalia MS, Reardon DA, Abad AP, Curry WT, Wong ET, Figel SA. et al. Phase IIa Study of SurVaxM Plus Adjuvant Temozolomide for Newly Diagnosed Glioblastoma. J Clin Oncol. 2023;41:1453-65
35. Withers HG, Figel SA, Qiu J, Ahluwalia MS, Reardon D, Abad AP. et al. Intratumoral B cell and interferon signatures in newly diagnosed glioblastoma are associated with longer survival in patients treated with SurVaxM. Cancer Immunol Immunother. 2025;74:332
36. Fu S, Piccioni DE, Liu H, Lukas RV, Kesari S, Aregawi D. et al. A phase I study of the WT2725 dosing emulsion in patients with advanced malignancies. Sci Rep. 2021;11:22355
37. Migliorini D, Dutoit V, Allard M, Grandjean Hallez N, Marinari E, Widmer V. et al. Phase I/II trial testing safety and immunogenicity of the multipeptide IMA950/poly-ICLC vaccine in newly diagnosed adult malignant astrocytoma patients. Neuro Oncol. 2019;21:923-33
38. Tsuboi A, Hashimoto N, Fujiki F, Morimoto S, Kagawa N, Nakajima H. et al. A phase I clinical study of a cocktail vaccine of Wilms' tumor 1 (WT1) HLA class I and II peptides for recurrent malignant glioma. Cancer Immunol Immunother. 2019;68:331-40
39. Tamura R, Morimoto Y, Kosugi K, Sato M, Oishi Y, Ueda R. et al. Clinical and histopathological analyses of VEGF receptors peptide vaccine in patients with primary glioblastoma - a case series. BMC Cancer. 2020;20:196
40. Okada H, Butterfield LH, Hamilton RL, Hoji A, Sakaki M, Ahn BJ. et al. Induction of robust type-I CD8+ T-cell responses in WHO grade 2 low-grade glioma patients receiving peptide-based vaccines in combination with poly-ICLC. Clin Cancer Res. 2015;21:286-94
41. Fenstermaker RA, Ciesielski MJ, Qiu J, Yang N, Frank CL, Lee KP. et al. Clinical study of a survivin long peptide vaccine (SurVaxM) in patients with recurrent malignant glioma. Cancer Immunol Immunother. 2016;65:1339-52
42. Sloan AE, Winter K, Gilbert MR, Aldape K, Choi S, Wen PY. et al. NRG-BN002: Phase I study of ipilimumab, nivolumab, and the combination in patients with newly diagnosed glioblastoma. Neuro Oncol. 2024;26:1628-37
43. Lassman AB, Polley MC, Iwamoto FM, Sloan AE, Wang TJC, Aldape KD. et al. Dual Immune Check Point Blockade in MGMT-Unmethylated Newly Diagnosed Glioblastoma: NRG Oncology BN007, a Randomized Phase II/III Clinical Trial. J Clin Oncol. 2025;43:3032-40
44. Narita Y, Arakawa Y, Yamasaki F, Nishikawa R, Aoki T, Kanamori M. et al. A randomized, double-blind, phase III trial of personalized peptide vaccination for recurrent glioblastoma. Neuro Oncol. 2019;21:348-59
45. Hilf N, Kuttruff-Coqui S, Frenzel K, Bukur V, Stevanović S, Gouttefangeas C. et al. Actively personalized vaccination trial for newly diagnosed glioblastoma. Nature. 2019;565:240-5
46. Muragaki Y, Ishikawa E, Maruyama T, Nitta M, Saito T, Ikuta S. et al. A multicenter, randomized, placebo-controlled phase IIb trial of an autologous formalin-fixed tumor vaccine for newly diagnosed glioblastomas. J Neurosurg. 2023;139:344-54
47. Wang Y, Li G, Su J, Liu Y, Zhang X, Zhang G. et al. Tumor-Associated Macrophages Nano-Reprogrammers Induce "Gear Effect" to Empower Glioblastoma Immunotherapy. Small. 2025;21:e2406839
48. Lutz J, Feist RK, Sonntag T, Peguero-Sánchez E, Wolter K, Bick R. et al. Preclinical development of an mRNA-based multiepitope immunotherapeutic for glioblastoma. Cancer Immunol Immunother. 2025;74:329
49. Shi Y, Sun Y, Zhao S, Sun Z, Xia M, Zhong Z. et al. Intranasal and Intravenous Sequential Administration of Survivin Peptide-CpG Nanovaccines Elicits Potent Immunity Toward Glioblastoma. Adv Mater. 2025;37:e2420630
50. Wang C, Feng W, Li J, Wang J, Liu L, Ye SH. et al. Enhanced Nano-Vaccine Utilizing Biomineralized Virus-like Particles for Efficient Glioblastoma Immunotherapy via the Nose-To-Brain Delivery Pathway. ACS Nano. 2025;19:21154-68
51. Jiang S, Lu Z. mRNA-LNP vaccines: rational design, delivery optimization, and clinical translation. J Mater Chem B. 2025;13:15447-67
52. Fang RH, Gao W, Zhang L. Targeting drugs to tumours using cell membrane-coated nanoparticles. Nat Rev Clin Oncol. 2023;20:33-48
53. Kuang L, Han M, Wu X, Deng Z, Liu T, Yin Y. et al. Starting the Engine and Releasing the Brakes of T-Cell Responses: A Biomimetic Dendritic Cell Nanoplatform for Improved Glioblastoma Immunotherapy. ACS Nano. 2025;19:21365-84
54. Wang W, Zou C, Liu X, He L, Cao Z, Zhu M. et al. Biomimetic Dendritic Cell-Based Nanovaccines for Reprogramming the Immune Microenvironment to Boost Tumor Immunotherapy. ACS Nano. 2024;18:34063-76
55. Cheng W, Yang J, Pan Y, Qu H, Duan Z, Wu J. et al. Noninvasive Activation of Local and Systemic Immunity with a Sequential-Targeting Sonodynamic Nanovaccine to Treat Glioblastoma. ACS Nano. 2025;19:27804-24
56. Brüßeler MMT, Zam A, Moreno-Zafra VM, Rouatbi N, Hassuneh OWM, Marrocu A. et al. Polyinosinic/Polycytidylic Lipid Nanoparticles Enhance Immune Cell Infiltration and Improve Survival in the Glioblastoma Mouse Model. Mol Pharm. 2024;21:6339-52
57. Yin Q, Li J, Zhang J, Leng J, Zhang K, Gao X. et al. Nanoshield Architecture Harnessing Neoantigen-Targeting Peptides Enables Durable Post-surgical Glioma Immunotherapy. Nano Lett. 2025;25:13629-38
58. Tülümen D, Aydemir E, Ayaz F. Immunoinformatics-based multi-epitope vaccine design using transforming growth factor beta-2 proprotein (TGFB2) for glioblastoma multiforme (GBM): GVac. Methods. 2025;242:38-53
59. Mirzaie S, Yuan KD, Ni H, Wu XY. Design of a novel multiepitope vaccine against glioblastoma by in silico approaches. Sci Rep. 2025;15:24046
60. Kosianova A, Pak O, Zaitsev S, Smirnova P, Bryukhovetskiy I. Clofazimine enhances anti-glioma effect of immunotherapy. Int Immunopharmacol. 2025;145:113738
61. Zhang X, Zhou W, Yu J, Jiang R, Han J, Li H. et al. Reprogramming Tumor-Associated Macrophage by Ornithine Decarboxylase Inhibitor and Immune Checkpoint for Orthotopic Glioblastoma Photothermal Immunotherapy. ACS Appl Mater Interfaces. 2025;17:35155-67
62. Puleo J, Polyak K. A Darwinian perspective on tumor immune evasion. Biochim Biophys Acta Rev Cancer. 2022;1877:188671
63. Ishida Y, Agata Y, Shibahara K, Honjo T. Induced expression of PD-1, a novel member of the immunoglobulin gene superfamily, upon programmed cell death. Embo j. 1992;11:3887-95
64. Brunet JF, Denizot F, Luciani MF, Roux-Dosseto M, Suzan M, Mattei MG. et al. A new member of the immunoglobulin superfamily-CTLA-4. Nature. 1987;328:267-70
65. Leach DR, Krummel MF, Allison JP. Enhancement of antitumor immunity by CTLA-4 blockade. Science. 1996;271:1734-6
66. Omuro A, Vlahovic G, Lim M, Sahebjam S, Baehring J, Cloughesy T. et al. Nivolumab with or without ipilimumab in patients with recurrent glioblastoma: results from exploratory phase I cohorts of CheckMate 143. Neuro Oncol. 2018;20:674-86
67. Reardon DA, Brandes AA, Omuro A, Mulholland P, Lim M, Wick A. et al. Effect of Nivolumab vs Bevacizumab in Patients With Recurrent Glioblastoma: The CheckMate 143 Phase 3 Randomized Clinical Trial. JAMA Oncol. 2020;6:1003-10
68. Kesari S, Wojcinski A, Pabla S, Seager RJ, Gill JM, Carrillo JA. et al. Pre-radiation Nivolumab plus ipilimumab in patients with newly diagnosed high-grade gliomas. Oncoimmunology. 2024;13:2432728
69. McFaline-Figueroa JR, Sun L, Youssef GC, Huang R, Li G, Kim J. et al. Neoadjuvant anti-PD1 immunotherapy for surgically accessible recurrent glioblastoma: clinical and molecular outcomes of a stage 2 single-arm expansion cohort. Nat Commun. 2024;15:10757
70. Weathers SP, Li X, Zhu H, Damania AV, Knafl M, McKinley B. et al. Improved overall survival in an anti-PD-L1 treated cohort of newly diagnosed glioblastoma patients is associated with distinct immune, mutation, and gut microbiome features: a single arm prospective phase I/II trial. Nat Commun. 2025;16:3950
71. Louis DN, Perry A, Reifenberger G, von Deimling A, Figarella-Branger D, Cavenee WK. et al. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: a summary. Acta Neuropathol. 2016;131:803-20
72. Wang D, Zhang J, Bu C, Liu G, Guo G, Zhang Z. et al. Dynamics of tumor in situ fluid circulating tumor DNA in recurrent glioblastomas forecasts treatment efficacy of immune checkpoint blockade coupled with low-dose bevacizumab. J Cancer Res Clin Oncol. 2024;150:466
73. Guo G, Zhang Z, Zhang J, Wang D, Xu S, Liu G. et al. Predicting recurrent glioblastoma clinical outcome to immune checkpoint inhibition and low-dose bevacizumab with tumor in situ fluid circulating tumor DNA analysis. Cancer Immunol Immunother. 2024;73:193
74. Omuro A, Brandes AA, Carpentier AF, Idbaih A, Reardon DA, Cloughesy T. et al. Radiotherapy combined with nivolumab or temozolomide for newly diagnosed glioblastoma with unmethylated MGMT promoter: An international randomized phase III trial. Neuro Oncol. 2023;25:123-34
75. Lim M, Weller M, Idbaih A, Steinbach J, Finocchiaro G, Raval RR. et al. Phase III trial of chemoradiotherapy with temozolomide plus nivolumab or placebo for newly diagnosed glioblastoma with methylated MGMT promoter. Neuro Oncol. 2022;24:1935-49
76. Pol JG, Lizarralde-Guerrero M, Checcoli A, Kroemer G. Targeted opening of the blood-brain barrier facilitates doxorubicin/anti-PD-1-based chemoimmunotherapy of glioblastoma. Oncoimmunology. 2024;13:2385124
77. Lin SW, Yu CP, Tsai JC, Shyong YJ. Delivery of extracellular vesicles loaded with immune checkpoint inhibitors for immunotherapeutic management of glioma. Mater Today Bio. 2024;28:101244
78. Zhang Z, Xu X, Du J, Chen X, Xue Y, Zhang J. et al. Redox-responsive polymer micelles co-encapsulating immune checkpoint inhibitors and chemotherapeutic agents for glioblastoma therapy. Nat Commun. 2024;15:1118
79. Aytekin E, Maksoud S, Tannous BA, Pehlivan SB, Badr CE. Dual-action nanotherapy: Temozolomide-loaded, anti-PD-L1 scFv-functionalized lipid nanocarriers for targeted glioblastoma therapy. Eur J Pharm Sci. 2025;213:107255
80. Chaudhari AP, Budayr OM, Bonacquisti EE, Kussatz CC, Bannon MS, Law KJ. et al. The status of extracellular vesicles as drug carriers and therapeutics. Nature Reviews Bioengineering. 2026
81. Liu C, Gong S, Du X, Liu Y. Micelle morphology phase diagram in a phospholipid, PEGylated lipid, and peptide amphiphiles ternary system. Chemical Engineering Research and Design. 2022;181:354-62
82. Fesenmeier DJ, Park S, Kim S, Won YY. Surface mechanical behavior of water-spread poly(styrene)-poly(ethylene glycol) (PS-PEG) micelles at the air-water interface: Effect of micelle size and polymer end/linking group chemistry. J Colloid Interface Sci. 2022;617:764-77
83. Abdul Rub M, Shafi Sheikh M, Khan F, Azum N, Alghamdi YG, Asiri AM. Impact on micellization between promethazine hydrochloride and ester bonded gemini surfactant in distinct solvents: A multi-faceted procedure. Journal of Molecular Liquids. 2021;342:117477
84. Klika Škopić M, Gramse C, Oliva R, Pospich S, Neukirch L, Manisegaran M. et al. Towards DNA-Encoded Micellar Chemistry: DNA-Micelle Association and Environment Sensitivity of Catalysis. Chemistry - A European Journal. 2021;27:10048-57
85. Lang X, Thumu U, Yuan L, Zheng C, Zhang H, He L. et al. Chemical fuel-driven transient polymeric micelle nanoreactors toward reversible trapping and reaction acceleration. Chem Commun (Camb). 2021;57:5786-9
86. Xu Z, Xie Y, Chen W, Deng W. Nanocarrier-Based Systems for Targeted Delivery: Current Challenges and Future Directions. MedComm (2020). 2025;6:e70337
87. De Martino M, Daviaud C, Lira MC, Hernandez-Zirofsky K, Vanpouille-Box C. Dual blockade of PD-1 and CTLA-4 generates long-lasting immunity against irradiated glioblastoma. Cancer Lett. 2025;628:217856
88. Ahn M, Na Y, Choi H, Lee S, Lee J, Park SA. et al. Photoimmuno-Lure Nanoplatform for Enhancing T Cell Expansion in Glioblastoma via Synergistic Treatment of Photodynamic Therapy and Immune Checkpoint Inhibition. Adv Healthc Mater. 2025;14:e2500880
89. Strassheimer F, Elleringmann P, Ludmirski G, Roller B, Macas J, Alekseeva T. et al. CAR-NK cell therapy combined with checkpoint inhibition induces an NKT cell response in glioblastoma. Br J Cancer. 2025;132:849-60
90. Hou D, Wang S, Castro BA, Katz JL, Dapash M, Arrieta VA. et al. Dual aVß8 Integrin and PD-1 Blockade Overcomes TGFβ-Mediated B-Cell Suppression to Enhance Anti-Tumor Immunity. Neuro Oncol. 2025;27:2355-69
91. Seetharam D, Chandar J, Ramsoomair CK, Desgraves JF, Alvarado Medina A, Hudson AJ. et al. Activating antiviral immune responses potentiates immune checkpoint inhibition in glioblastoma models. J Clin Invest. 2025 135
92. Liu T, Sun T, Chen X, Wu J, Sun X, Liu X. et al. Targeting ARPC1B Overcomes Immune Checkpoint Inhibitor Resistance in Glioblastoma by Reversing Protumorigenic Macrophage Polarization. Cancer Res. 2025;85:1236-52
93. Lee J, Kang Y, Lee H, Saravanakumar G, Park SA, Ahn S. et al. Amplifying glioblastoma immunotherapy: T cell shielding through Nitric oxide/reactive oxygen species scavenging nanoparticles Potentiates anti-PD-1. Biomaterials. 2025;315:122904
94. Królikowska A, Tarnowski M. CAR-T cells immunotherapy in the treatment of glioblastoma. Cancer Immunol Immunother. 2025;74:363
95. Nagai M. [Advances of BRM therapy of malignant brain tumors]. Gan To Kagaku Ryoho. 1991;18:188-94
96. Jacobs SK, Wilson DJ, Kornblith PL, Grimm EA. Interleukin-2 and autologous lymphokine-activated killer cells in the treatment of malignant glioma. Preliminary report. J Neurosurg. 1986;64:743-9
97. Boiardi A, Silvani A, Ruffini PA, Rivoltini L, Parmiani G, Broggi G. et al. Loco-regional immunotherapy with recombinant interleukin-2 and adherent lymphokine-activated killer cells (A-LAK) in recurrent glioblastoma patients. Cancer Immunol Immunother. 1994;39:193-7
98. Sankhla SK, Nadkarni JS, Bhagwati SN. Adoptive immunotherapy using lymphokine-activated killer (LAK) cells and interleukin-2 for recurrent malignant primary brain tumors. J Neurooncol. 1996;27:133-40
99. Lin Y, Okada H. Cellular immunotherapy for malignant gliomas. Expert Opin Biol Ther. 2016;16:1265-75
100. Arruda LCM, Karbach J, Kiselicki D, Altmannsberger HM, Sinelnikov E, Gustavus D. et al. Tumor-infiltrating lymphocytes-derived CD8(+) clonotypes infiltrate the tumor tissue and mediate tumor regression in glioblastoma. Oncoimmunology. 2025;14:2559784
101. Cherkassky L, Morello A, Villena-Vargas J, Feng Y, Dimitrov DS, Jones DR. et al. Human CAR T cells with cell-intrinsic PD-1 checkpoint blockade resist tumor-mediated inhibition. J Clin Invest. 2016;126:3130-44
102. Chheda ZS, Kohanbash G, Okada K, Jahan N, Sidney J, Pecoraro M. et al. Novel and shared neoantigen derived from histone 3 variant H3.3K27M mutation for glioma T cell therapy. J Exp Med. 2018;215:141-57
103. Walton CM, Bell M, O'Neil R, Sahin O, Choi BD, Fecci PE. et al. Chimeric antigen receptor (CAR) T-cell therapy for glioblastoma (GBM): current clinical insights, challenges, and future directions. J Immunother Cancer. 2025 13
104. Weathers SP, Penas-Prado M, Pei BL, Ling X, Kassab C, Banerjee P. et al. Glioblastoma-mediated Immune Dysfunction Limits CMV-specific T Cells and Therapeutic Responses: Results from a Phase I/II Trial. Clin Cancer Res. 2020;26:3565-77
105. Lim J, Park Y, Ahn JW, Sim J, Kang SJ, Hwang S. et al. Autologous adoptive immune-cell therapy elicited a durable response with enhanced immune reaction signatures in patients with recurrent glioblastoma: An open label, phase I/IIa trial. PLoS One. 2021;16:e0247293
106. Jaramillo M, Sarfraz Z, Ganiyani MA, Mustafayev FNA, Qidwai K, Mustafayev K. et al. CTIM-26. TOXICITY PROFILE OF CAR-T CELL THERAPY IN GLIOBLASTOMA: A META-ANALYSIS OF CYTOKINE RELEASE SYNDROME, NEUROTOXICITY, AND ADVERSE EVENTS. Neuro-Oncology. 2025;27:v120-v
107. Geraghty AC, Acosta-Alvarez L, Rotiroti MC, Dutton S, O'Dea MR, Kim W. et al. Immunotherapy-related cognitive impairment after CAR T cell therapy in mice. Cell. 2025;188:3238-58.e25
108. Graham CE, Velasco R, Alarcon Tomas A, Stewart OP, Dachy G, del Bufalo F. et al. Non-ICANS neurological complications after CAR T-cell therapies: recommendations from the EBMT Practice Harmonisation and Guidelines Committee. The Lancet Oncology. 2025;26:e203-e13
109. Karschnia P, Dietrich J. Neurological complications of CAR T cell therapy for cancers. Nature Reviews Neurology. 2025;21:422-31
110. Bagley SJ, Desai AS, Fraietta JA, Silverbush D, Chafamo D, Freeburg NF. et al. Intracerebroventricular bivalent CAR T cells targeting EGFR and IL-13Rα2 in recurrent glioblastoma: a phase 1 trial. Nat Med. 2025;31:2778-87
111. Brown CE, Badie B, Barish ME, Weng L, Ostberg JR, Chang WC. et al. Bioactivity and Safety of IL13Rα2-Redirected Chimeric Antigen Receptor CD8+ T Cells in Patients with Recurrent Glioblastoma. Clin Cancer Res. 2015;21:4062-72
112. Brown CE, Rodriguez A, Palmer J, Ostberg JR, Naranjo A, Wagner JR. et al. Off-the-shelf, steroid-resistant, IL13Rα2-specific CAR T cells for treatment of glioblastoma. Neuro Oncol. 2022;24:1318-30
113. Liu Z, Zhou J, Yang X, Liu Y, Zou C, Lv W. et al. Safety and antitumor activity of GD2-Specific 4SCAR-T cells in patients with glioblastoma. Mol Cancer. 2023;22:3
114. Barish ME, Aftabizadeh M, Hibbard J, Blanchard MS, Ostberg JR, Wagner JR. et al. Chlorotoxin-directed CAR T cell therapy for recurrent glioblastoma: Interim clinical experience demonstrating feasibility and safety. Cell Rep Med. 2025;6:102302
115. Kourtesakis A, Bailey E, Chow HNH, Rohdjeß H, Mussnig N, Agardy DA. et al. Utilization of universal-targeting mSA2 CAR-T cells for the treatment of glioblastoma. Oncoimmunology. 2025;14:2518631
116. Zhai Y, Li G, Pan C, Yu M, Hu H, Wang D. et al. The development and potent antitumor efficacy of CD44/CD133 dual-targeting IL7Rα-armored CAR-T cells against glioblastoma. Cancer Lett. 2025;614:217541
117. Liu L, He P, Wang Y, Ma F, Li D, Bai Z. et al. Engineering sonogenetic EchoBack-CAR T cells. Cell. 2025;188:2621-36.e20
118. Cook DR, Boesteanu AC, Yin Y, Reid R, Roccograndi L, Dahmane N. et al. Checkpoint antibody receptor modified ARMed CAR T circumvents the suppressive immunome in GBM. Front Immunol. 2025;16:1579925
119. Liu J, Dai K, Saliu MA, Salisu MD, Gan J, Afolabi LO. et al. Sodium valproate enhances efficacy of NKG2D CAR-T cells against glioblastoma. Front Immunol. 2024;15:1519777
120. Wu BX, Kreatsoulas D, Cam H, Bolyard C, Chang Y, Mandula J. et al. Targeting TGFβ docking receptor Glycoprotein A Repetitions Predominant (GARP) via novel chimeric antigen receptor (CAR)-T cell platform to treat glioblastoma. Neuro Oncol. 2025
121. Sætersmoen M, Kotchetkov IS, Torralba-Raga L, Mansilla-Soto J, Sohlberg E, Krokeide SZ. et al. Targeting HLA-E-overexpressing cancers with a NKG2A/C switch receptor. Med. 2025;6:100521
122. Foster JB, Madsen PJ, Harvey K, Griffin C, Stern A, Patterson L. et al. Transient mRNA CAR T cells targeting GD2 provide dose-adjusted efficacy against diffuse midline glioma and high-grade glioma models. Neuro Oncol. 2025;27:2684-96
123. Nehama D, Di Ianni N, Musio S, Du H, Patané M, Pollo B. et al. B7-H3-redirected chimeric antigen receptor T cells target glioblastoma and neurospheres. EBioMedicine. 2019;47:33-43
124. Yin P, Yang J, Jiang Y, Han L, Xiong T, Liu H. et al. Enhanced FOS expression improves tumor clearance and resists exhaustion in NR4A3-deficient CAR T cells under chronic antigen exposure. Sci Adv. 2025;11:eadw3571
125. Stemer G, Mittermayr T, Schnell-Inderst P, Wild C. Costs, challenges and opportunities of decentralised chimeric antigen receptor T-cell production: a literature review and clinical experts' interviews. Eur J Hosp Pharm. 2025;32:202-8
126. Ma HL, Garcia LSA, Ishiba R, Dametto LC, da Silva Moraes KA, Oliveira Ferreira R. et al. Modeling glioblastoma in 3D hydrogels enables investigation of Zika virus targeting and immune modulation in oncolytic virotherapy. Biomater Adv. 2026;179:214492
127. Markert JM, Razdan SN, Kuo HC, Cantor A, Knoll A, Karrasch M. et al. A phase 1 trial of oncolytic HSV-1, G207, given in combination with radiation for recurrent GBM demonstrates safety and radiographic responses. Mol Ther. 2014;22:1048-55
128. Kicielinski KP, Chiocca EA, Yu JS, Gill GM, Coffey M, Markert JM. Phase 1 clinical trial of intratumoral reovirus infusion for the treatment of recurrent malignant gliomas in adults. Mol Ther. 2014;22:1056-62
129. Freeman AI, Zakay-Rones Z, Gomori JM, Linetsky E, Rasooly L, Greenbaum E. et al. Phase I/II trial of intravenous NDV-HUJ oncolytic virus in recurrent glioblastoma multiforme. Mol Ther. 2006;13:221-8
130. Lang FF, Conrad C, Gomez-Manzano C, Yung WKA, Sawaya R, Weinberg JS. et al. Phase I Study of DNX-2401 (Delta-24-RGD) Oncolytic Adenovirus: Replication and Immunotherapeutic Effects in Recurrent Malignant Glioma. J Clin Oncol. 2018;36:1419-27
131. Desjardins A, Gromeier M, Herndon JE 2nd, Beaubier N, Bolognesi DP, Friedman AH. et al. Recurrent Glioblastoma Treated with Recombinant Poliovirus. N Engl J Med. 2018;379:150-61
132. Nassiri F, Patil V, Yefet LS, Singh O, Liu J, Dang RMA. et al. Oncolytic DNX-2401 virotherapy plus pembrolizumab in recurrent glioblastoma: a phase 1/2 trial. Nat Med. 2023;29:1370-8
133. Tur-Planells V, Bykov Y, Dawodu G, García-Romero N, Izpura-Luis S, Pérez-Rodríguez L. et al. Infectious bursal disease virus (IBDV) as a novel oncolytic virotherapy in glioblastoma. J Immunother Cancer. 2025 13
134. Guo C, Long Z, Lin P, Shen Y, Zhong Y, Qian J. et al. BRD9 inhibition overcomes oncolytic virus therapy resistance in glioblastoma. Cell Rep Med. 2025;6:102258
135. Sahu U, Mullarkey MP, Murphy SA, Anderson JC, Putluri V, Kamal AHM. et al. IDH status dictates oHSV mediated metabolic reprogramming affecting anti-tumor immunity. Nat Commun. 2025;16:3874
136. Galanis E, Dooley KE, Keith Anderson S, Kurokawa CB, Carrero XW, Uhm JH. et al. Carcinoembryonic antigen-expressing oncolytic measles virus derivative in recurrent glioblastoma: a phase 1 trial. Nat Commun. 2024;15:493
137. Ning W, Qian X, Dunmall LC, Liu F, Guo Y, Li S. et al. Non-secreting IL12 expressing oncolytic adenovirus Ad-TD-nsIL12 in recurrent high-grade glioma: a phase I trial. Nat Commun. 2024;15:9299
138. Fares J, Ahmed AU, Ulasov IV, Sonabend AM, Miska J, Lee-Chang C. et al. Neural stem cell delivery of an oncolytic adenovirus in newly diagnosed malignant glioma: a first-in-human, phase 1, dose-escalation trial. Lancet Oncol. 2021;22:1103-14
139. Todo T, Ito H, Ino Y, Ohtsu H, Ota Y, Shibahara J. et al. Intratumoral oncolytic herpes virus G47∆ for residual or recurrent glioblastoma: a phase 2 trial. Nat Med. 2022;28:1630-9
140. Todo T, Ino Y, Ohtsu H, Shibahara J, Tanaka M. A phase I/II study of triple-mutated oncolytic herpes virus G47∆ in patients with progressive glioblastoma. Nat Commun. 2022;13:4119
141. He Y, Li W, Zhang X, Cui Z. Oncolytic Virus Targeted Therapy for Glioma via Intravenous Delivery. Adv Healthc Mater. 2025;14:e2404965
142. Weller SK, Coen DM. Herpes simplex viruses: mechanisms of DNA replication. Cold Spring Harb Perspect Biol. 2012;4:a013011
143. Ferreira RS, Jandrey EHF, Granha I, Endo AK, Ferreira RO, Araujo BHS. et al. Differential Replication and Oncolytic Effects of Zika Virus in Aggressive CNS Tumor Cells: Insights from Organoid and Tumoroid Models. Viruses. 2024 16
144. Jiang H, Nace R, Ferguson C, Zhang L, Peng KW, Russell SJ. Oncolytic cytomegaloviruses expressing EGFR-retargeted fusogenic glycoprotein complex and drug-controllable interleukin 12. Cell Rep Med. 2025;6:101874
145. Jazowiecka-Rakus J, Pogoda-Mieszczak K, Rahman MM, McFadden G, Sochanik A. Adipose-Derived Stem Cells as Carrier of Pro-Apoptotic Oncolytic Myxoma Virus: To Cross the Blood-Brain Barrier and Treat Murine Glioma. Int J Mol Sci. 2024 25
146. Tang F, Zhang Z, Xu J, Wang Z, Chen Y, Huang T. et al. A chimeric oncolytic adenovirus carried by macrophages for glioma immunotherapy. Biochem Biophys Res Commun. 2025;790:152852
147. Niu A, Lv Y, Chen Y, Liu Y, Luo C, Zheng M. et al. Cholesterol surface-modified oncolytic adenovirus enriched with apolipoprotein E penetrates the blood-brain barrier to target glioblastoma immunotherapy. Mater Today Bio. 2025;35:102319
148. Nazarenko AS, Shkirdova AO, Orlova EA, Biryukova YK, Vorovitch MF, Kolyasnikova NM. et al. Viral-Porphyrin Combo: Photodynamic and Oncolytic Viral Therapy for Potent Glioblastoma Treatment. Int J Mol Sci. 2024 25
149. Wang S, Wei J. Distinguishing the pros and cons of metabolic reprogramming in oncolytic virus immunotherapy. Int J Cancer. 2022;151:1654-62
150. Chen Y, Bian S, Zhang J, Luan Y, Yin B, Dai W. et al. HSV-1-induced N6-methyladenosine reprogramming via ICP0-mediated suppression of METTL14 potentiates oncolytic activity in glioma. Cell Rep. 2024;43:114756
151. Li Y, Qin X, Liang C, Wang L. Progress in the research and development of oncolytic virus therapies. Front Pharmacol. 2026;17:1751206
152. Quail DF, Joyce JA. The Microenvironmental Landscape of Brain Tumors. Cancer Cell. 2017;31:326-41
153. Habashy KJ, Mansour R, Moussalem C, Sawaya R, Massaad MJ. Challenges in glioblastoma immunotherapy: mechanisms of resistance and therapeutic approaches to overcome them. British Journal of Cancer. 2022;127:976-87
154. Sampson JH, Gunn MD, Fecci PE, Ashley DM. Brain immunology and immunotherapy in brain tumours. Nat Rev Cancer. 2020;20:12-25
155. Pardridge WM. Blood-Brain Barrier and Delivery of Protein and Gene Therapeutics to Brain. Front Aging Neurosci. 2019;11:373
156. Arvanitis CD, Ferraro GB, Jain RK. The blood-brain barrier and blood-tumour barrier in brain tumours and metastases. Nat Rev Cancer. 2020;20:26-41
157. Koyama S, Akbay EA, Li YY, Herter-Sprie GS, Buczkowski KA, Richards WG. et al. Adaptive resistance to therapeutic PD-1 blockade is associated with upregulation of alternative immune checkpoints. Nat Commun. 2016;7:10501
158. Jackson CM, Choi J, Lim M. Mechanisms of immunotherapy resistance: lessons from glioblastoma. Nat Immunol. 2019;20:1100-9
159. Herrera GJ, Sun L, Lee AH, Everson RG, Nathanson DA, Liau LM. et al. 819 Temporal influence of PD-1 blockade after vaccination on macrophage-driven CD8+ T cell exhaustion within the glioblastoma microenvironment. Journal for ImmunoTherapy of Cancer. 2024 12
160. Friedrich M, Hahn M, Michel J, Sankowski R, Kilian M, Kehl N. et al. Dysfunctional dendritic cells limit antigen-specific T cell response in glioma. Neuro Oncol. 2023;25:263-76
161. Raphael I, Kumar R, McCarl LH, Shoger K, Wang L, Sandlesh P. et al. TIGIT and PD-1 Immune Checkpoint Pathways Are Associated With Patient Outcome and Anti-Tumor Immunity in Glioblastoma. Front Immunol. 2021;12:637146
162. Losurdo A, Di Muzio A, Cianciotti BC, Dipasquale A, Persico P, Barigazzi C. et al. T Cell Features in Glioblastoma May Guide Therapeutic Strategies to Overcome Microenvironment Immunosuppression. Cancers (Basel). 2024 16
163. Lin C, Teng W, Tian Y, Li S, Xia N, Huang C. Immune landscape and response to oncolytic virus-based immunotherapy. Front Med. 2024;18:411-29
164. Johnson AL, Lopez-Bertoni H. Cellular diversity through space and time: adding new dimensions to GBM therapeutic development. Front Genet. 2024;15:1356611
165. Brown NF, Carter TJ, Ottaviani D, Mulholland P. Harnessing the immune system in glioblastoma. Br J Cancer. 2018;119:1171-81
166. Dong W, Luo Y, He D, Zhang M, Zeng J, Chen Y. Oncolytic virotherapy against lung cancer: key receptors and signaling pathways of viral entry. Front Immunol. 2024;15:1473288
167. Kollmann CF, van Montfoort N, Cordelier P, Pol J, Olagnier D. Oncolytic virotherapy: Sparking durable anti-tumor immunity through microenvironment modulation. Semin Immunol. 2025;80:101994
168. Fu M, Xue B, Miao X, Gao Z. Overcoming immunotherapy resistance in glioblastoma: challenges and emerging strategies. Front Pharmacol. 2025;16:1584688
169. Xing Y, Liu C, Feng Y, Li S, Chen Y. Vaccine therapies for glioma: clinical frontiers and potential breakthrough. Front Oncol. 2025;15:1613332
170. Skadborg SK, Maarup S, Draghi A, Borch A, Hendriksen S, Mundt F. et al. Nivolumab Reaches Brain Lesions in Patients with Recurrent Glioblastoma and Induces T-cell Activity and Upregulation of Checkpoint Pathways. Cancer Immunol Res. 2024;12:1202-20
171. Sari G, Rock KL. Tumor immune evasion through loss of MHC class-I antigen presentation. Curr Opin Immunol. 2023;83:102329
172. Badrinath S, Dellacherie MO, Li A, Zheng S, Zhang X, Sobral M. et al. A vaccine targeting resistant tumours by dual T cell plus NK cell attack. Nature. 2022;606:992-8
173. Xu J, Xia Z, Wang S, Xia Q. Resistance to oncolytic virotherapy: Multidimensional mechanisms and therapeutic breakthroughs (Review). Int J Mol Med. 2025 56
174. Ouyang G, Liu Y, Liu J, Huang L, Luo F, Li L. Efficacy and safety of reduced-dose chemotherapy plus immunotherapy in patients with lung squamous cell carcinoma: A real-world observational study. Cancer Med. 2023;12:18679-90
175. Pickl M, Ruge E, Venturi M. Predictive markers in early research and companion diagnostic developments in oncology. N Biotechnol. 2012;29:651-5
176. Hegi ME, Diserens AC, Gorlia T, Hamou MF, de Tribolet N, Weller M. et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. N Engl J Med. 2005;352:997-1003
177. Ballo MT, Conlon P, Lavy-Shahaf G, Kinzel A, Vymazal J, Rulseh AM. Association of Tumor Treating Fields (TTFields) therapy with survival in newly diagnosed glioblastoma: a systematic review and meta-analysis. J Neurooncol. 2023;164:1-9
178. Giunco S, Padovan M, Angelini C, Cavallin F, Cerretti G, Morello M. et al. Prognostic role and interaction of TERT promoter status, telomere length and MGMT promoter methylation in newly diagnosed IDH wild-type glioblastoma patients. ESMO Open. 2023;8:101570
179. Eckel-Passow JE, Lachance DH, Molinaro AM, Walsh KM, Decker PA, Sicotte H. et al. Glioma Groups Based on 1p/19q, IDH, and TERT Promoter Mutations in Tumors. N Engl J Med. 2015;372:2499-508
180. Vuong HG, Nguyen TQ, Ngo TNM, Nguyen HC, Fung KM, Dunn IF. The interaction between TERT promoter mutation and MGMT promoter methylation on overall survival of glioma patients: a meta-analysis. BMC Cancer. 2020;20:897
181. Stichel D, Ebrahimi A, Reuss D, Schrimpf D, Ono T, Shirahata M. et al. Distribution of EGFR amplification, combined chromosome 7 gain and chromosome 10 loss, and TERT promoter mutation in brain tumors and their potential for the reclassification of IDHwt astrocytoma to glioblastoma. Acta Neuropathol. 2018;136:793-803
182. Makawi A, Khalafallah SA, Faris IM, Alfaki M. Comprehensive Analysis Reveals Epithelial Growth Factor Receptor as a Potential Diagnostic Biomarker in Glioblastoma Multiforme. Cureus. 2024;16:e64506
183. Ermoian RP, Furniss CS, Lamborn KR, Basila D, Berger MS, Gottschalk AR. et al. Dysregulation of PTEN and protein kinase B is associated with glioma histology and patient survival. Clin Cancer Res. 2002;8:1100-6
184. Davalan W, Alkins R. Prognostic and predictive determinants in high-grade gliomas: integrating tumor-intrinsic biology with patient and system-level factors. Front Neurol. 2025;16:1664458
185. Franceschi E, De Biase D, Di Nunno V, Pession A, Tosoni A, Gatto L. et al. IDH1 Non-Canonical Mutations and Survival in Patients with Glioma. Diagnostics (Basel). 2021 11
186. Takahashi Y, Nakamura H, Makino K, Hide T, Muta D, Kamada H. et al. Prognostic value of isocitrate dehydrogenase 1, O6-methylguanine-DNA methyltransferase promoter methylation, and 1p19q co-deletion in Japanese malignant glioma patients. World J Surg Oncol. 2013;11:284
187. Hariharan S, Whitfield BT, Pirozzi CJ, Waitkus MS, Brown MC, Bowie ML. et al. Interplay between ATRX and IDH1 mutations governs innate immune responses in diffuse gliomas. Nat Commun. 2024;15:730
188. Haase S, Garcia-Fabiani MB, Carney S, Altshuler D, Núñez FJ, Méndez FM. et al. Mutant ATRX: uncovering a new therapeutic target for glioma. Expert Opin Ther Targets. 2018;22:599-613
189. Yip S, Butterfield YS, Morozova O, Chittaranjan S, Blough MD, An J. et al. Concurrent CIC mutations, IDH mutations, and 1p/19q loss distinguish oligodendrogliomas from other cancers. J Pathol. 2012;226:7-16
190. Brat DJ, Verhaak RG, Aldape KD, Yung WK, Salama SR, Cooper LA. et al. Comprehensive, Integrative Genomic Analysis of Diffuse Lower-Grade Gliomas. N Engl J Med. 2015;372:2481-98
191. Appay R, Dehais C, Maurage CA, Alentorn A, Carpentier C, Colin C. et al. CDKN2A homozygous deletion is a strong adverse prognosis factor in diffuse malignant IDH-mutant gliomas. Neuro Oncol. 2019;21:1519-28
192. Mousavi Salehi A, Khavanin A, Azizidoost S, Cheraghzadeh M, Khombi Shooshtari M, Farzaneh M. et al. Targeting XBP1 in Cancer: A Review of Therapeutic Approaches and Strategies. Anticancer Agents Med Chem. 2026
193. Elias MG, Hadjiyiannis H, Vafaee F, Scott KF, de Souza P, Becker TM. et al. The Quest for Non-Invasive Diagnosis: A Review of Liquid Biopsy in Glioblastoma. Cancers (Basel). 2025 17
194. Baldini C, Porte M, Rouleau E, Touat M. Liquid biopsy in gliomas-are we there yet? Annals of Oncology. 2025;36:606-8
195. Bae WH, Maraka S, Daher A. Challenges and advances in glioblastoma targeted therapy: the promise of drug repurposing and biomarker exploration. Frontiers in Oncology. 2024; Volume 14 -. 2024
196. Jo Y, Lee SM, Hong C. From splicing noise to therapeutic signaling: RCAN1-4 as a neoepitope in glioblastoma. Cellular & Molecular Immunology. 2026;23:344-6
197. Taori S, Habib A, Adida S, Gecici NN, Sharma N, Calcaterra M. et al. Circulating biomarkers in high-grade gliomas: current insights and future perspectives. Journal of Neuro-Oncology. 2025;172:41-9
198. Qi Y, Hu L, Ji C, Yang X, Yao J, Chen D. et al. B7-H4 reduces the infiltration of CD8+T cells and induces their anti-tumor dysfunction in gliomas. Neoplasia. 2024;54:101007
199. Xiao D, Zhang H, Liu Y, Li Y, Li G, Ning Y. Oncolytic viruses: advanced strategies in cancer therapy. Signal Transduct Target Ther. 2026;11:45
200. Alwithenani A, Hengswat P, Chiocca EA. Oncolytic viruses as cancer therapeutics: From mechanistic insights to clinical translation. Mol Ther. 2025;33:2217-28
Author contact
![]() Corresponding authors: Jintao Gu, MD., State Key Laboratory of Cancer Biology, Biotechnology Center, School of Pharmacy, The Fourth Military Medical University, Xi'an, 710000, China (gujintaoedu.cn). Yalong He, MD., Department of Neurosurgery, Xijing Hospital, Xi'an, 710000, China (heyl.fmmucom).
Corresponding authors: Jintao Gu, MD., State Key Laboratory of Cancer Biology, Biotechnology Center, School of Pharmacy, The Fourth Military Medical University, Xi'an, 710000, China (gujintaoedu.cn). Yalong He, MD., Department of Neurosurgery, Xijing Hospital, Xi'an, 710000, China (heyl.fmmucom).