Impact Factor ISSN: 1449-1907

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2019; 16(2):212-220. doi:10.7150/ijms.29930 This issue Cite
Research Paper
MicroRNA-29a is a key regulon that regulates BRD4 and mitigates liver fibrosis in mice by inhibiting hepatic stellate cell activation
Ying-Hsien Huang1, Hsing-Chun Kuo2,3,4,5, Ya-Ling Yang6 ![]() , Feng-Sheng Wang7
, Feng-Sheng Wang7 ![]()
1. Department of Pediatrics, Kaohsiung Chang Gung Memorial Hospital and Chang Gung University College of Medicine, Kaohsiung, Taiwan
2. Department of Nursing, Chang Gung University of Science and Technology, Chiayi, Taiwan
3. Reseach Fellow, Chiayi Chang Gung Memorial Hospital, Chiayi, Taiwan
4. Research Center for Industry of Human Ecology and Research Center for Chinese Herbal Medicine, College of Human Ecology, Chang Gung University of Science and Technology, Taoyuan, Taiwan
5. Chronic Diseases and Health Promotion Research Center, CGUST, Chiayi, Taiwan
6. Department of Anesthesiology, Kaohsiung Chang Gung Memorial Hospital and Chang Gung University College of Medicine, Kaohsiung, Taiwan, 833
7. Core Laboratory for Phenomics & Diagnostics, Department of Medical Research, Kaohsiung Chang Gung Memorial Hospital and Chang Gung University College of Medicine, Kaohsiung, Taiwan, 833
Received 2018-9-14; Accepted 2018-12-5; Published 2019-1-1
Abstract
MicroRNA-29a is a key regulon that regulates hepatic stellate cells (HSCs) and mitigates liver fibrosis. However, the mechanism by which it does so remains largely undefined. The inhibition of bromodomain-4 protein (BRD4) represents a novel therapeutic target in hepatic fibrosis. Therefore, the purpose of this study is to investigate the miR-29a regulation of BRD4 signaling in a bile duct-ligation (BDL) animal model with regard to developing cholestatic liver fibrosis. Hepatic tissue in miR-29a transgenic mice (miR-29aTg mice) displayed weak fibrotic matrix, as shown by α-smooth muscle actin staining within affected tissues compared to wild-type mice. miR-29a overexpression reduced the BDL exaggeration of BRD4 and SNAI1 expression. Increased miR-29a signaling caused the downregulation of EZH2, MeCP2, and SNAI1, as well as the upregulation of PPAR-γ expression, in primary HSCs. We further demonstrated that the administration of JQ1, a BRD4 inhibitor, could inhibit BRD4, C-MYC, EZH2, and SNAI1 expression, while both JQ1 and a miR-29a mimic could inhibit the migration and proliferation of HSCs. In short, our research demonstrates that miR-29a negatively regulates HSC activation by inhibiting BRD4 and EZH2 function, thus making it a promising target for the pharmacologic treatment of hepatic fibrosis.
Keywords: miR-29a, bile duct ligation, cholestasis, liver fibrosis, BRD4
Introduction
Chronic liver damage caused by any form of hepatitis or cholestasis can cause liver fibrosis, which is a complex process controlled by a series of signaling pathways [1]. When hepatic stellate cells (HSC) are activated and undergo morphologic and functional trans-differentiation [1-3], they not only secrete profibrogenic mediators, such as transforming growth factor-β (TGF-β) signaling, but also generate ECM components.
MicroRNAs (miRNAs) are small single-stranded non-coding RNAs that can suppress endogenous mRNA transcripts [4]. Cumulative evidence has shown that miR-29 levels are significantly decreased in fibrotic livers and that their downregulation influences HSC activation [5-7]. Furthermore, an increase in miR-29 in murine HSCs has been shown to inhibit collagen expression [6, 8] by directly targeting the mRNA expression of ECM genes. In our previous studies [9-15], we have already demonstrated that miR-29a overexpression in cholestatic mice significantly inhibited hepatocellular damage and liver fibrosis, and the multiple pathways of apoptosis, autophagy, endoplasmic reticulum stress, and toll-like receptors were all included.
The field of epigenetics consists of changing both the chromatin structure and the DNA methylation and acetylation patterns of a genome [16]. Histones require the addition of a functional group, such as methylation, acetylation, phosphorylation, sumoylation, or ubiquitination [17]. We have previously found that miR-29a normalizes histone deacetylase 4 expression, increases the acetylation status of H3K9 in HSCs, and mitigates HSC activation [11]. However, histone methylation is reversible, and its dynamic nature is controlled by a balance between histone methyltransferases and demethylases [18]. A growing amount of evidence has implied that inhibiting the function of Enhancer of Zeste Homolog 2 (EZH2), a catalytic sub-unit of the Polycomb Repressive Complex 2 and histone methyltransferases that catalyze the addition of methyl groups to histone H3 at lysine 27 [18], can lessen liver fibrosis by blocking HSC function [19, 20]. Furthermore, suppression of BRD4 has been demonstrated to decrease the expression of EZH2 through the upregulation of C-MYC [21]. In a recent study, TGF-β1 was observed to promote HSC activation via the BRD4/C-MYC/EZH2 pathway in liver fibrosis [22]. The interaction of SNAI1 and EZH2 can also repress E-cadherin expression, which is essential for triggering epithelial-mesenchymal transition (EMT) [23]. Therefore, in this study, we decided to investigate the miR-29a regulation of BRD4/ EZH2 signaling in a cholestatic animal with regard to liver fibrosis and HSC activation.
Materials and Methods
Ethics statement
The Institutional Animal Care and Use Committee of Chang Gung Memorial Hospital reviewed and approved all protocols related to animal uses (#2017091801). We acquired male C57BL/6 mice (body weight 25- 35 g) from BioLASCO Taiwan Co., Ltd. and housed them in an animal facility at 22 °C, with a relative humidity of 55%, in a 12 h light/12 h dark cycle, where they were given both sterile tap water and food ad libitum.
Construction and breeding of the miR-29a transgenic mouse colony
Transgenic mice that overexpressed miR-29a driven by the PGK promoter were bred and housed in a specific pathogen-free rodent barrier, as described in a previous study [14]. The genotype of the transgenic mice was typed with PCR and primers (forward: 5'-GAGGATCCCCTCAAGGATACCAAGGGATGAAT-3' and reverse 5'-CTTCTAGAAGGAGTGTTTCTAGGTATCCGTCA-3'). We obtained wild-type mice from littermates that did not carry the construct.
Animal model and experimental protocol
Six to eight mice were used for each of our experiments. The mice were categorized into either the “BDL” group or the “sham” group in accordance with whether it had received an actual ligation or a sham ligation of the common bile duct, the method of which has been previously described [11]. All the mice were euthanized one week after the operation, at which point liver tissues were dissected, snap-frozen, and processed to isolate total RNA and proteins. All specimens were stored at -80 °C until biochemical analysis.
Primary HSC isolation and culture
We isolated primary HSCs from fresh livers in mice using the following procedure: Hepatic specimens were digested by pronase and collagenase. The digested mixtures were subjected to density gradient centrifugation in 8.5% Nycodenz (Sigma-Aldrich, St. Louis, MO) as previously described in another study [24, 25]. HSCs expressed autofluorescence of retinoids in the lipid droplets of cell cultures, and HSC lipid droplets were verified under a fluorescence microscope. Trypan blue exclusion assays demonstrated that the viability of cell culture was more than 95%. 95-99% cells were positive for Oil red O staining [25]. Cells were incubated in Dulbecco's modified Eagle's medium supplemented with 5% newborn calf serum. After one day in culture, the HSCs exhibited a dormant phenotype, which was followed by an activated phenotype 7-14 days after incubation. The cell cultures were incubated until confluence, and those within 2-6 passages were used for study.
RNAi transfection
The hepatic stellate cells (HSCs) were maintained in DMEM supplemented with 10% fetal bovine serum, glutamax, and antibiotic-antimycotic in a 5% CO2 humidified incubator at 37℃. Cells were seeded at a density of 9×105 cells per 6-cm culture dish for western blot, 3×104 cells/well in the ibidi Culture-Insert 2 Well for wound healing assay, and 8×103 cells/well in the 96-well culture microplate for cell proliferation assay. Twenty-four hours after initial seeding, we transfected the HSCs with a concentration of 25 nM of miR-29a precursor (mimic-miR-29a, GE Healthcare Dharmacon, IN), miR control (GE Healthcare Dharmacon, IN), or miR-29a antisense oligonucleotide inhibitor (inhibitor-miR-29a GE Healthcare Dharmacon, IN) for 24 h with Lipofectamine™ RNAiMAX Transfection Reagent (Invitrogen, CA) according to the manufacturer's instructions. Infected cells were incubated for 24 h at 37˚C and then used for further experiments [26].
Immunohistological Analysis
For immunohistochemical analysis, formalin-fixed paraffin-embedded blocks of the mice's liver tissues were cut into 2-μm sections. After deparaffinization and rehydration, we heated sections in a citrate buffer (10 mM, pH 6, Thermo Fisher Scientific, Waltham, MA, USA) in a microwave for 30 min to retrieve the antigens. Endogenous peroxidase activity was blocked with a 3% hydrogen peroxide (UltraVision Hydrogen Peroxide Block; Thermo Fisher Scientific) for 10 min. The sections were incubated with α-SMA antibody (ab5694, abcam, JHY) for 1 h at room temperature and then visualized using HRP polymer (UltraVision Quanto Detection System; Thermo Fisher Scientific) and DAB chromogen (DAB Peroxidase Substrate Kit; Vector Laboratories, Burlingame, CA, USA). The sections were counterstained with Mayer's hematoxylin (ScyTek Laboratories, Logan, UT, USA), dehydrated, and then mounted using a mounting medium. The staining intensity of sections was measured by independent color channel of an image J analysis [27]. Each group had six to eight samples.
JQ1 treatment for hepatic stellate cells
The HSCs were seeded in a 10-cm culture dish with a density of 1.5×106 cells /dish and incubated overnight at 37˚C. Cells were then maintained in DMEM medium with JQ1(100 nM) for 24 h. Proteins were extracted from the cells for western blot analysis.
Western blot analysis
We used approximately 20 mg of liver tissue in a 500 ml protein lysis buffer (iNtRON, Seongnam-si), homogenized by the MagNA Lyser system (Roche, Germany). The extraction protein (40 μg) from the supernatant of each sample was mixed with 4x sample buffer (Bio-Rad) and boiled for 10 min. All samples, including protein markers (TM-PM10170, TOOLS, NTPC), were fractionated by 6~15 % SDS-PAGE and transferred to a PVDF membrane. After incubation with 10 % nonfat milk in TBST (10 mM Tris, pH 8.0, 150 mM NaCl, 0.05% Tween 20) for 60 min at room temperature, they were washed with TBST and incubated with primary antibodies against BRD4 (ab128874, abcam, JHY), SNAI1 (#3895, cell signaling, MA ), EZH2 (#4905, cell signaling, MA), MeCP2 (#10861-1-AP, PROTEINTECH, IL), or PPAR-γ (#16643-1-AP, PROTEINTECH, IL), as well as Nucleolin (ab134164, abcam, JHY) or GAPDH, used as the internal control (#60004-1-Ig, PROTEINTECH, IL) at 4 °C overnight. We washed membranes three times for 10 min and incubated them with horseradish peroxidase-coupled anti-rabbit immunoglobulin-G antibodies (dilution: 1:5000) and HRP anti-mouse immunoglobulin-G antibodies (dilution: 1:10,000) at room temperature for 1 h. Blots were washed with TBST three times and developed with enhanced chemiluminescence detection (GE Healthcare Biosciences AB, Uppsala, Sweden) and exposed to film, and we quantified the signals using densitometry.
Cell proliferation analysis
Cell proliferation was determined by the WST-1 assay. After infection by the miR-29a precursor or being treated with 100 nM JQ1(Sigma-Aldrich Chemie GmbH) for 24 h, we added 10 μL WST-1 reagent (Roche Diagnostics, Laval, Quebec, Canada) to each well and incubated for another 60 minutes at 37° C. The absorbance was determined using a microplate reader (Hidex Sense microplate reader) at a test wavelength of 450 nm and reference wavelength of 630 nm.
Wound healing assay
We adopted the wound healing assay to detect cell migration. HSCs were seeded in a culture-insert (ibidi culture-insert 2 well, ibidi GmbH, Martinsried, Germany) at a density of 3×104 cells per well. After allowing the cells to attach overnight, we removed the culture-insert and washed the cells with PBS to remove non-adherent cells. We then provided fresh medium containing JQ1(100nM) or miR-29a(25nM) mimic and photographed the plate at 0, 5, and 8 h to capture the two different fields at each time point on each plate. The number of cells that migrated into the wound space were manually counted in three fields per well under a light microscope at 50× magnification. We then quantified the areas using image J analysis.
Statistical analysis
All values in the figures and tables are expressed as mean ± standard error. Quantitative data were analyzed using the one-way analysis of variance [28] when appropriate, and we adopted the least significant difference (LSD) test for post-hoc testing. A two-sided p-value less than 0.05 was considered statistically significant.
Results
Overexpression of miR-29a considerably decreases liver fibrosis in cholestatic livers
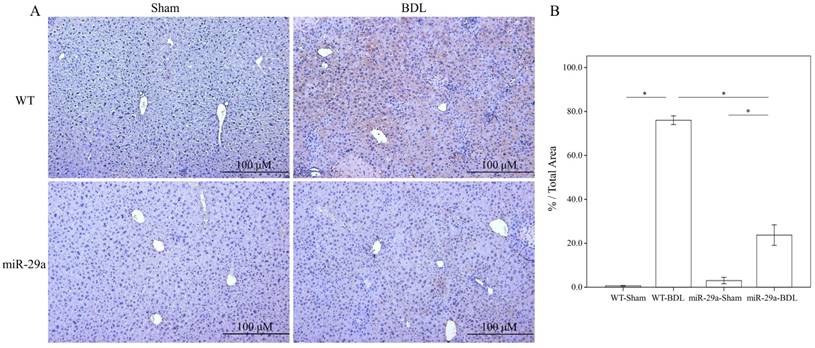
To determine the influence of overexpressed miR-29a on the cholestatic liver injury process, we studied α-SMA expression by immunohistochemistry. As shown in Figure 1, α-SMA expression revealed abundant brown staining around the portal regions in the BDL-affected liver specimens in the wild-type mice compared to the sham group (p < 0.001). This histopathology was clearly alleviated in the BDL-affected miR-29Tg mice (p < 0.001) (Figure 1).
Overexpression of miR-29a in the murine model resulted in the downregulation of fibrosis in mice livers after BDL. (A) Histomorphometric analyses of alpha-smooth muscle actin (α-SMA) demonstrated strongα-SMA immunoreactivity (brown staining) in wild-type (WT) mice compared to those in miR-29aTg mice. (B) We measured the histomorphometric analyses of alpha-smooth muscle actin by independent color channel of an image J analysis. Data are expressed as the mean ± SE of six to eight samples per group. *indicates a p < 0.05 between the groups.
miR-29a overexpression reduced hepatic BRD4 and SNAI1 expression in cholestatic livers
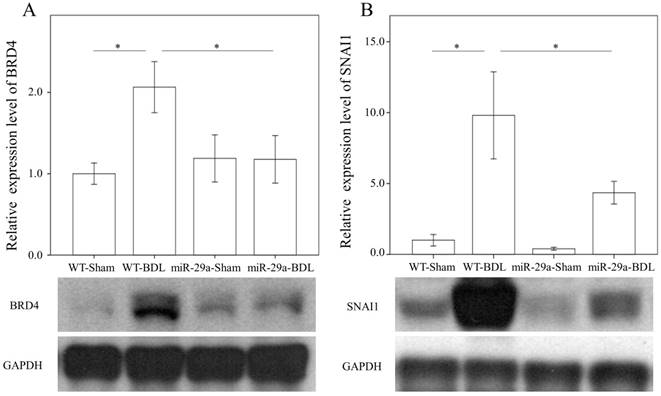
We first examined whether miR-29a signaling could alter BRD4, EZH2, and SNAI1 expressions in liver tissues injured by BDL. In the wild-type group, BRD4 (p = 0.011) and SNAI1 (p < 0.001) were significantly increased after BDL (Fig. 2). The BDL increase of BRD4 (p=0.029) and SNAI1 (p=0.014) levels was significantly reduced in the miR-29aTg mice (Fig. 2). However, the signal intensity of EZH2 was too weak to detect between the two groups.
Comparison of the protein expression of BRD4 (A) and SNAI1 (B) in WT and miR-29Tg mice livers following BDL. Data from the six to eight samples per group are expressed as mean ± SE. *indicates a p < 0.05 between the groups.
Increased miR-29a function reduced EZH2, MeCP2, and SNAI1 and increased PPAR-γ expressions in HSCs
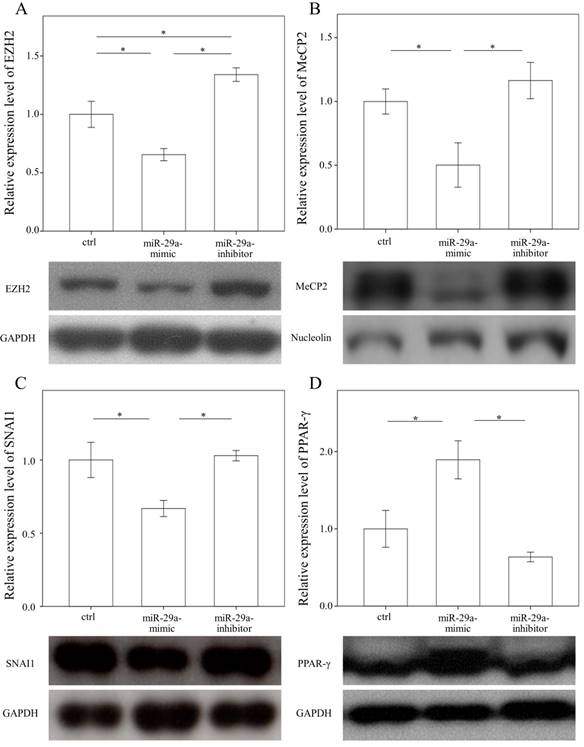
In a previous study, we found that the expressions in BRD4 were significantly reduced in the miR-29 mimic-transfected HSCs [14]. Therefore, we examined whether miR-29a signaling could regulate EZH2, MeCP2, SNAI1, and PPAR-γ expressions in primary HSCs. As shown in Figure 3 A, B, and C, the expressions in EZH2, MeCP2, and SNAI1 were significantly reduced in the miR-29 mimic-transfected cell cultures (p = 0.007; p = 0.048; and p = 0.016, respectively). In contrast, after treatment with a miR-29 mimic, PPAR-γ expression was significantly upregulated in the HSCs (p = 0.012, Fig. 3D).
Comparison of protein expression of EZH2 (A), MeCP2(B), SNAI1(C), and PPAR-γ (D) in primary hepatic stellate cells after being treated with or without a miR-29a mimic or inhibitor or only a miRNA scramble for 24 hours. Data are expressed as mean ± SE of the four to six samples per group. *indicates a p < 0.05 between the groups.
Administering JQ1 reduced BRD4, C-MYC, EZH2, and SNAI1 expressions in HSCs
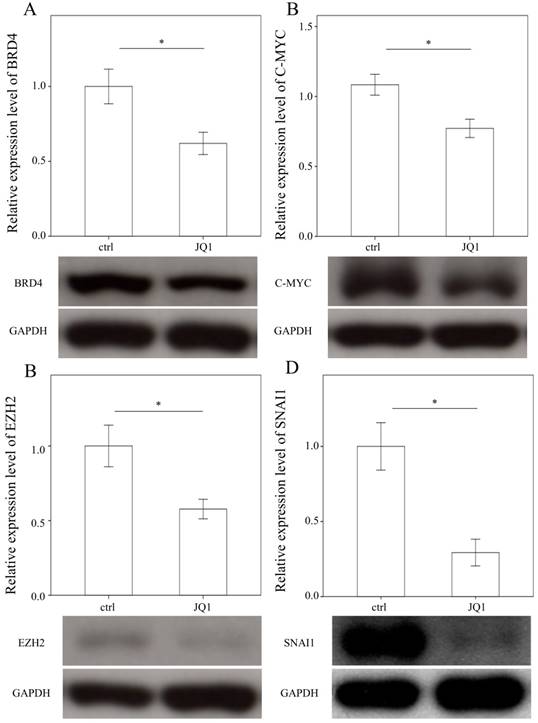
To examine the effects of JQ1 on the expression of BRD4, C-MYC, EZH2, and SNAI1, we treated primary HSCs with JQ1, which is a BRD4 inhibitor. As expected, JQ1 significantly downregulated the expression of BRD4, C-MYC, EZH2, and SNAI1 in primary HSCs (p = 0.05; p= 0.035; p= 0.033 and p= 0.005, Fig. 4).
Comparison of protein expression of BRD4 (A), C-MYC (B), EZH2 (C), and SNAI1(D) in primary hepatic stellate cells after being treated with JQ1 for 24 hours. Data are expressed as mean ± SE of the four to five samples per group. *indicates a p < 0.05 between the groups.
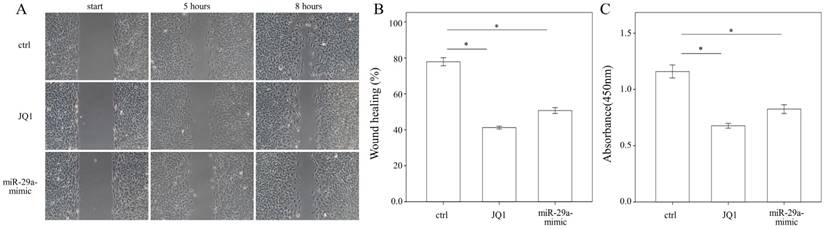
Administering JQ1 and miR-29a inhibits HSC migration and proliferation
To evaluate whether JQ1 may regulate HSC migration, a wound healing assay was performed using primary HSCs. The results of the present study showed that both JQ1 and miR-29a mimic were capable of inhibiting the migration of primary HSCs (both p < 0.001; Fig. 5A, B). We then conducted a cell proliferation assay to examine the effects of JQ1 and miR-29a on cell proliferation. As shown in Fig. 5C, either a JQ1 or miR-29a mimic could inhibit the proliferation of primary HSCs (both p < 0.001).
We measured the migration and proliferation of primary activated HSCs using wound healing and WST-1 assay, respectively. A miR-29a mimic or JQ1 significantly inhibited the migration of primary HSCs of WT mice. Data are expressed as the mean ± SE of three independent experiments. *indicates a p < 0.05 between the groups.
Discussion
The trans-differential activation of liver injury and HSCs is responsible for the pathogenesis of liver fibrosis. Increased miR-29a function in cholestatic mice significantly inhibited liver fibrosis, and this study is the first to report that miR-29a could directly modulate BRD4 expression of the profibrogenic phenotype of HSCs in an obstructive jaundice mouse model. Furthermore, we discovered that the expression of BRD4 is associated miR-29a, along with EZH2, MeCP2, and SNAI1, as well as upregulation of PPAR-γ expression, in activating primary HSCs. Finally, we have demonstrated that overexpression of miR29a or inhibition of BRD4 can significantly weaken activated HSC migration and proliferation.
Although studies have implicated BRD4, a bromodomain protein and extra-terminal member, can delicately control inflammatory gene expression through epigenetic pathways [29]. We have also uncovered that overexpression of miR-29a in cholestatic mice significantly obstructed TLR2 and TLR4 signaling in liver tissues while significantly decreasing the proinflammatory cytokines expression [30]. Sun et al. also showed that inhibition of BRD4 with JQ1 suppressed activation of TLR4 signaling and decreased acute myocardial infarction damage [31]. Meanwhile, JQ1 has been clearly shown to effectively suppress HSC proliferation and differentiation into myofibroblasts and to ameliorate pathological fibrotic responses [32]. EZH2 is the key catalytic sub-unit of Polycomb repressive complex 2 and has emerged as a crucial regulator of EMT, wound healing, fibrogenesis, and tumor metastasis [33, 34]. In this study, we demonstrated that miR-29a positively regulated EZH2 expression by upregulating C-MYC in primary HSCs. In line with our study, overexpression of miR-29a inhibits BDL-provoked excessive fibrogenesis, which is partially mediated by the inhibition of BRD4/C-MYC/EZH2 signaling. SNAI1 is the key transcription factor best known for its ability to trigger EMT to influence fibrosis development [35]. Scarpa et al. demonstrated an upregulated expression of SNAI1 in human hepatic fibrosis and murine experimental models of liver injury [36]. SNAI1 knockdown resulted in the improvement of hepatic fibrosis and its involvement in the HSC trans-differentiation process [36]. Furthermore, SNAI1 has been reported to recruit Polycomb Repressive Complex 2 to the E-cadherin promoter to repress E-cadherin expression [23]. Recently, Oh et al. also reported that high glucose levels enhance the formation of EZH2/Snail complex to facilitate E-cadherin repression and stimulate human mesenchymal stem cell migration [37]. All of these studies agree with our finding that increased miR-29a function significantly inhibits the upregulation of SNAI1 in cholestatic liver and primary HSCs.
The MeCP2 (methyl CpG binding protein 2) dependent epigenetic pathway facilitates the activation of HSCs by regulating the suppression of PPARγ, which opposes myofibroblast trans-differentiation expression by targeting DNA methyltransferases to maintain the HSCs in a quiescent stage [38]. MeCP2 also promotes EZH2 expression and H3K27 methylation to constitute a repressive chromatin structure in the promoter of PPARγ [5]. Meanwhile, the inhibition of EZH2 or the activation of PPARγ blocked the exacerbation of liver fibrosis in mice [20]. Therefore, the EZH2-mediated repression of PPARγ may be involved in the aggravated liver fibrosis driven by increased miR-29a in HSCs. In short, our data suggest that miR-29a plays an essential role in guiding the transition of HSC phenotypes and contributing to the regulation of cellular proliferation and the migration of activated HSCs.
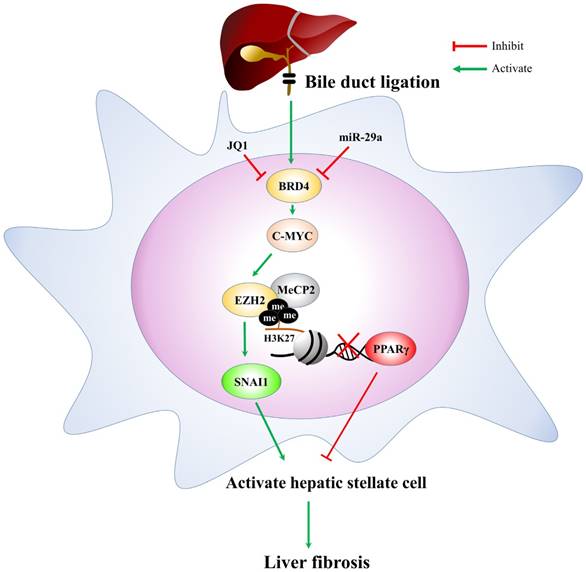
In conclusion, based on the miR-29a signaling protection model (miR-29aTg mice) in liver fibrosis, we demonstrated a new mechanism for liver damage via inhibiting BRD4/EZH2/SNAI1 pathway. The bile duct-ligation model and liver fibrosis correspond to the effects of BRD4, EZH2, SNAI1, and PPRP-γ expressions at cholestatic liver locations, thereby triggering livers injury. Increased miR-29a function hinders BRD4, EZH2, SNAI1, and PPRP-γ expressions, thus inhibiting HSC activation (Fig. 6). This study provides additional evidence that the possibility of finding novel therapeutic agents targeting miR-29a-based BRD4 for the improvement of hepatic fibrosis would play an important role in liver injury diseases. This study highlights that the epigenetic mechanism by miR-29a signaling may modulate their profibrogenic phenotype in the activation of HSCs. Further studies are required in the future to delineate the use of miR-29a agonists as novel therapeutics for treating liver fibrosis.
The proposed miR-29a signaling protection model in liver fibrosis by inhibiting BRD4/EZH2/SNAI1. miR-29a is a vital regulator of the profibrogenic phenotype of HSCs. Increased miR-29a function hinders BRD4, EZH2, SNAI1, and PPRP-γexpressions, thus inhibiting HSC activation.
Acknowledgements
The authors would like to thank Miss Yuan-Ting Chuang and Chia-Ling Wu for assisting with this article.
Funding
This study was supported by grants from the National Health Research Institute (NHRI-EX107-10736SI), Ministry of Science and Technology, Taiwan (106-2314-B-182A-141 -MY3) and Chang Gung Memorial Hospital, Taiwan (CMRPG8G1341, 8G1342 and 8F1562). However, these organizations had no part in the study design, data collection and analysis, publication decisions, or preparation of the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Friedman SL. Evolving challenges in hepatic fibrosis. Nature reviews Gastroenterology & hepatology. 2010;7:425-36
2. Bataller R, Brenner DA. Liver fibrosis. The Journal of clinical investigation. 2005;115:209-18
3. Friedman SL. Mechanisms of hepatic fibrogenesis. Gastroenterology. 2008;134:1655-69
4. Cha W, Fan R, Miao Y, Zhou Y, Qin C, Shan X. et al. MicroRNAs as novel endogenous targets for regulation and therapeutic treatments. Medchemcomm. 2018;9:396-408
5. Mann J, Chu DC, Maxwell A, Oakley F, Zhu NL, Tsukamoto H. et al. MeCP2 controls an epigenetic pathway that promotes myofibroblast transdifferentiation and fibrosis. Gastroenterology. 2010;138:705-14 14 e1-4
6. Roderburg C, Urban GW, Bettermann K, Vucur M, Zimmermann H, Schmidt S. et al. Micro-RNA profiling reveals a role for miR-29 in human and murine liver fibrosis. Hepatology. 2011;53:209-18
7. Sekiya Y, Ogawa T, Yoshizato K, Ikeda K, Kawada N. Suppression of hepatic stellate cell activation by microRNA-29b. Biochemical and biophysical research communications. 2011;412:74-9
8. Bandyopadhyay S, Friedman RC, Marquez RT, Keck K, Kong B, Icardi MS. et al. Hepatitis C virus infection and hepatic stellate cell activation downregulate miR-29: miR-29 overexpression reduces hepatitis C viral abundance in culture. J Infect Dis. 2011;203:1753-62
9. Ghavami S, Hashemi M, Ande SR, Yeganeh B, Xiao W, Eshraghi M. et al. Apoptosis and cancer: mutations within caspase genes. Journal of medical genetics. 2009;46:497-510
10. Li SC, Wang FS, Yang YL, Tiao MM, Chuang JH, Huang YH. Microarray Study of Pathway Analysis Expression Profile Associated with MicroRNA-29a with Regard to Murine Cholestatic Liver Injuries. International journal of molecular sciences. 2016;17:324
11. Tiao MM, Wang FS, Huang LT, Chuang JH, Kuo HC, Yang YL. et al. MicroRNA-29a protects against acute liver injury in a mouse model of obstructive jaundice via inhibition of the extrinsic apoptosis pathway. Apoptosis. 2014;19:30-41
12. Huang YH, Yu-Hsieh H, Huang CC, Shin-Mu VT, Tai MH, Chen CL. et al. Liver hepcidin and stainable iron expression in biliary atresia. Pediatr Res. 2006;59:662-6
13. Huang YH, Shih HH, Tiao MM, Huang CC, Kuo KC, Huang FC. et al. Toll-like receptor 7 agonist induces hypoplasia of the biliary system in a neonatal mouse model. J Microbiol Immunol Infect. 2018;51:166-73
14. Huang YH, Yang YL, Huang FC, Tiao MM, Lin YC, Tsai MH. et al. MicroRNA-29a mitigation of endoplasmic reticulum and autophagy aberrance counteracts in obstructive jaundice-induced fibrosis in mice. Exp Biol Med (Maywood). 2018;243:13-21
15. Huang YH, Yang YL, Wang FS. The Role of miR-29a in the Regulation, Function, and Signaling of Liver Fibrosis. International journal of molecular sciences. 2018:19
16. Vallejo D, Crespo I, San-Miguel B, Alvarez M, Prieto J, Tunon MJ. et al. Autophagic response in the Rabbit Hemorrhagic Disease, an animal model of virally-induced fulminant hepatic failure. Veterinary research. 2014;45:15
17. Sano R, Reed JC. ER stress-induced cell death mechanisms. Biochim Biophys Acta. 2013;1833:3460-70
18. Iredale JP. Models of liver fibrosis: exploring the dynamic nature of inflammation and repair in a solid organ. The Journal of clinical investigation. 2007;117:539-48
19. Zhao H, Wang Z, Tang F, Zhao Y, Feng D, Li Y. et al. Carnosol-mediated Sirtuin 1 activation inhibits Enhancer of Zeste Homolog 2 to attenuate liver fibrosis. Pharmacological research: the official journal of the Italian Pharmacological Society. 2018;128:327-37
20. Li M, Hong W, Hao C, Li L, Wu D, Shen A. et al. SIRT1 antagonizes liver fibrosis by blocking hepatic stellate cell activation in mice. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2018;32:500-11
21. Wu X, Liu D, Tao D, Xiang W, Xiao X, Wang M. et al. BRD4 Regulates EZH2 Transcription through Upregulation of C-MYC and Represents a Novel Therapeutic Target in Bladder Cancer. Mol Cancer Ther. 2016;15:1029-42
22. Cai X, Li Z, Zhang Q, Qu Y, Xu M, Wan X. et al. CXCL6-EGFR-induced Kupffer cells secrete TGF-beta1 promoting hepatic stellate cell activation via the SMAD2/BRD4/C-MYC/EZH2 pathway in liver fibrosis. J Cell Mol Med. 2018;22:5050-61
23. Herranz N, Pasini D, Diaz VM, Franci C, Gutierrez A, Dave N. et al. Polycomb complex 2 is required for E-cadherin repression by the Snail1 transcription factor. Mol Cell Biol. 2008;28:4772-81
24. Friedman SL, Roll FJ, Boyles J, Bissell DM. Hepatic lipocytes: the principal collagen-producing cells of normal rat liver. Proc Natl Acad Sci U S A. 1985;82:8681-5
25. Chou MH, Huang YH, Lin TM, Du YY, Tsai PC, Hsieh CS. et al. Selective activation of Toll-like receptor 7 in activated hepatic stellate cells may modulate their profibrogenic phenotype. Biochem J. 2012;447:25-34
26. Kwiecinski M, Noetel A, Elfimova N, Trebicka J, Schievenbusch S, Strack I. et al. Hepatocyte growth factor (HGF) inhibits collagen I and IV synthesis in hepatic stellate cells by miRNA-29 induction. PLoS One. 2011;6:e24568
27. Chung YH, Huang YH, Chu TH, Chen CL, Lin PR, Huang SC. et al. BMP-2 restoration aids in recovery from liver fibrosis by attenuating TGF-beta1 signaling. Lab Invest. 2018
28. Wang H, Bloom O, Zhang M, Vishnubhakat JM, Ombrellino M, Che J. et al. HMG-1 as a late mediator of endotoxin lethality in mice. Science. 1999;285:248-51
29. Bao Y, Wu X, Chen J, Hu X, Zeng F, Cheng J. et al. Brd4 modulates the innate immune response through Mnk2-eIF4E pathway-dependent translational control of IkappaBalpha. Proc Natl Acad Sci U S A. 2017;114:E3993-E4001
30. Lin YC, Wang FS, Yang YL, Chuang YT, Huang YH. MicroRNA-29a mitigation of toll-like receptor 2 and 4 signaling and alleviation of obstructive jaundice-induced fibrosis in mice. Biochem Biophys Res Commun. 2018;496:880-6
31. Sun Y, Huang J, Song K. BET protein inhibition mitigates acute myocardial infarction damage in rats via the TLR4/TRAF6/NF-kappaB pathway. Exp Ther Med. 2015;10:2319-24
32. Ding N, Hah N, Yu RT, Sherman MH, Benner C, Leblanc M. et al. BRD4 is a novel therapeutic target for liver fibrosis. Proc Natl Acad Sci U S A. 2015;112:15713-8
33. Liu CA, Chang CY, Hsueh KW, Su HL, Chiou TW, Lin SZ. et al. Migration/Invasion of Malignant Gliomas and Implications for Therapeutic Treatment. International journal of molecular sciences. 2018:19
34. Zhang Q, Dong P, Liu X, Sakuragi N, Guo SW. Enhancer of Zeste homolog 2 (EZH2) induces epithelial-mesenchymal transition in endometriosis. Scientific reports. 2017;7:6804
35. Zhang J, Tian XJ, Xing J. Signal Transduction Pathways of EMT Induced by TGF-beta, SHH, and WNT and Their Crosstalks. J Clin Med. 2016:5
36. Scarpa M, Grillo AR, Brun P, Macchi V, Stefani A, Signori S. et al. Snail1 transcription factor is a critical mediator of hepatic stellate cell activation following hepatic injury. Am J Physiol Gastrointest Liver Physiol. 2011;300:G316-26
37. Oh JY, Choi GE, Lee HJ, Jung YH, Ko SH, Chae CW. et al. High Glucose-Induced Reactive Oxygen Species Stimulates Human Mesenchymal Stem Cell Migration Through Snail and EZH2-Dependent E-Cadherin Repression. Cellular physiology and biochemistry: international journal of experimental cellular physiology, biochemistry, and pharmacology. 2018;46:1749-67
38. Yang MD, Chiang YM, Higashiyama R, Asahina K, Mann DA, Mann J. et al. Rosmarinic acid and baicalin epigenetically derepress peroxisomal proliferator-activated receptor gamma in hepatic stellate cells for their antifibrotic effect. Hepatology. 2012;55:1271-81
Author contact
![]() Corresponding authors: Ya-Ling Yang, M.D., Department of Anesthesiology, Kaohsiung Chang Gung Memorial Hospital and Chang Gung University College of Medicine, Kaohsiung, Taiwan, 833, 123 Ta-Pei Road, Niao-Sung District, Kaohsiung, Taiwan 833, R.O.C. E-mail: yaling453com.tw. Feng-Sheng Wang, PhD, Department of Medical Research, Kaohsiung Chang Gung Memorial Hospital, 123 Ta-Pei Road, Niao-Sung District, Kaohsiung, Taiwan 833, R.O.C. Tel: +886-7-7317123 ext. 8795; Fax: +886-7-7338009; E-mail: wangfshinet.net
Corresponding authors: Ya-Ling Yang, M.D., Department of Anesthesiology, Kaohsiung Chang Gung Memorial Hospital and Chang Gung University College of Medicine, Kaohsiung, Taiwan, 833, 123 Ta-Pei Road, Niao-Sung District, Kaohsiung, Taiwan 833, R.O.C. E-mail: yaling453com.tw. Feng-Sheng Wang, PhD, Department of Medical Research, Kaohsiung Chang Gung Memorial Hospital, 123 Ta-Pei Road, Niao-Sung District, Kaohsiung, Taiwan 833, R.O.C. Tel: +886-7-7317123 ext. 8795; Fax: +886-7-7338009; E-mail: wangfshinet.net