Impact Factor ISSN: 1449-1907
- Issue 9; 2026
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Volume 23; 2026
- Past Issues
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
1. Classification and...
2. TAMs in HCC surveillance
3. TAMs promoting HCC progress
4. Treatment targeting...
5. Conclusions
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2021; 18(10):2109-2116. doi:10.7150/ijms.56003 This issue Cite
Review
The role of tumor-associated macrophages in primary hepatocellular carcinoma and its related targeting therapy
Lu Deng*, Kang He*, Yixiao Pan, Hai Wang, Yi Luo ![]() , Qiang Xia
, Qiang Xia ![]()
Department of Liver Surgery, Renji Hospital, School of Medicine, Shanghai Jiao Tong University, Shanghai, China.
* Lu Deng and Kang He contributed equally to this article.
Received 2020-11-16; Accepted 2021-3-3; Published 2021-3-15
Abstract

Liver macrophages consist of ontogenically distinct populations termed Kupffer cells and monocyte-derived macrophages. Tumor-associated macrophages (TAMs) inhepatocellularcarcinoma (HCC) play a prominent role in tumormicroenvironment by presenting M1(induced by IFN γ along with LPS) and M2(induced by IL-4 and IL13) polarization. Although TAMs are involved in tumor immune surveillance during the course of HCC, they contribute to tumour progression at different levels by inhibiting the anti-tumor immune response, promoting the generation of blood vessels and lymphatic vessels, and supporting the proliferation and survival of tumor cells. In this paper, the multiple functions of TAMs in HCC were reviewed to provide assistance for future researches about therapeutic approaches.
Keywords: Tumor-associated macrophages, hepatocellular carcinoma, therapy
Introduction
HCC accounts for 75-85% of primary liver cancer, making it the sixth most common cancer in the world and the fourth leading cause of cancer-related death [1]. Macrophages, as the main component of leukocyte infiltration, play an important role in different types of tumor-related inflammation and are closely related to the outcome of treatment. Analyzingfunctions ofTAMsin the process ofprimary HCC can not only help with treatment of HCC, but also promote researches about functions of other inflammatory cells and factors in tumor-related inflammation. Herein, we provide a brief overview of the role of TAMs in the development and progression of HCC, and review the current emerging macrophage targeting therapies associated with TAMs.
1. Classification and polarization of TAMs in HCC
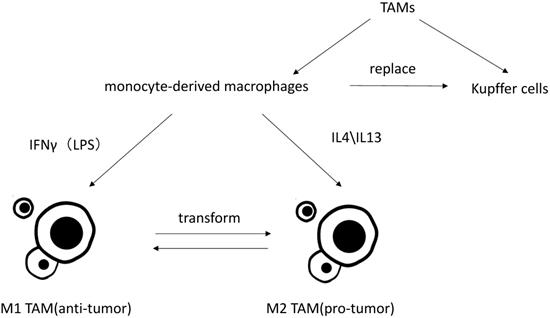
Macrophages are particularly rich in the liver, and mainly divide into liver-resident macrophages, termed Kupffer cells, and blood/bone marrow-derived macrophages, termed monocyte-derived macrophages. Under the condition of liver injury leading to the reduction of Kupffer cells or experimental removal of Kupffer cells, monocyte-derived macrophage can replace Kupffer cells [2, 3]. Monocyte-derived macrophages can then acquire a phenotype that is virtually indistinguishable from Kupffer cells [4].
TAM is an important part in tumor associated inflammation and can be polarized to disparate functional phenotypes, as M1 (induced by IFN γ along with LPS) and M2 (induced by IL-4 and IL13) macrophages [5]. IL-10, corticosteroids, andmolecules and immune complexes released by apoptotic cells canalsoinfluence functions of monocyte-derived macrophages.These signals induce macrophages to express some of the same functional phenotypes as M2 cells (such as expression of scavenger receptors), but these cells are different from M2 cells (such as chemokines). This group of cells is termed M2 like [6]. Present studies do not distinguish between M2-TAMs and M2-like TAMs, nor do M1-TAMs and M1-like TAMs.Studies have shown that the more infiltration of M2-TAMs, the worse its clinical prognosis will be by inducing tumor cell proliferation, angiogenesis, tumor metastasis and epithelial-mesenchymal transition (EMT) [7-9]. Though it has reported that CD68+HLA-DR + M1-like TAMs may also enhance motility of HCC cells via NF-κB/FAK pathway [10], it is generally believed that M2 TAMs play pro-tumor role while M1 TAMs exhibit anti-tumor functions [11]. Kupffer cells are considered as specific TAMs in HCC microenvironment. Kupffer cells and monocytes-derived macrophages are both highly plastic. They can adjust their functional phenotypes according to signals from HCC microenvironment [12, 13]. Most researches on macrophage polarization are simply implemented in vitro, which neglect the tumor microenvironment in vivo and rarely distinguish Kupffer cells from monocyte-derived macrophages. Thus, the polarization and activation of TAMs may involve more stimuli agents and more complex mechanisms in vivo [14].
2. TAMs in HCC surveillance
Cellular senescence is a stress response mechanism that induces cells at risk of malignant transformation to stop proliferating, and is therefore widely recognized as an anti-tumor mechanism. Senescent cells can also secrete specific cytokines, including IL-1 and IL-6, known as senescence-associated secretory phenotype (SASP). These cytokines can affect adjacent tissues and immune cells [15]. Because specific SASP can induce changes of chromatin, the cell cycle of most senescent hepatocytes stop [16]. In liver, cellular senescence mechanism is associated with inhibition of liver cancer progression [17, 18]. Under the stimulation of oncogenic stress, the activation of the p53/p21 and p16/Rb anti-tumor pathways is an important trigger factor of senescence. These pathways promote cellular senescence by activating genes that inhibit cell cycle progression and promote senescence [19-22]. If additional mutations of the proto-oncogene P53 are induced, resulting in the cessation of cellular senescence, liver cancer will develop progressively [23, 24].
Cells that have undergone oncogenic transformation, precancerous liver cells and senescent liver cells coexist in cancerous liver [25]. Senescent hepatocytes, especially senescent precancerous cells, produce many chemokines, among which C-C motif chemokine 2 (CCL2) can recruit NK cells to achieve the clearance of tumor cells [23, 24]. CCL2 can also recruit lots of monocyte-derived macrophages, likeC-C chemokine receptortype 2(CCR2)+ macrophages. CCR2+ macrophages and T cells work together to clear senescent liver cells and prevent the progression of liver cancer [26]. However, in cells that have progressed to HCC, recruitment of CCR2+ macrophages leads to aggregation of M2-TAMs or myeloid-derived suppressor cells(MDSCs) benefiting tumor growth. M2-TAMs and MDSCs inhibit CD8 T cells and NK cells, thus promoting the growth of HCC and worsening the prognosis and survival of HCC patients [26, 27].
Classification of TAMs and differently polarization of TAMs in vitro.
3. TAMs promoting HCC progress
3.1. TAM promotes angiogenesis and lymphogenesis
Tumor angiogenesis provides essential nutrients and oxygen to tumor tissues, which is one of the basic requirements for tumor growth and proliferation, and also provides vascular pathways for tumor metastasis. Some clinical studies have shown that the degree of vascular distribution in HCC tissues is related to the severity of the disease [28]. TAMs can stimulate angiogenesis by secreting angiogenic factors, or indirectly angiogenesis by secreting angiogenic factors, or indirectly by producing extracellular matrix-degrading proteases, which in turn release sequestered angiogenic factors [29]. Theseangiogenesis factors can stimulate chemotaxis ofendothelial cells and macrophages [30, 31], one of which is Tyrosine kinase with Ig and EGF homology domains 2(TIE2). TIE2 receptor expressing monocytes (TEMs) increase a lot in peripheral blood and the liver of HCC patients, which is of diagnostic value for HCC. In addition, the density of TEMs in HCC tissues is related to the degree of angiogenesis. Therefore, TEMs may represent a new cell marker for the diagnosis of HCC that reflects the degree of hepatic angiogenesis [31].
A majority of studies mainly explored the interaction between HCC cells and TAMs under normoxic conditions, whereas in patients, HCC tumor mass is exposed to varying degrees of hypoxia. Inflammation and hypoxia can reinforce each other in tumor microenvironment. On one hand, hypoxia enhances the proinflammatory activity of innate immune cells such as neutrophils and macrophage [32]. On the other hand, oxygen consumption of innate immune cells increases in response to the sudden burst of cytokine release. At the same time, NF-κB and Hypoxia Inducing Factor-1(HIF-1) are also mutually activated [33].
The distribution and function of TAMs are sensitive to hypoxia-related stress. For example, TAMs tend to cluster in the hypoxic areas of the tumor [34] and have the stronger ability to promote angiogenesis under hypoxia [35]. HIF-1 is also stable under hypoxia [36].It was found that TAMs secreted more Interleukin-1β (IL-1β) due to increased stability of HIF-1α in moderate hypoxia. Under persistent and severe hypoxia, the necrotic debris of HCC cells induced potent IL-1β release by TAMs with an M2 phenotype through Toll-like receptor 4(TLR4)/TIR domain-containing adapter-inducing interferon-β (TRIF)/NF-κB signaling pathway. Following the increase of IL-1β in the local microenvironment, the synthesis of HIF-1α was up-regulated by IL-1β in HCC cells through cyclooxygenase-2. Overexpression of HIF‐1α enhances EMT of HCC cells and IL‐1β promotes HCC metastasis [8].
TAM promotes lymphangiogenesis mainly through two mechanisms. One is secretion of lymphangiogenic factors, suchas vascular endothelial growth factor-C and -D(VEGF-C and VEGF-D) [37]. Angiogenic factors, vascular endothelial growth factor-A (VEGF-A) and matrix metalloproteinase 9(MMP9), secreted by macrophages also promote lymphangiogenesis [38, 39]. The other is promoting tumor lymphangiogenesis through trans-differentiation to lymphatic endothelial cells. TAM can express LYVE-1, one of the markers of lymphatic endothelial cells [40], but there are still some doubts about this theory [41]. However, the mechanism of TAMspromotinglymphangiogenesis has not been confirmed in HCC.
3.2. TAMs andsuppression of antitumoral immune responses
3.2.1. Factors inducing M2 polarization of TAMs
Current studies have shown that IFN-γ and LPS induce TAMs to polarize toward M1, which mediates Th1 response, and are associated with inducing death of tumor cells. In contrast, M2-TAMs, which mediate Th2 response, is polarized by IL-4 and IL-13 and has an immunosuppressive effect, promoting tumor progression [5, 9, 11]. The role of IL-10 in TAM polarization is still in question. Some studies have shown that IL-10 can induce TAMs to transform into the M2 phenotype [42], but there are also studies suggesting that it just plays a supporting role [43].
Oxidored Nitro Domain Containing Protein 1(NOR1) can overexpress in human HCC tissues and is associated with poor prognosis. However, the overexpression of NOR1 protein in human HCC cell lines has no effect on cell proliferation and migration. The poor prognosis of HCC is mainly related to the overexpression of NOR1 protein in CD163+ M2-TAMs. The reason is that overexpression of NOR1 gene in M2-TAMs promotes the development of N-nitrosodiethylamine (DEN) induced HCC by promoting the M2-selective polarization of TAMs [44].
In addition, the inflammatory cytokines produced by TAMs can promote the differentiation and amplification of IL-17 producing T cells [45, 46]. After direct interaction with IL-17 + cells at the edge of tumor invasion, tumor cells acquired the ability to induce CXCR3+ B cell recruitment and maturation. CXCR3+ B cells can induce M2b polarization ofmacrophages through IgG-dependent pathway, thus creating conditions for tumor growth [47].
Agents enhancing M2 polarization of TAMs in vivo. In tumor microenvironment, more agents can promote TAMs transform to M2 phenotype such as CSF1,Wnt,HIFs,IL8,NOR1,IL17.

Treatment with CSF-1R inhibitor PLX3397 delayed tumor growth in both xenograft and allograft models, and this delay was probably mediated by the transition from M2 macrophages to M1 macrophages in a TAM population induced by blockade of the CSF1/CSF-1R signal pathway [48].
3.2.2. Mechanisms of M2-TAMssuppressing antitumoral immune responses
3.2.2.1. Relationships between TAMs and T cells
T cells and NK cells play a key role in the elimination of tumor cells and are closely related to the polarization of TAMs. Overall, TAMs infiltration is proportional to CD4+CD25 + FoxP3 T cells (Tregs) and inversely proportional to CD8+T cells (cytotoxic T cells) in the HCC environment. Blocking CSF1R on TAMs will increase the infiltration of CD8+ T cells and decrease the infiltration of CD4+ T cells [48]. Cantharidin can induce a decrease in CD4+CD25 + FoxP3 T cells by promoting the polarization of TAMs towards M2 [49]. Increased infiltration of Tregs (CCR4+) may be due to TAMsincreasing secretion of CCL17. In turn, CD69+T cells release IFN, which will cause TAMs to produce inhibitor 1-methyl-DL-tryptophan (IDO) and lead to tumor promoting function [50]. IDO protein is a tryptophan degrading enzyme, which has a strong control on T cell response. Experiments in vitro show that suppressive macrophages contacting autologous T cells can restore the ability to produce IL-12, activate T cells and produce IFN-γ. IFN-γ, in turn, lead macrophages to express IDO, and finally suppress anti-tumor functions of T-cells. TAMs can also produce immunosuppressive cytokines (such as IL-10 and TGFβ) that affect the proliferation and differentiation of T cells, and also produce CCL5, CCL20, and CCL22 to recruit nTreg cells. The reduction of T cells may be related to the FASL/FAS pathway [51].
3.2.2.2. Relationships between TAMs and NK cells
In patients with advanced HCC, the number of NK cells decreases significantly, accompanied by decreased production of TNF-α and IFN-γ. The high infiltration of TAMs is positively correlated with impaired functional activity of NK cells in HCC tissues. CD48 protein is highly expressed in TAMs in HCC tissues, which binds to the 2B4 receptor on NK cells, leading to rapid activation of NK cells, followed by failure and death [52]. Sorafenib can mediate the activation of NK cells, enhance the cytotoxicity of NK cells to tumor cells and promote anti-tumor function of NK cells. However, TAMs are crucial for sorafenib activating NK cells. Blocking experiments confirmed the presence of cytokine signaling between two types of cells, which is related to cell contact. Non-secreted surface cytokines on the surface of macrophages may explain the activation of NK cells related to cell contact [53].
3.2.2.3. Other immunosuppressive reactions related to TAMs in HCC
Current studies have reported that the inhibition of Kupffer cells on CD8+ T cells in HCC is mediated by PD-1/PD-L1 interaction [54]. However, the overexpression of PD-L1 in HCC is also associated with TAMs [55]. TAMs also express PD-1, which is negatively correlated with the ability to engulf tumors. Blocking PD-1/PD-L1 signaling pathway in vivo can improve the phagocytosis of macrophages, reduce tumor growth and prolong the survival time of mice [56]. IL-17 secreted by monocytes or macrophages may also inhibit T cell function by promoting the expression of PD-L1 in HCC [55].
Major histocompatibility complex class I(MHC class I) also plays an important role in the control of macrophage reactions. Tumor cells express common MHC class I component β2-microglobulin (β2M), and TAMs up-regulate expression of suppressive receptor, leukocyte immunoglobulin like receptor B1 (LILRB1), on the cell surface. This signaling pathway can directly block phagocytosis of tumor cells. In vivo or in vitro experiments, blocking MHC class I or LILRB1 activates phagocytosis of tumor cells, indicating the importance of this signaling pathway in suppressing anti-tumor immune response [57].
3.3. TAMspromote proliferation and survivalof tumor cells
3.3.1. The pathways that M2-TAMs promote transformation of HCC cells into CSCs
Cancer stem cells or cancer-initiating cells (CSCs) are a kind of tumor cells with self-renewal ability and pluripotency. Many studies believe that CSCs with multiple characteristics of stem cells may be the reason for tumor recurrence after treatment, development and metastasis [58, 59]. CSCs may not be a fixed group of cells, and have plasticity subject to the regulation of tumor microenvironmental factors. Many cell surface markers have been identified to define human HCC CSCS, including CD24, CD44, CD90, CD117, CD133, and epithelial cell adhesion molecule (EpCAM), of which CD44 is an important marker [60-62].
It has shown that M2-TAMs promoted CSCs amplification and tumorigenesis through the IL-6/STAT3 signaling pathway [63], which can transform HCC cells into CSCs from the perspective of cell surface markers. M2-TAMs promote migration and EMT of HCC cells through TLR4/STAT3 signaling pathway [64]. Other hormones and growth factors also participate in this pathway by activating Janus kinase (JAK), and then, downstream STAT3 is phosphorylated to form dimers that mediate nuclear translocation and DNA binding associated with cell proliferation and survival. TAMs can also promote HCC cells to gain CSC-like features through EMT mediated by TGF-β1 [65, 66].
3.3.2. Other pathways of TAMs promoting proliferation and survival of tumor cells
TAMs can lead to deterioration of HCC by producing other proteins. Allograft inflammatory factor-1 (AIF-1) is a cytoskeletal related protein that can increase proliferation and migration of macrophages [67]. CSF1 induces the expression of AIF1 in macrophages by activating the CSF1R-MEK1/2-Erk1/2-c-Jun signaling pathway [68].It was found that CSF1 can also induce activation of CSF1R-PI3K-PLCγ signaling pathway. AIF1, together with phospholipase Cγ (PLCγ), further activates RAC signaling pathway related to cytoskeleton rearrangement, leading to proliferation and migration of macrophages [69]. The expression of AIF1 in TAMs also increases proliferation and migration of HCC cells in vitro, and promotes growth of tumor in animal models [68]. At the same time, AIF1 over-expressed macrophages are accompanied by increased secretion of CXCL16, which is also one of the mechanisms by which AIF1 promotes migration and infiltration of HCC [70].
A study has demonstrated that secretion of M2 macrophage-derived CCL22 in HCC could be induced by tumor cells, and in return that CCL22 could facilitate HCC cells during migration and dissemination via EMT activation [9]. The transcript level of CCL22 was significantly over-expressed in the peritumoral but not in the intratumoral region as well as in normal liver tissues, while expression of the corresponding receptor CCR4 was higher in most HCC cell lines as well as in clinical tumoral tissues compared with hepatocytes and peritumoral tissues. M2 macrophage-derived CCL22 was proved to be a potent chemoattractant for HCC cells and the CCL22-CCR4 mediated migration was inhibited in MHCC97L cells treated with the CCR4 antagonist C-021. Immunoblot analysis revealed that the induction of EMT was through Smad2/3 and Smad1/5/8 activation and Snail upregulation in MHCC97L cells. Based on such evidence, it can be speculated that HCC cells could possibly employ CCR4 to migrate from the bulk tumor towards peritumoral regions where CCL22-producing M2 macrophages predominantly reside.
CXCL8 is one of chemokines of proinflammatory CXC (C-X-C Motif) [71]. Macrophages activated by co-cultured HCC cells can produce high levels of CXCL8. Under stimulation of CXCL8, expression of miR-18a and miR-19a (belonging to miR-17 group) increases in HCC and leads to enhanced proliferation and metastasis of HCC cells [72].Besides, as previously mentioned, CCL2/CCR2 axis promotes the development of HCC in cells that have advanced to tumor cells.
4. Treatment targeting macrophages
4.1. Treatment targeting suppressing functions of M2-TAMs
A number of drugs targeting inhibiting functions of M2-TAMs have been approved for clinical trials against different types of cancer, such as the CSF‐1R inhibitor and antibody for the treatment of diffuse giant cell tumor of the tendon sheath [73], CCL2 antibody and antagonist for treatment of metastatic prostate cancer and so on[74]. However, in the field of HCC, apart from researches on TAMs by using sorafenib, no other drugs related to the inhibition of M2-TAMs have been put into clinical trials.
Combretastatin a-1 phosphate (CA1P) is a microtubule polymerization inhibitor that binds to colchicine binding sites of microtubuloproteins. In addition to inducing apoptosis of HCC cells, CA1P shows anti-TAMs activity in mice. In vitro studies have shown that CA1P induces microtubule depolymerization-mediated AKT inactivation, which resulted in GSK-3β activation, Wnt/β-Catenin pathway inhibition, and Mcl-1 down-regulation. The Wnt/β-Catenin signaling pathway is reported to regulate the polarization of macrophages into M2, while the down-regulation of Mcl-1 will cause the accumulation of ROS and lead to cell apoptosis. Experiments invivohave demonstrated that CA1P reduces TAMs in the tumor microenvironment and reduces the expression of TGF-β and TNF-α [75].
As previously mentioned, the level of IL6 in human HCC samples is related to tumor stage and CSCs level. Upregulating receptor-interacting protein 140 (RIP140) can inhibit NF-κB/IL-6 axis in TAMs to suppress HCC [76]. Tocilizumab, a drug for treatment of rheumatoid arthritis approved by FDA, can block IL6 signaling and inhibit CSCs activity stimulated by TAMs. It was suggested that this drug may be effective in the treatment of HCC [63].
4.2. Modulating TAMs to assist other anti-tumor drugs
Sorafenib is a multikinase inhibitor that has been widely used in the treatment of HCC. Currently, sorafenib is one of the first-line systematic standard treatment for patients with advanced HCC, and the patient's average survival benefit is 3 months [77]. Sorafenib is not that effective, which may be related to infiltration of TAMs [78]. Sorafenib-induced hypoxia may increase the levels of CSF1, HIF, and CCR4, thereby affecting TAM invasion and leading to tumor progression [8, 68, 79]. As previously mentioned, the infiltration of macrophages leads to the overexpression of PD-L1 on the surface of HCC cells, which may also be one of the reasons for sorafenib resistance [55].It was found that a natural product from Abies georgei, structurally related to kaempferol, can be used as a CCR2 antagonist. In mice model with HCC, inhibiting the recruitment of TAMs by inhibiting CCR2 can enhance the efficacy of sorafenib [80]. Compared with Sorafenib, CA1P is likely to be more effective while reducing drug resistance [75].
Zoledronic acid (ZA) can inhibit bone resorption and is mainly used to treat pain caused by bone metastasis of metastatic tumors [81]. Some scholars applied ZA in liver tumors and found that ZA, as TAM inhibitor, improved the anti-tumor effect of Sorafenib [82]. Other studies have shown that combined treatment with ZA can enhance the inhibitory effect of hepatic transcatheter arterial chemoembolization (TACE) on HCC tumor growth, in which ZA had a significant inhibitory effect on the infiltration of TAM after TACE. In addition, ZA can also inhibit the secretion of VEGF after TACE [83].
Although the application of CAR-T in primary HCC has been rarely reported, it has been confirmed that partial fusion of CAR-T can delay tumor progression in mice with metastatic liver cancer, but immunosuppression in liver regulated by MDSC prevents complete tumor clearance. Therefore, further study of CAR-T and TAMs combined target therapy may solve this problem [84].
4.3. Immune checkpoint therapy
Nivolumab is an IgG4 antibody that causes immune checkpoint blockade by diminishing inhibitory signaling through the programmed death receptor-1 pathway [85]. Based on the encouraging results in Phase I/II trials, nivolumab was approved by the FDA for liver cancer as a second line treatment after failure of sorafenib. Anti-PD-1 therapy can enhance the anti-tumor immune response of HCC models. However, this therapy only shows efficacy with targeted drugs aiming hypoxia like CCR4 inhibitors under sorafenib treatment background [79]. As previously mentioned, the elimination of TAMs provides a new approach for dealing with HCC afterPD-1 therapy tolerance. The relationship between the expression level of PD-1/PD-L1 on TAM and Nivolumab responsiveness remains to be elucidated and carefully assessed [86, 87].
Lymphocyte-activation gene 3(LAG3) is also an important immune checkpoint. Fibrinogen-like protein 1(FGL1), a liver-secreted protein, is a major immunosuppression ligand of LAG3. FGL1 inhibits antigen-specific T cell activation, and ablation of FGL1 in mice promotes T cell immunity. FGL1 is overexpressed in tumor cells, and knockout of FGL1 can reduce the density of TAMs [88].
5. Conclusions
TAM inHCC has dual function owning to their different polarization. On the one hand, TAM is involved in tumor immune surveillance during the course of HCC. On the other hand, TAMs can promote the progression of HCC by inhibiting their anti-tumor immune response, promoting the generation of blood vessels and lymphatic vessels, and promoting the proliferation and survival of tumor cells. TAMs targeting therapy focuses on inhibiting tumor-promoting functions of M2 cells, modulating TAMs to assist other anti-tumor drugs and immune checkpoint therapy. TAMs may be an important potential target for the treatment of HCC due to its impact on the development of HCC and subsequent chemotherapy.
Plasticity and flexibility are key characteristics of mononuclear phagocytes and their activation states [77]. Since macrophages are susceptible to the microenvironment, the polarized M1 or M2 phenotypes of macrophages can to some extent reverse to each other in vivo or in vitro [89]. Macrophages that have induced polarization in vitro may not remain polarized after being transferred to the body. All these bring difficulties for TAMs targeting therapy.
Acknowledgements
This study was supported by the Project of the Shanghai Municipal Health Commission (20204Y0012), Innovative Research Team of High-Level Local Universities in Shanghai (SSMU-ZDCX20180802), National Natural Science Foundation of China (81972205), and the Project of Shanghai key clinical specialties (shslczdzk05801).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Ferlay J, Colombet M, Soerjomataram I, Mathers C, Parkin DM, Pineros M. et al. Estimating the global cancer incidence and mortality in 2018: GLOBOCAN sources and methods. International journal of cancer. 2019;144:1941-53
2. David BA, Rezende RM, Antunes MM, Santos MM, Freitas Lopes MA, Diniz AB. et al. Combination of Mass Cytometry and Imaging Analysis Reveals Origin, Location, and Functional Repopulation of Liver Myeloid Cells in Mice. Gastroenterology. 2016;151:1176-91
3. Klein I, Cornejo JC, Polakos NK, John B, Wuensch SA, Topham DJ. et al. Kupffer cell heterogeneity: functional properties of bone marrow derived and sessile hepatic macrophages. Blood. 2007;110:4077-85
4. Scott CL, Zheng F, De Baetselier P, Martens L, Saeys Y, De Prijck S. et al. Bone marrow-derived monocytes give rise to self-renewing and fully differentiated Kupffer cells. Nature communications. 2016;7:10321
5. Murray PJ. Macrophage Polarization. Annual review of physiology. 2017;79:541-66
6. Biswas SK, Mantovani A. Macrophage plasticity and interaction with lymphocyte subsets: cancer as a paradigm. Nat Immunol. 2010;11:889-96
7. Yang Y, Ye YC, Chen Y, Zhao JL, Gao CC, Han H. et al. Crosstalk between hepatic tumor cells and macrophages via Wnt/beta-catenin signaling promotes M2-like macrophage polarization and reinforces tumor malignant behaviors. Cell death & disease. 2018;9:793
8. Zhang J, Zhang Q, Lou Y, Fu Q, Chen Q, Wei T. et al. Hypoxia-inducible factor-1alpha/interleukin-1beta signaling enhances hepatoma epithelial-mesenchymal transition through macrophages in a hypoxic-inflammatory microenvironment. Hepatology. 2018;67:1872-89
9. Yeung OW, Lo CM, Ling CC, Qi X, Geng W, Li CX. et al. Alternatively activated (M2) macrophages promote tumour growth and invasiveness in hepatocellular carcinoma. Journal of hepatology. 2015;62:607-16
10. Wang H, Wang X, Li X, Fan Y, Li G, Guo C. et al. CD68(+)HLA-DR(+) M1-like macrophages promote motility of HCC cells via NF-kappaB/FAK pathway. Cancer Lett. 2014;345:91-9
11. Mantovani A, Sica A. Macrophages, innate immunity and cancer: balance, tolerance, and diversity. Curr Opin Immunol. 2010;22:231-7
12. Beattie L, Sawtell A, Mann J, Frame TCM, Teal B, de Labastida Rivera F. et al. Bone marrow-derived and resident liver macrophages display unique transcriptomic signatures but similar biological functions. Journal of hepatology. 2016;65:758-68
13. Ramachandran P, Pellicoro A, Vernon MA, Boulter L, Aucott RL, Ali A. et al. Differential Ly-6C expression identifies the recruited macrophage phenotype, which orchestrates the regression of murine liver fibrosis. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:E3186-95
14. Li Z, Wu T, Zheng B, Chen L. Individualized precision treatment: Targeting TAM in HCC. Cancer Lett. 2019;458:86-91
15. Acosta JC, Banito A, Wuestefeld T, Georgilis A, Janich P, Morton JP. et al. A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat Cell Biol. 2013;15:978-90
16. Kuilman T, Michaloglou C, Mooi WJ, Peeper DS. The essence of senescence. Genes Dev. 2010;24:2463-79
17. Kang TW, Yevsa T, Woller N, Hoenicke L, Wuestefeld T, Dauch D. et al. Senescence surveillance of pre-malignant hepatocytes limits liver cancer development. Nature. 2011;479:547-51
18. Lujambio A, Akkari L, Simon J, Grace D, Tschaharganeh DF, Bolden JE. et al. Non-cell-autonomous tumor suppression by p53. Cell. 2013;153:449-60
19. Narita M, Nũnez S, Heard E, Narita M, Lin AW, Hearn SA. et al. Rb-mediated heterochromatin formation and silencing of E2F target genes during cellular senescence. Cell. 2003;113:703-16
20. Michaloglou C, Vredeveld LCW, Soengas MS, Denoyelle C, Kuilman T, van der Horst CMAM. et al. BRAFE600-associated senescence-like cell cycle arrest of human naevi. Nature. 2005;436:720-4
21. Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell. 1997;88:593-602
22. Ventura A, Kirsch DG, McLaughlin ME, Tuveson DA, Grimm J, Lintault L. et al. Restoration of p53 function leads to tumour regression in vivo. Nature. 2007;445:661-5
23. Iannello A, Thompson TW, Ardolino M, Lowe SW, Raulet DH. p53-dependent chemokine production by senescent tumor cells supports NKG2D-dependent tumor elimination by natural killer cells. The Journal of experimental medicine. 2013;210:2057-69
24. Xue W, Zender L, Miething C, Dickins RA, Hernando E, Krizhanovsky V. et al. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature. 2007;445:656-60
25. Mudbhary R, Hoshida Y, Chernyavskaya Y, Jacob V, Villanueva A, Fiel MI. et al. UHRF1 overexpression drives DNA hypomethylation and hepatocellular carcinoma. Cancer Cell. 2014;25:196-209
26. Eggert T, Wolter K, Ji J, Ma C, Yevsa T, Klotz S. et al. Distinct Functions of Senescence-Associated Immune Responses in Liver Tumor Surveillance and Tumor Progression. Cancer Cell. 2016;30:533-47
27. Li X, Yao W, Yuan Y, Chen P, Li B, Li J. et al. Targeting of tumour-infiltrating macrophages via CCL2/CCR2 signalling as a therapeutic strategy against hepatocellular carcinoma. Gut. 2017;66:157-67
28. El-Assal ON, Yamanoi A, Soda Y, Yamaguchi M, Igarashi M, Yamamoto A. et al. Clinical significance of microvessel density and vascular endothelial growth factor expression in hepatocellular carcinoma and surrounding liver: possible involvement of vascular endothelial growth factor in the angiogenesis of cirrhotic liver. Hepatology. 1998;27:1554-62
29. De Palma M, Venneri MA, Galli R, Sergi Sergi L, Politi LS, Sampaolesi M. et al. Tie2 identifies a hematopoietic lineage of proangiogenic monocytes required for tumor vessel formation and a mesenchymal population of pericyte progenitors. Cancer Cell. 2005;8:211-26
30. Lewis CE, De Palma M, Naldini L. Tie2-expressing monocytes and tumor angiogenesis: regulation by hypoxia and angiopoietin-2. Cancer Res. 2007;67:8429-32
31. Matsubara T, Kanto T, Kuroda S, Yoshio S, Higashitani K, Kakita N. et al. TIE2-expressing monocytes as a diagnostic marker for hepatocellular carcinoma correlates with angiogenesis. Hepatology. 2013;57:1416-25
32. Snodgrass RG, Boß M, Zezina E, Weigert A, Dehne N, Fleming I. et al. Hypoxia Potentiates Palmitate-induced Pro-inflammatory Activation of Primary Human Macrophages. J Biol Chem. 2016;291:413-24
33. Eltzschig HK, Carmeliet P. Hypoxia and inflammation. N Engl J Med. 2011;364:656-65
34. Laoui D, Van Overmeire E, Di Conza G, Aldeni C, Keirsse J, Morias Y. et al. Tumor hypoxia does not drive differentiation of tumor-associated macrophages but rather fine-tunes the M2-like macrophage population. Cancer Res. 2014;74:24-30
35. Staples KJ, Sotoodehnejadnematalahi F, Pearson H, Frankenberger M, Francescut L, Ziegler-Heitbrock L. et al. Monocyte-derived macrophages matured under prolonged hypoxia transcriptionally up-regulate HIF-1α mRNA. Immunobiology. 2011;216:832-9
36. Werno C, Menrad H, Weigert A, Dehne N, Goerdt S, Schledzewski K. et al. Knockout of HIF-1α in tumor-associated macrophages enhances M2 polarization and attenuates their pro-angiogenic responses. Carcinogenesis. 2010;31:1863-72
37. Schoppmann SF, Birner P, Stöckl J, Kalt R, Ullrich R, Caucig C. et al. Tumor-associated macrophages express lymphatic endothelial growth factors and are related to peritumoral lymphangiogenesis. Am J Pathol. 2002;161:947-56
38. Scavelli C, Vacca A, Di Pietro G, Dammacco F, Ribatti D. Crosstalk between angiogenesis and lymphangiogenesis in tumor progression. Leukemia. 2004;18:1054-8
39. Gomes FG, Nedel F, Alves AM, Nör JE, Tarquinio SBC. Tumor angiogenesis and lymphangiogenesis: tumor/endothelial crosstalk and cellular/microenvironmental signaling mechanisms. Life Sci. 2013;92:101-7
40. Zumsteg A, Baeriswyl V, Imaizumi N, Schwendener R, Rüegg C, Christofori G. Myeloid cells contribute to tumor lymphangiogenesis. PloS one. 2009;4:e7067
41. Gordon EJ, Rao S, Pollard JW, Nutt SL, Lang RA, Harvey NL. Macrophages define dermal lymphatic vessel calibre during development by regulating lymphatic endothelial cell proliferation. Development. 2010;137:3899-910
42. Li L, Sun P, Zhang C, Li Z, Cui K, Zhou W. MiR-98 modulates macrophage polarization and suppresses the effects of tumor-associated macrophages on promoting invasion and epithelial-mesenchymal transition of hepatocellular carcinoma. Cancer Cell Int. 2018;18:95
43. Lang R, Patel D, Morris JJ, Rutschman RL, Murray PJ. Shaping gene expression in activated and resting primary macrophages by IL-10. J Immunol. 2002;169:2253-63
44. Chen S, Zheng P, Wang W, Yi M, Chen P, Cai J. et al. Abberent expression of NOR1 protein in tumor associated macrophages contributes to the development of DEN-induced hepatocellular carcinoma. J Cell Physiol. 2018;233:5002-13
45. Kuang D-M, Peng C, Zhao Q, Wu Y, Chen M-S, Zheng L. Activated monocytes in peritumoral stroma of hepatocellular carcinoma promote expansion of memory T helper 17 cells. Hepatology. 2010;51:154-64
46. Kuang D-M, Peng C, Zhao Q, Wu Y, Zhu L-Y, Wang J. et al. Tumor-activated monocytes promote expansion of IL-17-producing CD8+ T cells in hepatocellular carcinoma patients. J Immunol. 2010;185:1544-9
47. Liu R-X, Wei Y, Zeng Q-H, Chan K-W, Xiao X, Zhao X-Y. et al. Chemokine (C-X-C motif) receptor 3-positive B cells link interleukin-17 inflammation to protumorigenic macrophage polarization in human hepatocellular carcinoma. Hepatology. 2015;62:1779-90
48. Ao J-Y, Zhu X-D, Chai Z-T, Cai H, Zhang Y-Y, Zhang K-Z. et al. Colony-Stimulating Factor 1 Receptor Blockade Inhibits Tumor Growth by Altering the Polarization of Tumor-Associated Macrophages in Hepatocellular Carcinoma. Mol Cancer Ther. 2017;16:1544-54
49. Lu S, Gao Y, Huang X, Wang X. Cantharidin exerts anti-hepatocellular carcinoma by miR-214 modulating macrophage polarization. Int J Biol Sci. 2014;10:415-25
50. Zhao Q, Kuang D-M, Wu Y, Xiao X, Li X-F, Li T-J. et al. Activated CD69+ T cells foster immune privilege by regulating IDO expression in tumor-associated macrophages. J Immunol. 2012;188:1117-24
51. Noy R, Pollard JW. Tumor-associated macrophages: from mechanisms to therapy. Immunity. 2014;41:49-61
52. Wu Y, Kuang D-M, Pan W-D, Wan Y-L, Lao X-M, Wang D. et al. Monocyte/macrophage-elicited natural killer cell dysfunction in hepatocellular carcinoma is mediated by CD48/2B4 interactions. Hepatology. 2013;57:1107-16
53. Sprinzl MF, Reisinger F, Puschnik A, Ringelhan M, Ackermann K, Hartmann D. et al. Sorafenib perpetuates cellular anticancer effector functions by modulating the crosstalk between macrophages and natural killer cells. Hepatology. 2013;57:2358-68
54. Wu K, Kryczek I, Chen L, Zou W, Welling TH. Kupffer cell suppression of CD8+ T cells in human hepatocellular carcinoma is mediated by B7-H1/programmed death-1 interactions. Cancer Res. 2009;69:8067-75
55. Chen J, Li G, Meng H, Fan Y, Song Y, Wang S. et al. Upregulation of B7-H1 expression is associated with macrophage infiltration in hepatocellular carcinomas. Cancer Immunol Immunother. 2012;61:101-8
56. Gordon SR, Maute RL, Dulken BW, Hutter G, George BM, McCracken MN. et al. PD-1 expression by tumour-associated macrophages inhibits phagocytosis and tumour immunity. Nature. 2017;545:495-9
57. Barkal AA, Weiskopf K, Kao KS, Gordon SR, Rosental B, Yiu YY. et al. Engagement of MHC class I by the inhibitory receptor LILRB1 suppresses macrophages and is a target of cancer immunotherapy. Nat Immunol. 2018;19:76-84
58. Sun Y-F, Xu Y, Yang X-R, Guo W, Zhang X, Qiu S-J. et al. Circulating stem cell-like epithelial cell adhesion molecule-positive tumor cells indicate poor prognosis of hepatocellular carcinoma after curative resection. Hepatology. 2013;57:1458-68
59. Zeng Z, Ren J, O'Neil M, Zhao J, Bridges B, Cox J. et al. Impact of stem cell marker expression on recurrence of TACE-treated hepatocellular carcinoma post liver transplantation. BMC Cancer. 2012;12:584
60. Yamashita T, Ji J, Budhu A, Forgues M, Yang W, Wang H-Y. et al. EpCAM-positive hepatocellular carcinoma cells are tumor-initiating cells with stem/progenitor cell features. Gastroenterology. 2009;136:1012-24
61. Yang ZF, Ho DW, Ng MN, Lau CK, Yu WC, Ngai P. et al. Significance of CD90+ cancer stem cells in human liver cancer. Cancer Cell. 2008;13:153-66
62. Lee TKW, Castilho A, Cheung VCH, Tang KH, Ma S, Ng IOL. CD24(+) liver tumor-initiating cells drive self-renewal and tumor initiation through STAT3-mediated NANOG regulation. Cell Stem Cell. 2011;9:50-63
63. Wan S, Zhao E, Kryczek I, Vatan L, Sadovskaya A, Ludema G. et al. Tumor-associated macrophages produce interleukin 6 and signal via STAT3 to promote expansion of human hepatocellular carcinoma stem cells. Gastroenterology. 2014;147:1393-404
64. Yao R-R, Li J-H, Zhang R, Chen R-X, Wang Y-H. M2-polarized tumor-associated macrophages facilitated migration and epithelial-mesenchymal transition of HCC cells via the TLR4/STAT3 signaling pathway. World J Surg Oncol. 2018;16:9
65. Fan Q-M, Jing Y-Y, Yu G-F, Kou X-R, Ye F, Gao L. et al. Tumor-associated macrophages promote cancer stem cell-like properties via transforming growth factor-beta1-induced epithelial-mesenchymal transition in hepatocellular carcinoma. Cancer Lett. 2014;352:160-8
66. Gupta DK, Singh N, Sahu DK. TGF-β Mediated Crosstalk Between Malignant Hepatocyte and Tumor Microenvironment in Hepatocellular Carcinoma. Cancer Growth Metastasis. 2014;7:1-8
67. Tian Y, Kelemen SE, Autieri MV. Inhibition of AIF-1 expression by constitutive siRNA expression reduces macrophage migration, proliferation, and signal transduction initiated by atherogenic stimuli. Am J Physiol, Cell Physiol. 2006;290:C1083-C91
68. Cai H, Zhu X-D, Ao J-Y, Ye B-G, Zhang Y-Y, Chai Z-T. et al. Colony-stimulating factor-1-induced AIF1 expression in tumor-associated macrophages enhances the progression of hepatocellular carcinoma. Oncoimmunology. 2017;6:e1333213
69. Imai Y, Kohsaka S. Intracellular signaling in M-CSF-induced microglia activation: role of Iba1. Glia. 2002;40:164-74
70. Liu J, Chen S, Wang W, Ning B-F, Chen F, Shen W. et al. Cancer-associated fibroblasts promote hepatocellular carcinoma metastasis through chemokine-activated hedgehog and TGF-β pathways. Cancer Lett. 2016;379:49-59
71. David JM, Dominguez C, Hamilton DH, Palena C. The IL-8/IL-8R Axis: A Double Agent in Tumor Immune Resistance. Vaccines (Basel). 2016 4
72. Yin Z, Huang J, Ma T, Li D, Wu Z, Hou B. et al. Macrophages activating chemokine (C-X-C motif) ligand 8/miR-17 cluster modulate hepatocellular carcinoma cell growth and metastasis. Am J Transl Res. 2017;9:2403-11
73. Cassier PA, Italiano A, Gomez-Roca CA, Le Tourneau C, Toulmonde M, Cannarile MA. et al. CSF1R inhibition with emactuzumab in locally advanced diffuse-type tenosynovial giant cell tumours of the soft tissue: a dose-escalation and dose-expansion phase 1 study. Lancet Oncol. 2015;16:949-56
74. Pienta KJ, Machiels J-P, Schrijvers D, Alekseev B, Shkolnik M, Crabb SJ. et al. Phase 2 study of carlumab (CNTO 888), a human monoclonal antibody against CC-chemokine ligand 2 (CCL2), in metastatic castration-resistant prostate cancer. Invest New Drugs. 2013;31:760-8
75. Mao J, Wang D, Wang Z, Tian W, Li X, Duan J. et al. Combretastatin A-1 phosphate, a microtubule inhibitor, acts on both hepatocellular carcinoma cells and tumor-associated macrophages by inhibiting the Wnt/β-catenin pathway. Cancer Lett. 2016;380:134-43
76. Hu Y-C, Yi Z-J, Zhou Y, Li P-Z, Liu Z-J, Duan S-G. et al. Overexpression of RIP140 suppresses the malignant potential of hepatocellular carcinoma by inhibiting NF-κB-mediated alternative polarization of macrophages. Oncol Rep. 2017;37:2971-9
77. Bruix J, Gores GJ, Mazzaferro V. Hepatocellular carcinoma: clinical frontiers and perspectives. Gut. 2014;63:844-55
78. Lin Y-Y, Tan C-T, Chen C-W, Ou D-L, Cheng A-L, Hsu C. Immunomodulatory Effects of Current Targeted Therapies on Hepatocellular Carcinoma: Implication for the Future of Immunotherapy. Semin Liver Dis. 2018;38:379-88
79. Chen Y, Ramjiawan RR, Reiberger T, Ng MR, Hato T, Huang Y. et al. CXCR4 inhibition in tumor microenvironment facilitates anti-programmed death receptor-1 immunotherapy in sorafenib-treated hepatocellular carcinoma in mice. Hepatology. 2015;61:1591-602
80. Yao W, Ba Q, Li X, Li H, Zhang S, Yuan Y. et al. A Natural CCR2 Antagonist Relieves Tumor-associated Macrophage-mediated Immunosuppression to Produce a Therapeutic Effect for Liver Cancer. EBioMedicine. 2017;22:58-67
81. Zekri J, Mansour M, Karim SM. The anti-tumour effects of zoledronic acid. J Bone Oncol. 2014;3:25-35
82. Zhang W, Zhu X-D, Sun H-C, Xiong Y-Q, Zhuang P-Y, Xu H-X. et al. Depletion of tumor-associated macrophages enhances the effect of sorafenib in metastatic liver cancer models by antimetastatic and antiangiogenic effects. Clin Cancer Res. 2010;16:3420-30
83. Zhou D-Y, Qin J, Huang J, Wang F, Xu G-P, Lv Y-T. et al. Zoledronic acid inhibits infiltration of tumor-associated macrophages and angiogenesis following transcatheter arterial chemoembolization in rat hepatocellular carcinoma models. Oncol Lett. 2017;14:4078-84
84. Burga RA, Thorn M, Point GR, Guha P, Nguyen CT, Licata LA. et al. Liver myeloid-derived suppressor cells expand in response to liver metastases in mice and inhibit the anti-tumor efficacy of anti-CEA CAR-T. Cancer Immunol Immunother. 2015;64:817-29
85. Rajan A, Kim C, Heery CR, Guha U, Gulley JL. Nivolumab, anti-programmed death-1 (PD-1) monoclonal antibody immunotherapy: Role in advanced cancers. Human vaccines & immunotherapeutics. 2016;12:2219-31
86. Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJ, Robert L. et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature. 2014;515:568-71
87. Herbst RS, Soria JC, Kowanetz M, Fine GD, Hamid O, Gordon MS. et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature. 2014;515:563-7
88. Wang J, Sanmamed MF, Datar I, Su TT, Ji L, Sun J. et al. Fibrinogen-like Protein 1 Is a Major Immune Inhibitory Ligand of LAG-3. Cell. 2019 176
89. Saccani A, Schioppa T, Porta C, Biswas SK, Nebuloni M, Vago L. et al. p50 nuclear factor-kappaB overexpression in tumor-associated macrophages inhibits M1 inflammatory responses and antitumor resistance. Cancer Res. 2006;66:11432-40
Author contact
![]() Corresponding authors: Yi Luo (Email: luoyicom), Qiang Xia (Email: xiaqiangedu.cn).
Corresponding authors: Yi Luo (Email: luoyicom), Qiang Xia (Email: xiaqiangedu.cn).