Impact Factor ISSN: 1449-1907

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2018; 15(8):832-839. doi:10.7150/ijms.23270 This issue Cite
Research Paper
Proscillaridin A induces apoptosis, inhibits STAT3 activation and augments doxorubicin toxicity in prostate cancer cells
Yangyang He1, Muhammad Khan2, Jingbo Yang3, Min Yao1, Shili Yu1, Hongwen Gao1 ![]()
1. Department of Pathology, the Second Hospital of Jilin University, Changchun 130041, P.R. China.
2. Department of Zoology, University of the Punjab, Quaid-e-Azam Campus Lahore 54590, Pakistan.
3. Jilin Provincial Key Laboratory on Molecular and Chemical Genetics, the Second Hospital of Jilin University, Changchun 130041, P.R. China.
Received 2017-10-11; Accepted 2018-4-12; Published 2018-5-22
Abstract
Cardiac glycosides are natural compounds used for the treatment of congestive heart failure and cardiac arrhythmias. Recently, they have been reported to exhibit anticancer activity. Proscillaridin A (PSN-A), a cardiac glycoside constituent of Urginea maritima has been shown to exhibit anticancer activity. However, the cellular targets and anticancer mechanism of PSN-A in various cancers including prostate cancer remain largely unexplored. In the present study, we have shown that PSN-A inhibits proliferation and induces apoptosis in prostate cancer cells in a dose-dependent manner. Further mechanistic study have shown that anticancer activity of PSN-A in prostate cancer cells is associated with ROS generation, Bcl-2 family proteins modulation, mitochondrial membrane potential disruption and ultimately activation of caspase-3 and cleavage of PARP. Moreover, we found that PSN-A inhibits JAK2/STAT3 signaling and augments doxorubicin toxicity in prostate cancer cells. Of note, LNCaP cells were found to be more sensitive to PSN-A treatment as compared to DU145 cells. Taken together, the data provided first evidence of the anticancer activity and possible molecular mechanism of PSN-A in prostate cancer. Further study is needed to develop PSN-A into a potential lead compound for the treatment of prostate cancer.
Keywords: Cardiac glycosides, Proscillaridin A, Bcl-2, STAT3, Prostate cancer
Introduction
Prostate cancer is one of the most commonly diagnosed malignancies and 2nd leading cause of death in men worldwide [1]. At present, surgery, radiotherapy, androgen deprivation therapy (ADT) and chemotherapy are the available treatment options for prostate cancer [1]. ADT is still the first line treatment; however, most patients develop highly aggressive castration-resistant prostate cancer 14-20 months post-ADT therapy for which the median survival rate is less than 19 months [2, 3]. Thus, chemotherapy remains the only choice for advanced stage castration-resistant prostate cancer. Although, chemotherapy with taxane improved the survival rate of patients with metastatic castration-resistant prostate cancer (MCRPC), the prognosis of MCRPC still remains very poor [4, 5]. Therefore, exploring novel bioactive molecules and their anticancer mechanism is urgently needed for improving the outcomes of prostate cancer treatment.
Cancer chemoprevention and treatment by natural compounds has gained increasing attention because it is believed to be safe, cheap and alternative form of modern healthcare system. Natural products have significantly contributed in discovery of anticancer drugs. At present, more than 60% FDA approved anticancer drugs have been derived from natural products including medicinal plants. Plants secondary metabolites such as vinca alkaloids and terpenes have been extensively studied for their potential use in the treatment of various cancers [6, 7]. Cardiac glycosides are plants' secondary metabolites which have long been used to treat cardiac congestions and cardiac arrhythmias. Although originally prescribed to treat cardiac failure, more recently they have been rediscovered for their potential anticancer activity. Cardiac glycosides have been reported to exhibit anticancer activity at non-toxic concentration in various in vitro and in vivo cancer models through multiple mechanisms including inhibition of cell proliferation, induction of apoptosis and augmentation of chemotherapy [8, 9]. In the present study, we have shown for the first time that PSN-A, a cardiac glycoside component of Urginea maritima [10] inhibits growth and induces mitochondrial apoptosis in prostate cancer cells. Moreover, we found that PSN-A inhibits JAK2/STAT3 signaling and enhances anticancer activity of doxorubicin in prostate cancer cells for the first time.
Materials and Methods
Antibodies and reagents
PSN-A (Figure 1A) was purchased from EXTRASYNTHESE (Genay, France). Dulbecco's Modified Eagle's Medium (DMEM) and fatal bovine serum (FBS) were purchased from Gibco (Eggenstein, Germany) while Penicillin/ Streptomycin antibiotics was obtained from Solarbio co., Ltd. (Beijing, China). Annexin V-FITC/PI double staining apoptosis detection kit, Reactive Oxygen Species (ROS) assay kit, mitochondrial membrane potential (MMP) assay kit, Crystal violet, 3-(4,5-Dimethylthiazol-2-yl)-2,5-Diphenyltetrazolium Bromide (MTT), dimethyl sulfoxide (DMSO), Fluo-3AM and doxorubicin were purchased from Beyotime Institute of Biotechnology (Haimen, Jiangsu, China). The primary antibodies for cleaved caspases-3, cleaved PARP, p-STAT3 (Tyr705), STAT3 and SHP-2 were purchased from Cell Signaling Technology (Beverly, MA) while primary antibodies for Bax, Bcl-2, SHP-2, PTEN and β-actin were purchased from Proteintech (Wuhan, China). The primary antibodies for p-JAK2 and JAK2 were purchased from abcam (Cambridge, MA). Horseradish peroxidase (HRP)-conjugated secondary antibodies (goat anti-rabbit, goat anti-mouse) were obtained from Sigma.
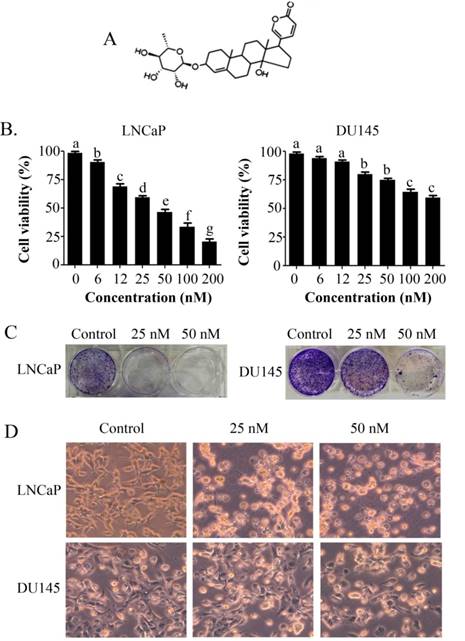
PSN-A inhibits cell proliferation and induces toxicity in prostate cancer cells. (A) Chemical structure of PSN-A. (B) LNCaP and DU145 prostate cancer cells were treated with different concentrations of PSN-A for 24 h as indicated and cell proliferation was determined by MTT assay. Data are expressed as Mean±SEM (n=3). Columns withdifferent superscript letter differ significantly (P<0.05). (C) LNCaP and DU145 cells were treated with 0, 25 and 50nM PSN-A for 24. The cells were harvested and 300 cells were seeded into 6 well plates and allowed to grow for one week to make colonies. The colonies were fixed with 4% PFA, stained with crystal violet and photographed. (D) LNCaP and DU145 cells were treated with 0, 25 and 50 nM PSN-A for 24 h in 96 well plates and cell morphology was observed under phase contrast microscope. Scale bar 100µm.
Cell culture and treatments
Human LNCaP and DU145 prostate cancer cell lines were purchased from American Type Culture Collection (Manassas VA) and were maintained in DMEM supplemented with 10% FBS, 100 units/mL penicillin and 100 μg/mL streptomycin at 37°C with 5% CO2 in humidified atmosphere. PSN-A was dissolved in DMSO to make 1mM stock solution and kept in the dark protected from light. Serial dilution of stock solution were prepared and cells were treated with PSN-A with final DMSO concentration of 0.5%. Control cells were treated with 0.5% DMSO.
Determination of cell viability
Cell viability was evaluated by MTT assay as described by us previously [11]. LNCaP and DU145 cells were cultured in 96 well plates in triplicates overnight at 37°C. The cells were incubated with different concentrations of PSN-A for 24 h with or without 10 µM BAPTA-AM. Following treatments, cells were incubated with 10 µL MTT reagent (5mg/mL) in the dark at 37°C for 4 h. Finally, medium was carefully removed and 150µL DMSO was added into each well to dissolve farmazan crystals. The plates were shaked gently to mix the color. The absorbance was then measured at 570 nm by a microplate reader (Synergy neo HTS multimode microplate reader, BioTek). The percentage of cell viability was calculated using following formula.
Cell viability (%) = (A570sample - A570blank)/(A570control - A570blank) × 100
Microscopic analysis of cell morphological changes
LNCap and DU145 cells were treated with 0, 25 and 50 nM PSN-A in 96 well culture plates in triplicates for 24 h. Aftre PSN-A treatment, the cells were observed for morphological changes and photographed by a phase contrast microscope (Leica, DMIL LED).
Clonogenic assay
LNCap and DU145 cells were cultured in 6 well culture plates and treated with 0, 25 and 50nM PSN-A for 24 h. The cells were harvested and seeded into 6 well cell culture plates (500 cells/well). The cells were incubated in CO2 incubator for 10 days for colony formation. The medium was removed and colonies were washed and fixed with 4% paraformaldehyde (PFA) for 15 min. After fixation, the colonies were stained with crystal violet solution and photographed.
Apoptosis assay
Apoptosis assay was performed using Annexin V-FITC/PI double staining kit. Briefly, LNCaP and DU145 cells were cultured in 6 well plates overnight. The cells were then treated with 0, 25 and 50 nM PSN-A for 24 h. Following treatments, floating and adherent cells were collected, and washed with PBS. The cell pallets were resuspended in 0.500 mL binding buffer. Finally, 5 μL annexin V-FITC and 10 μL PI was added into cell suspension and samples were incubated in the dark for 15 min according to kit instructions. After filtration, the cells samples were analyzed by flow cytometry (Beckman Coulter, Epics XL) for the evaluation of apoptosis.
Determination of ROS generation
The intracellular Reactive oxygen species (ROS) generation was determined by ROS assay kit as per manufacturer's instructions. Briefly, LNCaP and DU145 cells were cultured in 6 well plates overnight. The cells were treated with 0, 25 and 50 nM PSN-A for 24 h. Following treatments, the cells were incubated with DCFH-DA for 30 min at 37°C. Finally, the cells were collected by centrifugation and cell pallets were gently washed with PBS. After washing, the cell pellets were gently resuspended in PBS and filtered. The filtrates were analyzed by flow cytometry for (DCF) fluorescence.
Determination of mitochondrial membrane potential (MMP)
MMP was measured using rhodamine 123 as described previously [12]. Briefly, LNCaP and DU145 cells were cultured in 6 well plates overnight. The cells were treated with 0, 25 and 50 nM PSN-A for 24 h. After drug treatments, cells were harvested and washed with PBS. The cell pellets were resuspended in PBS solution and 10 µg rhodamine 123 was added to each sample. The samples were incubated in the dark for 30 min. Following incubation, the cell pellets were collected by centrifugation at 1000 rpm, washed with PBS once and then resuspended in 500 µL PBS. After filteration, the samples were analyzed by flow cytometry.
Western blotting
LNCaP and DU145 cells were treated with 0, 25 and 50 nM PSN-A for 24 h in 6 well plates. Following drug treatment, cells were collected, washed with cold PBS and proteins were extracted as described previously [13]. The protein concentration was determined by BCA protein assay kit (Beyotime) was used to determine protein concentration. A total of 30 μg protein was resolved on 10-12% sodium dodecylsulfate polyacrylamide gel electrophoresis (SDS-PAGE), and transferred to polyvinylidene difluoride (PVDF) membrane. The membrane were blocked with 5% skim milk at room temperature and incubated with Bax (1:500), Bcl-2 (1:500), cleaved caspase-3 (1:1000), PARP (1:1000), β-actin(1:1000) SHP-1(1:500), SHP-2 (1:500), PTEN (1:500), p-Jak2 (1:1000), Jak2 (1:1000), p-STAT3 (1:1000), and STAT3 (1:1000) antibodies overnight at 4°C. After washing with Tris-buffered saline-Tween (TBST) solution three times, the membranes were incubated with HRP-conjugated goat anti-rabbit IgG (1:5000) or goat anti-mouse IgG (1:5000) secondary antibodies with gentle shaking for 1 h at room temperature. Finally, the membrane were washed with TBST and Western blot bands were detected by Immobilon Western chemiluminescent HRP substrate (Millipore, Billerica, MA) using MicroChemi 4.2 imaging system (DNR Bio-Imaging system).
Measurement of intracellular free Ca++
The intracellular free Ca++ was determined by fluorescent probe Fluo-3AM. Briefly, LNCaP and DU145 cells were treated with indicated concentrations of PSN-A for 24 h. The cells were harvested, washed with PBS and resuspended in serum free medium. The samples were incubated with 5µM Fluo-3AM in the dark for 30 min. Finally, the cells were washed with PBS twice, and analyzed using a multimode microplate reader at 488nm excitation and 525 nm emission wavelengths.
Statistical analysis
The data are expressed as mean ± standard error mean (SEM) of 3 different experiments. One way ANOVA was used to compare means followed by Tukey's Multiple Comparison Test. P<0.05 was considered statistically significant.
Results
PSN-A inhibits proliferation and induces apoptosis in prostate cancer cells
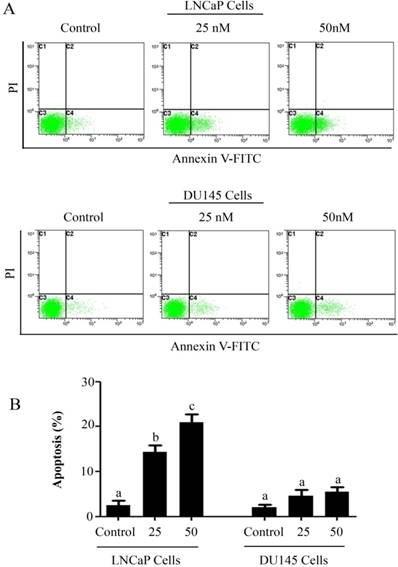
The anti-proliferative and apoptotic effect of PSN-A in prostate cancer was evaluated using LNCaP (androgen-dependent) and DU145 (androgen-independent) cell lines. Treatment of PSN-A for 24 h inhibited the proliferation of cells in a dose-dependent manner as evident from the results of MTT and colony forming assays (Figure 1B & C). However, anti-proliferative effect of PSN-A was remarkably higher in LNCaP cells compared to DU145 cells. We further examined the effect of PSN-A on cell morphology. PSN-A induced severe morphological changes characteristically associated with cell death in LNCaP cells in a dose-dependent manner, however; DU145 cells were found to be relatively resistant to PSN-A treatment (Figure 1D). In order to ascertain the nature of cell death, we performed cell apoptosis assay using Annexin V-FITC/PI double staining kit and flow cytometry. The data showed that PSN-A induced apoptosis in LNCaP cells in a dose-dependent manner while DU145 cells were found insensitive to PSN-A treatment as shown in figure 2.
PSN-A induces apoptosis in prostate cancer cells. (A) LNCaP and DU145 cells were treated with 0, 25 and 50 nM PSN-A for 24 h. Cell samples were prepared as described in methodology. The samples were analyzed by flow cytometry for the detection of apoptosis. (B) Statistical analysis of data from A. Columns with different superscript letter within the same group differ significantly (P<0.05).
PSN-A induces ROS generation and disrupts mitochondrial membrane potential in prostate cancer cells
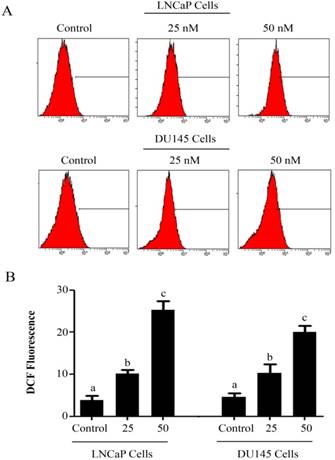
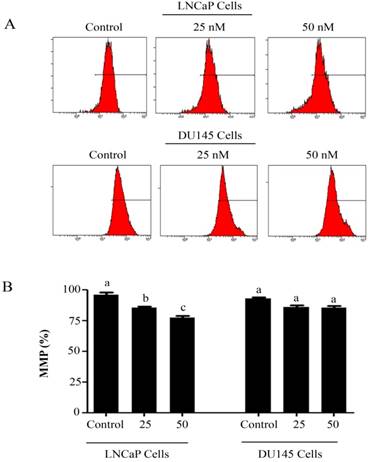
Intracellular Reactive Oxygen Species (ROS) generation and alterations in mitochondrial membrane potential (MMP) was evaluated by staining the cells with 2',7'-dichlorodihydrofluorescein diacetate (DCFH-DA) and rhodamine 123, respectively. As shown in figure 3, PSN-A treatment increased the level of ROS generation in both cell lines in a dose-dependent manner however, higher level of ROS was observed in LNCaP cells compared to DU145 cells. Next, we determined the effect of PSN-A treatment on MMP. The data demonstrated that PSN-A treatment dissipated MMP significantly in LNCaP cells in a dose-dependent manner (Figure 4). Although PSN-A disrupted MMP in DU145 cells, however, this effect was not significant (P<0.05) as shown in figure 3.
PSN-A induces ROS generation in prostate cancer cells. (A) LNCaP and DU145 cells were treated with 0, 25 and 50nM PSN-A for 24 h and ROS generation was measured by staining the cells with DCFH-DA according the instructions of kit. (B) Data are expressed as Mean±SEM (n=3). Columns with different superscript letter differ significantly (P<0.05)
PSN-A decreases mitochondrial membrane potential (MMP) in prostate cancer cells. (A) LNCaP and DU145 cells were treated with 0, 25 and 50 nM PSN-A for 24 h and MMP was checked by staining the samples with JC-1 as per kit's instructions. (B) Statistical analysis of data from figure A. Data are expressed as Mean±SEM (n=3). Columns with different superscript letter differ significantly (P<0.05).
PSN-A decreases Bcl-2/Bax ratio and induces caspase-3 and PARP cleavage
As PSN-A treatment disrupted MMP, therefore, we measured the expressions of Bcl-2 family proteins by immunobloting. The data demonstrated that PSN-A treatment inhibited the expression of anti-apoptotic Bcl-2 protein while increased the expression of pro-apoptotic Bax protein in a dose-dependent manner in LNCaP cells. This modulatory effect of PSN-A on Bcl-2 family proteins expression was found to be negligible in case of DU145 cells (Figure 5A). Flow cytometry analysis showed that PSN-A induces apoptotic cell death, therefore, we measured the expression of cleaved caspase-3 and PARP-1. The data demonstrated that PSN-A treatment increased the expressions of cleaved caspase-3 and cleaved PARP-1 in LNCaP cells dose-dependently (Figure 5A). Collective data indicated that PSN-A-induced-apoptosis is associated with mitochondrial dysfunction as evident from MMP dissipation and Bcl-2 family proteins modulation.
PSN-A modulates Bcl-2 family proteins and inhibits STAT3 signaling. (A) LNCaP and DU145 cells were treated with 0, 25 and 50 nM PSN-A for 24 h. Total proteins were extracted and subjected to immunobloting for the expressions of Bax, Bcl-2, cleaved caspases-3 and PARP-1. β-actin was used as loading control. (B) LNCaP and DU145 cells were treated with 0, 25 and 50 nM PSN-A for 24 h. Total cell lysates were extracted and subjected to Western blot for the expressions of SHP-1, SHP-2, PTEN, p-Jak2, Jak2, p-STAT3 and STAT3. β-actin was used as loading control.
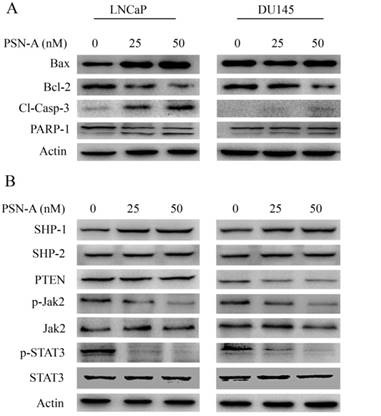
PSN-A inhibits JAK/STAT3 signaling in prostate cancer cells
Janus Kinase (JAK)-Signal transducer and activator of transcription 3 (STAT3) pathway is aberrantly activated in various cancers including prostate cancer and plays an important role in tumor development and progression [14]. STAT3 is activated by phosphorylation at Tyr705 by various up-stream regulators including Kinases (positive regulators) and protein tyrosine phosphatases (negative regulators) [13]. Therefore, we measured the effect of PSN-A treatment on STAT3 signaling. The data indicated that PSN-A effectively inhibited the phosphorylation of STAT3 at Tyr705 in a dose-dependent manner in both LNCaP and DU145 cell lines. Next, we determined the effect of PSN-A on STAT3 up-stream protein tyrosine phosphatases. As shown in figure 5B, PSN-A slightly increased the expression of SHP-1 while SHP-2 and PTEN remains unaffected. Next, we measured the expression and phosphorylation of JAK2. We found that PSN-A inhibited the phosphorylation of JAK2 in both cell lines in a dose-dependent manner (Figure 5B).
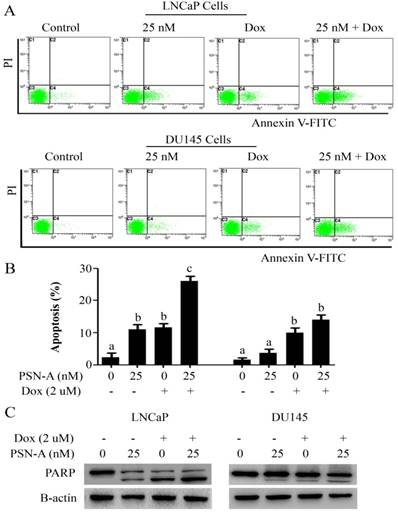
PSN-A augments doxorubicin toxicity in prostate cancer cells
Doxorubicin is one of the most important chemotherapeutic drugs used to treat various cancers including prostate cancer [15]. Enhancing the efficacy of existing clinical drugs is a rational approach to reduce the burden of cancer. We were interested to know if PSN-A could potentiate the anticancer activity of doxorubicin in prostate cancer cells. Therefore, we treated the cells either with PSN-A and doxorubicin as single agent or in combination of both and determined the effect on apoptosis. The data showed that PSN-A remarkably enhanced the apoptotic effect of doxorubicin in LNCaP cells compared to DU145 cells as evident from flow cytometry analysis of apoptosis and PARP-1 cleavage (Figure 6). Since doxorubicin has been reported to induce STAT3 activation in cancer cells [13], we were interested to know if PSN-A could inhibit doxorubicin-induced STAT3 activation in prostate cancer cells. For this, we incubated the cells with 2µM doxorubicin for 72 h followed by PSN-A treatment (25nM) for 24 h. The phosphorylation and expression of STAT3 was determined by Western blot. The data showed that doxorubicin increased the phosphorylation of STAT3 at Tyr705 while PSN-A effectively suppressed doxorubicin-induced STAT3 phosphorylation in prostate cancer cells as shown in Figure 7A.
PSN-A augments doxorubicin toxicity in prostate cancer cells. (A) LNCaP and DU145 cells were treated with PSN-A (25nM) and doxorubicin (2µM) either as a single agent or in combination for 24 h as indicated and apoptotic cell death was determined by flow cytometry. (B) Statistical analysis from A. Columns with different superscript letters differ significantly (P<0.05). (C) LNCaP and DU145 cells were treated with PSN-A and doxorubicin as single agent or in combination for 24 h as indicated. Total cell lysates were extracted and subjected to Western blot for the expression of cleaved PARP.
PSN-A inhibits doxorubicin-induced STAT3 activation and increases intracellular Ca++ level in prostate cancer cells. (A) LNCaP and DU145 cells were treated with 2µM doxorubicin for 72 h followed by PSN-A treatment (25nM) for another 24 h. The total cell lysates were extracted and expression of p-STAT3 and STAT3 were measured by Western blot. (B) Cell were treated with indicated concentrations of PSN-A for 24 h and intracellular free Ca++ was measured using FLUo-3AM. (C) Cells were treated with 50nM PSN-A in the presence or absence of BAPTA-AM (10 µM) for 24 h and cell viability was determined by MTT assay. Data (B and C) are expressed as Mean±SEM (n=3). Columns with different superscript letter differ significantly (P<0.05).
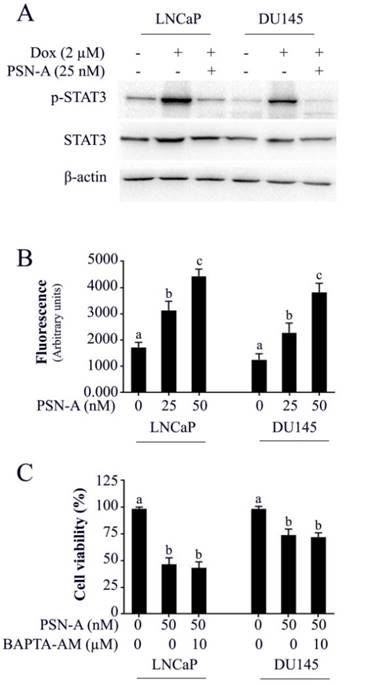
PSN-A increases intracellular free Ca++ in prostate cancer cells
The effect of PSN-A on intracellular free Ca++ was determined by Fluo-3AM. The data demonstrated that PSN-A increased intracellular Ca++ in both LNCaP and DU145 cells in a dose-dependent manner as shown in figure 7B. Next, we asked the question, if anticancer activity of PSN-A in prostate cancer cells is associated with increased level of intracellular Ca++. To answer this question, we measured the effect of PSN-A on cell viability in the presence/or absence of BAPTA-AM (10µM), a Ca++ chelator. We found that BAPTA-AM failed to protect the cells from toxic effect of PSN-A in prostate cancer cells indicating that increase in intracellular Ca++ is not associated with anticancer activity of PSN-A in our study model.
Discussion
Although cardiac glycosides have long been used to treat cardiac arrest in clinic, the anticancer activity of cardiac glycoside is rather novel. In the present study, we have investigated the anticancer activity of a cardiac glycoside, PSN-A in prostate cancer cells. PSN-A has been found to exert both anti-proliferative and cytotoxic effects in prostate cancer cells at extremely low concentrations (25-50 nM). The major mechanisms identified for the anticancer activity of PSN-A in prostate cancer cells in the present study are; growth inhibition, inhibition of STAT3 signaling, mitochondrial dysfunction, Bcl-2 family proteins modulation and caspase-3 and PARP cleavage which ultimately lead to apoptotic cell death.
Cardiac glycosides can be broadly categorized into two main groups; cardenolides and bufadienolides. Cardenolides have been extensively studied for their potential use in the treatment of cancer. Several cardenolides such as digitoxin and degoxin are now in cancer clinical trials [16]. In contrast to cardenolides, bufadienolides have not been well studied for their anticancer activity. Here in this study, we investigated the anticancer activity of a bufadienolide cardiac glycoside, PSN-A in prostate cancer cells using anderogen-dependent (LNCaP) and antdrogen-independent (DU145) cells. PSN-A effectively inhibited proliferation and induced apoptosis in LNCaP cells while DU145 cells exhibited much more resistance against PSN-A toxicity as evident from the results of MTT assay, colony forming assay and flow cytometry analysis of apoptosis. The data indicate that PSN-A-mediated anticancer activity might be associated with androgen sensitivity of prostate cancer cells. These finding are further supported by a previous study of Tian et al., who found that bufadienolide derivatives exhibit strong growth inhibitory effect in androgen-dependent prostate cancer cells as compared to androgen-independent prostate cancer cells [17].
It is widely accepted now that mitochondria play a vital role in proliferation and apoptosis of a cell. In contrast to death receptor pathway, mitochondrial apoptosis pathway is initiated by various non-receptor mediated stimuli. Bcl-2 family proteins are considered the main players of mitochondrial apoptosis. Bcl-2 family proteins contains both anti-apoptotic as well as pro-apoptotic proteins. In normal proliferating cells, bax, a pro-apoptotic protein resides in the cytosol, and is negatively regulated by bcl-2 an anti-apoptotic protein. Down-regulation of bcl-2 protein results in translocation of bax into mitochondrial membrane. Insertion of bax into mitochondrial membrane results in the opening of the mitochondrial permeability transition (MPT) pore and subsequent release of cytochrome c and other pro-apoptotic proteins into cytosol which initiates apoptotic cell death by activating caspase-cascade. Alterations in Bcl-2 family proteins expression, ROS generation and mitochondrial membrane potential disruption are the main features of intrinsic or mitochondrial apoptosis [6, 7, 18]. Here we found that PSN-A increased ROS generation and intracellular free Ca++, decreased bcl-2/bax ratio and decreased mitochondrial membrane potential in prostate cancer cells validating the involvement of mitochondrial apoptosis. We also found increased expressions of cleaved caspase-3 and PARP-1 in PSN-A treated prostate cancer cells which are the hallmarks of apoptotic cell death. Induction of mitochondrial apoptosis by PSN-A is in line with previous reports indicating that cardiac glycosides could effectively initiate mitochondrial apoptosis by modulating bcl-2 family protein in prostate cancer cells [19, 20, 21]. Although, PSN-A increased intracellular free Ca++ in prostate cancer cells, however, this Ca++ release has not been found to be associated with anticancer activity of PSN-A. The findings are in line with our previous report that PSN-A mediated increase in intracellular free Ca++ is not responsible for its anticancer activity in lung cancer cells [22].
STAT3 is constitutively activated in various human cancers including prostate cancer and has been implicated in cancer progression and drug resistance [14, 23, 24]. In the present study, we demonstrated for the first time that PSN-A inhibits STAT3 activation both in androgen-dependent and androgen-independent prostate cancer cells. STAT3 is activated by various up-stream positive regulators such as tyrosine kinases and negative regulators such as protein tyrosine phosphatases (PTPs). SHP-1, SHP-2 and PTEN are major PTPs that negatively regulate STAT3 activation [13]. PSN-A slightly increased the expression of SHP-1 while the expression of SHP-2 and PTEN remains unchanged. Among tyrosine kinases, JAK-2 is the most studied non-receptor tyrosin kinase which induces STAT3 activation [13]. Here in this study, PSN-A inhibited the phosphorylation of JAK2. Taken together, the data demonstrate that PSN-A inhibits STAT3 activation, at least, in part by inhibiting JAK-2 phosphorylation. We and others have previously reported that natural compounds with inhibitory effect on STAT3 could enhance the anticancer activity of doxorubicin in A549 lung adenocarcinoma and MDA-MB-231 breast carcinoma [13, 23]. Since doxorubicin is being used clinically in the treatment of prostate cancer, we tested the efficacy of doxorubicin in combination with PSN-A in prostate cancer cells. In line with previous findings, PSN-A enhanced the cytotoxicity of doxorubicin in prostate cancer cells.
Conclusion
In conclusion, PSN-A inhibited growth, induced mitochondrial apoptosis by altering bcl-2/bax ratio, disrupting mitochondrial membrane potential and ultimately activation of caspase-3 and cleavage of PARP-1 in a dose-dependent manner at extremely low concentrations. Moreover, PSN-A suppressed STAT3 activation at least in part, by inhibiting JAK2 phosphorylation and enhanced anticancer activity of doxorubicin in prostate cancer cells. LNCaP cells were found to be more sensitive to PSN-A treatment while DU145 cells exhibited much more resistance against PSN-A treatment. This differential effect of PSN-A might be associated with androgen sensitivity. Further study is needed to find out the exact molecular mechanism of this differential anticancer activity of PSN-A in androgen-dependent and androgen-independent prostate cancer cells.
Acknowledgements
This study was supported in part by a grant from the Science and Technology Development Project of Jilin Province (#3D515U393429) and Science and Technology of Jilin Province key laboratory (20170622006JC).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Yang JB, Khan M, He YY, Yao M, Li YM, Gao HW, Ma TH. Tubeimoside-1 induces oxidative stress-mediated apoptosis and G0/G1 phase arrest in human prostate carcinoma cells in vitro. Acta Pharmacol Sin. 2016;37:950-62
2. Ingersoll MA, Miller DR, Martinez O, Wakefield CB, Hsieh KC, Simha MV, Kao CL, Chen HT, Batra SK, Lin MF. Statin derivatives as therapeutic agents for castration-resistant prostate cancer. Cancer Lett. 2016;383:94-105
3. Wong KY, Liu J, Chan KW. KIF7 attenuates prostate tumor growth through LKB1-mediated AKT inhibition. Oncotarget. 2017;8:54558-54571
4. Schutz FA, Buzaid AC, Sartor O. Taxanes in the management of metastatic castration-resistant prostate cancer: efficacy and management of toxicity. Crit Rev Oncol Hematol. 2014;91:248-56
5. Tsao CK, Cutting E, Martin J, Oh WK. The role of cabazitaxel in the treatment of metastatic castration-resistant prostate cancer. Ther Adv Urol. 2014;6:97-104
6. Khan M, Maryam A, Zhang H, Mehmood T, Ma T. Killing cancer with platycodin D through multiple mechanisms. J Cell Mol Med. 2016;20:389-402
7. Khan M, Maryam A, Qazi JI, Ma T. Targeting Apoptosis and Multiple Signaling Pathways with Icariside II in Cancer Cells. Int J Biol Sci. 2015;11:1100-12
8. Cerella C1, Dicato M, Diederich M. Assembling the puzzle of anti-cancer mechanisms triggered by cardiac glycosides. Mitochondrion. 2013;13:225-34
9. Slingerland M, Cerella C, Guchelaar HJ, Diederich M, Gelderblom H. Cardiac glycosides in cancer therapy: from preclinical investigations towards clinical trials. Invest New Drugs. 2013;31:1087-94
10. El-Seedi HR, Burman R, Mansour A, Turki Z, Boulos L, Gullbo J, Göransson U. The traditional medical uses and cytotoxic activities of sixty-one Egyptian plants: discovery of an active cardiac glycoside from Urginea maritime. J Ethnopharmacol. 2013;145:746-57
11. Khan M, Ding C, Rasul A, Yi F, Li T, Gao H, Gao R, Zhong L, Zhang K, Fang X, Ma T. Isoalantolactone induces reactive oxygen species mediated apoptosis in pancreatic carcinoma PANC-1 cells. Int J Biol Sci. 2012;8:533-47
12. Khan M, Li T, Ahmad Khan MK, Rasul A, Nawaz F, Sun M, Zheng Y, Ma T. Alantolactone induces apoptosis in HepG2 cells through GSH depletion, inhibition of STAT3 activation, and mitochondrial dysfunction. Biomed Res Int. 2013;2013:719858
13. Maryam A, Mehmood T, Zhang H, Li Y, Khan M, Ma T. Alantolactone induces apoptosis, promotes STAT3 glutathionylation and enhances chemosensitivity of A549 lung adenocarcinoma cells to doxorubicin via oxidative stress. Sci Rep. 2017;7:6242
14. Wu Z, Huang W, Chen B, Bai PD, Wang XG, Xing JC. Up-regulation of miR-124 inhibits invasion and proliferation of prostate cancer cells through mediating JAK-STAT3 signaling pathway. Eur Rev Med Pharmacol Sci. 2017;21:2338-2345
15. Klippstein R, Bansal SS, Al-Jamal KT. Doxorubicin enhances curcumin's cytotoxicity in human prostate cancer cells in vitro by enhancing its cellular uptake. Int J Pharm. 2016;514:169-175
16. Calderón-Montaño JM, Burgos-Morón E, Orta ML, Maldonado-Navas D, García-Domínguez I, López-Lázaro M. Evaluating the cancer therapeutic potential of cardiac glycosides. Biomed Res Int. 2014
17. Tian HY, Yuan XF, Jin L, Li J, Luo C, Ye WC, Jiang RW. A bufadienolide derived androgen receptor antagonist with inhibitory activities against prostate cancer cells. Chem Biol Interact. 2014;207:16-22
18. Khan M, Yi F, Rasul A, Li T, Wang N, Gao H, Gao R, Ma T. Alantolactone induces apoptosis in glioblastoma cells via GSH depletion, ROS generation, and mitochondrial dysfunction. IUBMB Life. 2012;64:783-94
19. Pan L, Zhang Y, Zhao W, Zhou X, Wang C, Deng F. The cardiac glycoside oleandrin induces apoptosis in human colon cancer cells via the mitochondrial pathway. Cancer Chemother Pharmacol. 2017;80:91-100
20. Lee CH, Shih YL, Lee MH, Au MK, Chen YL, Lu HF, Chung JG. Bufalin Induces Apoptosis of Human Osteosarcoma U-2 OS Cells through Endoplasmic Reticulum Stress, Caspase- and Mitochondria-Dependent Signaling Pathways. Molecules. 2017 doi: 10.3390/molecules22030437
21. Chen Y, Li M, Li Z, Gao P, Zhou X, Zhang J. Bufalin induces apoptosis in the U2OS human osteosarcoma cell line via triggering the mitochondrial pathway. Mol Med Rep. 2016;13:817-22
22. Maryam A, Mehmood T, Yan Q, Li Y, Khan M, Ma T. Proscillaridin A promotes oxidative stress and ER stress, inhibits STAT3 activation, and induces apoptosis in A549 lung adenocarcinoma cells. Oxid Med Cell Longev. 2018;2018:3853409
23. Chun J, Li RJ, Cheng MS, Kim YS. Alantolactone selectively suppresses STAT3 activation and exhibits potent anticancer activity in MDA-MB-231 cells. Cancer Lett. 2015;357:393-403
24. Xu X, Zhu Y. Curcumin inhibits human non-small cell lung cancer xenografts by targeting STAT3 pathway. Am J Transl Res. 2017;9:3633-3641
Author contact
![]() Corresponding author: Hongwen Gao, Department of Pathology, the Second Hospital of Jilin University, Changchun 130041, P.R. China. Email: gaohongwenedu.cn
Corresponding author: Hongwen Gao, Department of Pathology, the Second Hospital of Jilin University, Changchun 130041, P.R. China. Email: gaohongwenedu.cn