Impact Factor ISSN: 1449-1907

 Global reach, higher impact
Global reach, higher impactInt J Med Sci 2017; 14(6):523-529. doi:10.7150/ijms.19033 This issue Cite
Research Paper
Synergistic Antitumor Effect of Sorafenib in Combination with ATM Inhibitor in Hepatocellular Carcinoma Cells
Jianhua Liu1, Yahui Liu1, Lingyu Meng1, Bai Ji1 ![]() , Daqing Yang2
, Daqing Yang2
1. Department of Hepatobiliary and Pancreatic Surgery, the First Hospital of Jilin University, Changchun 130021, China;
2. The Hormel Institute, University of Minnesota, Austin, MN 55912, USA.
Received 2017-1-3; Accepted 2017-3-5; Published 2017-4-9
Abstract
Background: Currently, sorafenib is the only systemic chemotherapy drug for advanced stage Hepatocellular carcinoma (HCC). However, emerging data from some clinical HCC patients indicate that sorafenib alone has only moderate antitumor efficacy, and could not inhibit disease metastasis and progression. KU-55933 is a specific ATM inhibitor, which has pro-apoptotic effect on tumor cells. In this study, we analyzed the synergistic effect of sorafenib and KU-55933 on the proliferation of HCC cell lines.
Methods: Three HCC cell lines were treated with sorafenib and KU-55933 alone or combination in vitro to investigate inhibitory effect by MTT and wound healing assay. Epithelial to mesenchymal transition (EMT) phenotype change was investigated after sorafenib and KU-55933 treatment by microscopy. Akt signaling pathway proteins including p-Akt, p-mTOR and p-p70S6K were examined by western blot. In addition, cleaved PARP and autophage-related proteins LC3A/B were detected by western blot.
Results: KU-55933 can enhance the effect of sorafenib in inhibiting cell proliferation and migration, overcoming EMT, inducing cell apoptosis via inactivating Akt signaling pathway and inducing autophage. The combination treatment with sorafenib and KU-55933 resulted in a strong synergistic effect in vitro.
Conclusion: Our results demonstrate that sorafenib combined with KU-55933 treatment does effectively inhibit proliferation of HCC cell lines synergistically. These data suggests that KU-55933 may be a promising chemosensitizer to sorafenib in the treatment of HCC.
Keywords: Hepatocellular carcinoma, sorafenib, KU-55933, EMT, migration, autophage.
Introduction
Hepatocellular carcinoma (HCC) is the third leading cause of cancer-related death worldwide, with an increasing incidence in the United States and China [1, 2]. In China, HCC commonly arises in patients with chronic liver diseases. Only early stage HCC patients are applicable to potentially curative therapies, such as surgical resection and liver transplantation. Today, the multi-kinase inhibitor sorafenib is the only systemic therapy to improve survival in those patients with advanced HCC [3, 4]. However, some patients show nature or acquired resistance to it. Therefore, prognosis of advanced HCC remains poor, and new effective therapeutic strategies are urgently needed. To find efficient targets, a number of large-scale molecular studies have been conducted in HCC, including Akt [5].
The AKT/mTOR signaling pathway is a promising target with respect to its frequent dysregulation in HCC and its key role in regulating cell proliferation, migration, survival and angiogenesis [6, 7]. Aberrant Akt signaling has been detected in nearly half of hepatocellular carcinoma, and a correlation between poor outcome and Akt signaling activation has been shown [8]. ATM, a protein deficient in patients with ataxia-telangiectasia disease, functions as a signal transducer in response to DNA damage [9]. It has recently been shown that ATM is also a cytoplasmic protein that mediates the full activation of Akt in response to insulin [10]. Li Y, et al [11] reported that a specific ATM inhibitor, KU-55933, blocks the phosphorylation of Akt induced by insulin in cancer cells that exhibit abnormal Akt activity. Moreover, KU-55933 inhibits cancer cell proliferation by inducing G1 cell cycle arrest In addition, KU-55933 treatment during serum starvation triggers apoptosis in these cancer cells. Furthermore, Li et al reported that combination of KU-55933 and rapamycin not only induces apoptosis, which is not seen in cancer cells treated only with rapamycin, but also shows better efficacy in inhibiting cancer cell proliferation than each drug alone. Based on this data, we hypothesize KU-55933 can enhance the effect of sorafenib.
Currently, sorafenib plays a critical role in treating patients with advanced stage HCC, contributing to an improved overall survival in treated patients in clinical trials [12, 13]. Unfortunately, some patients don't benefit from the treatment. Therefore, it is imperative to investigate the potential molecular mechanisms which lead to low survival benefits to help develop potential strategies aimed at increasing its efficacy against HCC. In this study, we show that ATM inhibitor can enhance sorafenib-induced apoptosis through downregulation of p-Akt (Thr308), p-mTOR and p-p70S6K and upregulation of cleaved PARP and LC3A/B II. In addition, they present a synergistic effect in inhibiting migration and EMT. These results suggest that KU-55933 may be a novel chemosensitizer to increase chemotherapeutic sensitivity of sorafenib on HCC cells.
Materials and methods
Chemicals and antibodies
Sorafenib purchased from Santa Cruz Co. KU-55933 was purchased from Calbiochem. They were both dissolved in DMSO to prepare the stock solution of 20mM and stored in aliquots at -20℃. Antibodies against PARP, LC3A/B, phospho-Akt (Thr308), phospho-mTOR, phospho-p70s6k (Thr 389), and β-actin were purchased from cell signaling Technology.
Cell lines and culture conditions
Hepatocellular carcinoma cell lines, HepG2, Huh7 and Hep3B purchased from ATCC were cultured in DMEM supplemented (Hyclone, Logan, UT, USA) with 10% FBS (Hyclone, Logan, UT, USA) and 1% of penicillin-streptomycin at 37℃, in humidified air containing 5% CO2.
MTT Cell Proliferation Assay
Cells were seeded in a 48-well plate and incubated overnight. Following treatment with sorafenib and/or KU-55933, the viable cells in each well were determined using a CellTiter Nonradioactive cell proliferation assay kit (Promega) following the manufacturer's instructions. Briefly, MTS dye solution in the kit was added to each well and incubated at 37℃ for 4 h. The absorbance at 490nm was recorded by a microplate reader.
Western blot
Cells were lysed with TGN lysis buffer containing protease inhibitor cocktails (Roche). The protein concentration was measured by the Lowry method. Equal amounts of protein were subjected to SDS-PAGE and then transferred to a nitrocellulose membrane. Primary antibody was added in milk and allowed to incubate overnight at 4℃, washed with TBST for 3 times (5 min per time) before the secondary antibody was added and then incubated for an hour at room temperature. The membrane was again washed 3 times before adding SuperSignal West Pico Chemiluminescent Substrate (Thermo Scientific, IL, USA) and then immediately developed by chemiluminescence.
Cell migration
A total of 200000 cells were seeded onto a six-well plate and allowed to reach full confluence. The monolayer was wounded using a 200µL tip. Cells were incubated with medium containing sorafenib and KU-55933 alone or combination. Digital images were taken at times of 0h and 48h. The results are representative of three individual experiments.
EMT phenotype change
A total of 40000 cells were seeded onto a six-well plate and incubated overnight. Following TGF-β1, KU-55933 and sorafenib treatment, digital images were taken at times of 48h. The results are representative of three individual experiments.
Statistical analysis
All data were presented as mean±SD. Student's t-Test (unpaired, 2-tailed) was used for comparison between two groups. One-way ANOVA was used to compare difference of multiple groups. P value less than 0.05 was considered statistically significant.
Results
Sorafenib inhibits HCC cell lines proliferation
In order to determine the 50% of inhibitory concentration (IC50) of sorafenib on three HCC cell lines, we analyzed the effects of the sorafenib on the HCC cell lines using MTT approach. The cell lines were exposed to sorafenib (0 µM, 2.5 µM, 5 µM, 10 µM, 20 µM), and cell viability was determined by MTS solution after 72h. In the experiment, we found that sorafenib led to a dose-dependent inhibition on cell proliferation (Figure 1), and IC50 was shown in Table 1.
MTT proliferation assays. After treatment with sorafenib at concentrations ranging from 0 to 20 µM on HCC cell lines HepG2, Huh7 and Hep3B for 72h, cell viability was determined using MTS dye solution. Results are presented as the median of 3 independent experiments.
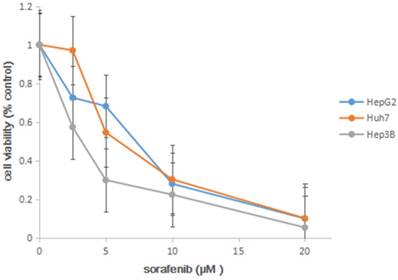
Inhibitory concentration 50% (IC50) of sorafenib
| cells | HepG2 | Huh7 | Hep3B |
|---|---|---|---|
| IC50(μM) | 7.42 | 5.97 | 3.31 |
KU-55933 and sorafenib inhibits hepatocellular carcinoma cell lines proliferation synergistically
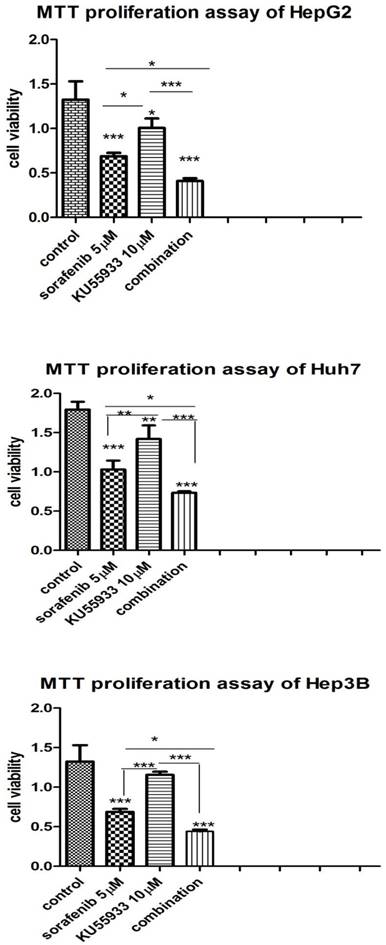
In order to examine synergistic effect of KU-55933 and sorafenib in vitro, we analyzed the effects of two drugs on three different HCC cell lines using MTS approach. The cell lines were grown in 24-well plate and was exposed to KU-55933(10 µmol/L), sorafenib (5 µmol/L) and combination respectively. 72h later, cell viability was examined by MTS. In the experiment we found that sorafenib and KU-55933 inhibit cell proliferation in a synergistic manner (Figure 2).
KU-55933 and sorafenib inhibit the proliferation of HepG2, Huh7 and Hep3B cells in a synergistic manner. Cells were seeded in a 24-well plate and were then treated with KU-55933 (10 µmol/L) and sorafenib (5 µmol/L) alone or combination for 3 d. The cell proliferation rate in each well was determined with the CellTiter 96 MTT cell proliferation assay kit (Promega) following the manufacturer's instructions. Columns, mean of absorbance from three separate experiments (*,p<0.05;**, p<0.01; ***, p<0.001);bars , SD.
Scratch wound healing assay
Cell scratch assay was used to analyze whether KU-55933 and sorafenib inhibit the migration of HCC cells. As shown in Figure 3, cell free area of the combination treatment was wider than that of single-drug treatment, and treatment groups were wider than that of control in three cell lines at 48 hours. This demonstrates that KU-55933 and sorafenib inhibit migration of three HCC cell lines in a synergistic manner.
Sorafenib (5 µmol/L) and KU-55933 (10 µmol/L) showed synergistic effect in inhibiting migration in wound healing assays. Images show the gap change of the scratched region of the different groups. Magnification of 25x.

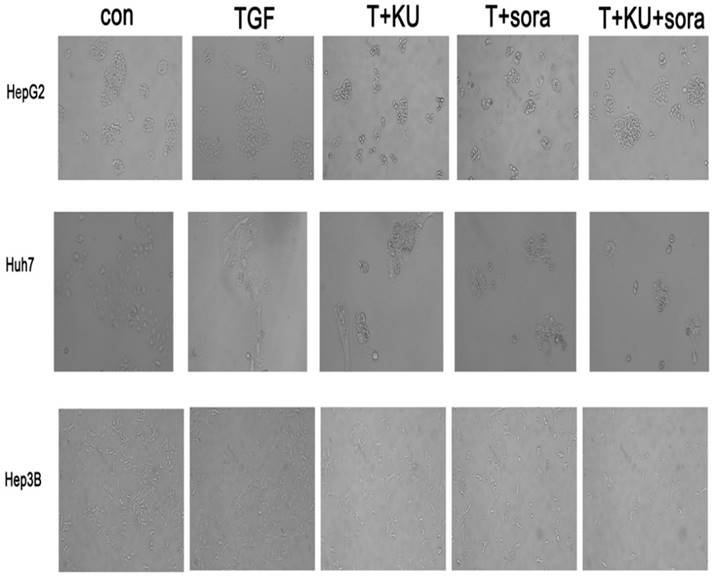
33 and sorafenib reverse EMT phenotype change induced by TGF-β1 in HCC cell lines
TGF treatment alone led to phenotype change from epithelium-like morphology to mesenchymal phenotype and grew separately in a more aggressive manner, which demonstrates that cells present metastatic and invasive characteristics. Sorafenib and KU-55933 combination can reverse EMT change more obviously than single treatment, and re-acquire the epithelium-like phenotype and proliferation in clusters (Figure 4). This revealed that sorafenib and KU-55933 can inhibit HCC cells metastasis and invasion synergistically. The results are representative of three individual experiments.
The effect of sorafenib and KU-55933 treatment on cell phenotype and marker change.cell phenotype changes resulting from different treatment with TGF-β1 (10 ng/ml), sorafenib (5 µmol/L) and KU-55933 (10 µmol/L). Magnification of 100x. Con, control; T, TGF-β1; KU, KU-55933; sora, sorafenib. The results are representative of three individual experiments.
Combination of KU-55933 and sorafenib induces stronger apoptosis in HCC cells by inhibiting PI3K/Akt pathway activation and inducing autophage
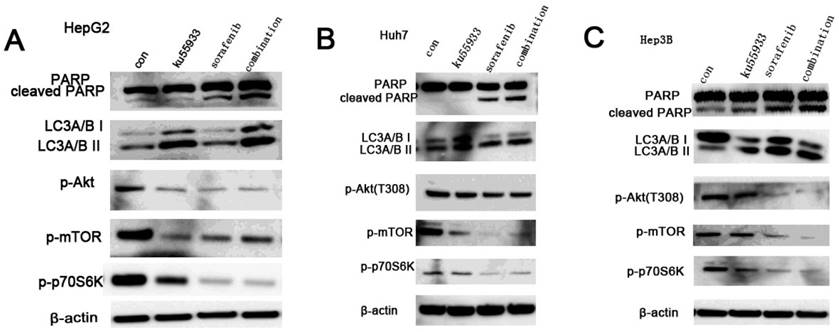
All three HCC cell lines were incubated with KU-55933 (10 µmol/L) and sorafenib (5 µmol/L) alone or combination for 24h, and the levels of p-Akt, p-mTOR and p-p70S6k which is a downstream target of Akt and cleaved PARP were detected. Our data showed that KU-55933 or sorafenib treatment not only resulted in reduced Akt phosphorylation at Thr308 and p70S6K phosphorylation, but also caused a dramatic increase in cleaved PARP levels (Fig. 5A). In addition, results in Fig. 5 indicate that inhibition of Akt and p70S6 kinase phosporylation caused by sorafenib in HCC cells was completely stronger than that is induced by KU-55933. And cells with combination treatment showed the lowest level of p-Akt, p-mTOR and p-p70S6K, which demonstrates that KU-55933 can work as chemosensitizer to sorafenib. Furthermore, although KU-55933 alone failed to induce apoptosis of Huh7 cells, cells treated with KU-55933 plus sorafenib present increased apoptosis by upregulation the cleaved PARP compared with sorafenib alone treatment. These results were also confirmed by an autophage related protein detection. Here, western blotting analysis of the expression of key autophagic proteins showed that both KU-55933 and sorafenib increased, and in combination further increased the expression of LC3A/B-II in three HCC cell lines. All these data suggest a synergistic effect of KU-55933 and sorafenib.
Sorafenib and KU-55933 inhibit cell proliferation and induce apoptosis by inactivating Akt signaling pathway in a synergistic manner. Both floating and attached cells were collected after treatment. Cells were lysed by TGN lysis buffer, and cell lysates were subjected to SDS-PAGE and immunoblotting. PARP, cleaved PARP, phospho-Akt at Thr308, phosph-mTOR, phospho-p70S6K, LC3A/B and β-actin were detected. A.B.C represent results of HepG2, Huh7 and Hep3B cell lines respectively treated by sorafenib (5 µmol/L) and KU-55933(10 µmol/L) alone or combination for 24h, β-actin was detected as a control. The results in A to C are representative of three individual experiments.
Discussion
Hepatocellular carcinoma is the third leading cause of cancer -related mortality worldwide. Surgical resection may provide curative treatment for patients with early stage HCC. Once the cancer becomes the advanced stage, sorafenib is the only systemic chemotherapeutic drug to postpone survival time because patients have lost the opportunity for curative therapies [3, 4]. However, studies of HCC cell lines have revealed that it is not fully effective in preventing recurrence and progression because of resistance [14]. Therefore, many research groups focus on the molecular mechanisms in sorafenib resistance in search of the established therapeutic agents that can overcome the resistance of HCC cells, and help develop potential strategies aimed at increasing its efficacy against HCC [15, 16].
Akt is a major component of the phosphoinositide 3-kinase (PI3K) signaling pathway. In normal cells, Akt acts as a single transducer of PI3K and promotes cell proliferation and cell survival [17]. However, upregulation of Akt leads to the development of cancer [18]. Therefore, it is a significant target in search for drugs that can be used as chemotherapeutic agents for cancer [19]. In our work, treatment with sorafenib and KU-55933 alone or combination results in decreased expression of p-Akt and downstream proteins, p-mTOR and p-p70S6K.
KU-55933 is a specific inhibitor of the ATM kinase [20]. Li et al [11] found that it can inhibit cell proliferation and induce apoptosis via preventing the activation of Akt and block the function of its downstream substrates. However, there is no report regarding its effect on HCC cells. In this study, we showed that KU-55933 alone treatment had only a minor effect on proliferation of three HCC cell lines, and sorafenib alone treatment had a moderate inhibition effect. However, combined sorafenib with KU-55933 treatment led to a strong synergistic effect on proliferation of HCC cells. Moreover, the same synergistic effect was shown on apoptosis, migration and EMT, which plays a key role in cancer progression and resistance to different therapeutic approaches. EMT data demonstrated that sorafenib and KU-55933 treatment revert mesenchymal change induced by TGF-β1 to epithelium cells. Further, The mechanism of synergistic effect of sorafenib and ku55933 in HCC cells was analyzed by western blot and the results showed that the expression of cleaved PARP and LC3A/B II were increased while p-Akt, p-p70S6K and p-mTOR were strongly decreased after treated with sorafenib combined with KU-55933, and facilitate the sorafenib-induced apoptosis in HCC cell lines. These data demonstrate that sorafenib and KU-55933 combination treatment exerts anticancer effect on HCC via inhibiting Akt signaling pathway and inducing autophage. We also found that sorafenib led to a dose-dependent cell apoptosis. Therefore, we hypothesize that KU-55933 may be a chemosensitizer to sorafenib for advanced HCC patients. This study has therefore provided a framework for the development of sorafenib-based combination therapies for HCC.
Conclusion
In conclusion, the results of this study demonstrate that combining sorafenib and KU-55933 shows a synergistic effect in HCC cell lines. Therefore, this combination treatment strategy may be a promising treatment option for patients with advanced HCC since KU-55933 is already being tested in clinical trials and reported to be well tolerated.
Acknowledgements
This work was in part supported by grants from Foundation of Jilin Provincial Development and Reform Commission (KY20160002, No. 3J115AJ73428 ) and Jilin University Research Fund for Excellent Young teachers (No.419080500355 ).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Wang S, Sun H, Xie Z. et al. Improved survival of patients with hepatocellular carcinoma and disparities by age, race, and socioeconomic status by decade, 1983-2012. Oncotarget. 2016 doi: 10.18632/oncotarget.10930
2. Altekruse SF1, McGlynn KA, Reichman ME. Hepatocellular carcinoma incidence, mortality, and survival trends in the United States from 1975 to 2005. J Clin Oncol. 2009;27:1485-91
3. Finn RS, Poon RT, Yau T. et al. Phase I study of everolimus in combination with sorafenib in patients with advanced hepatocellular carcinoma (HCC). J Clin Oncol. 2011;29(15_suppl):4074
4. Llovet JM, Hernandez-Gea V. Hepatocellular carcinoma: reasons for phase III failure and novel perspectives on trial design. Clin Cancer Res. 2014;20:2072-9
5. Grabinski N, Ewald F, Hofmann BT. et al. Combined targeting of AKT and mTOR synergistically inhibits proliferation of hepatocellular carcinoma cells. Mol Cancer. 2012;11:85
6. Engelman JA. Targeting PI3K signaling in cancer: opportunities, challenges and limitations. Nat Rev Cancer. 2009;9:550-62
7. Baik SH, Lee J, Lee YS. et al. ANT2 shRNA downregulates miR-19a and miR-96 through the PI3K/Akt pathway and suppresses tumor growth in hepatocellular carcinoma cells. Exp Mol Med. 2016;48:e222
8. Villanueva A, Chiang DY, Newell P. et al. Pivotal role of mTOR signaling in hepatocellular carcinoma. Gastroenterology. 2008;135:1972-83
9. Shiloh Y, Kastan MB.ATM. genome stability, neuronal development, and cancer cross paths. Adv Cancer Res. 2001;83:209-54
10. Viniegra JG1, Martínez N, Modirassari P. et al. Full activation of PKB/Akt in response to insulin or ionizing radiation is mediated through ATM. J Biol Chem. 2005;280:4029-36
11. Li Y, Yang DQ. The ATM inhibitor KU-55933 suppresses cell proliferation and induces apoptosis by blocking Akt incancer cells with overactivated Akt. Mol Cancer Ther. 2010;9:113-25
12. Llovet JM, Ricci S, Mazzaferro V. et al. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med. 2008;359:378-90
13. Cheng AL, Kang YK, Chen Z. et al. Efficacy and safety of sorafenib in patients in the Asia-Pacific region with advanced hepatocellular carcinoma: a phase III randomised, double-blind, placebo-controlled trial. Lancet oncol. 2009;10:25-34
14. Liu J, Cui X, Qu L. et al. Overexpression of DLX2 is associated with poor prognosis and sorafenib resistance in hepatocellular carcinoma. Exp Mol Pathol. 2016;101:58-65
15. Tang S, Tan G, Jiang X. et al. An artificial lncRNA targeting multiple miRNAs overcomes sorafenib resistance in hepatocellular carcinoma cells. Oncotarget. 2016 doi: 10.18632/oncotarget.12304
16. Nishida N, Kitano M, Sakurai T. et al. Molecular Mechanism and Prediction of Sorafenib Chemoresistance in Human Hepatocellular Carcinoma. Dig Dis. 2015;33:771-9
17. West KA, Castillo SS, Dennis PA. Activation of the PI3K/Akt pathway and chemotherapeutic resistance. Drug Resist Updat. 2002;5:234-48
18. Nicholson KM1, Anderson NG. The protein kinase B/Akt signalling pathway in human malignancy. Cell Signal. 2002;14:381-95
19. Wang XJ, Feng CW, Li M. ADAM17 mediates hypoxia-induced drug resistance in hepatocellular carcinoma cells through activation of EGFR/PI3K/Akt pathway. Mol Cell Biochem. 2013;380(1-2):57-66
20. Hickson I, Zhao Y, Richardson CJ. et al. Identification and characterization of a novel and specific inhibitor of the ataxia-telangiectasia mutated kinase ATM. Cancer Res. 2004;64:9152-9
Author contact
![]() Corresponding author: Bai Ji, MD, Department of Hepatobiliary and Pancreatic Surgery, the First Hospital of Jilin University, Changchun 130021, China (Tel: 86-431-81875160; Email:jirulincom)
Corresponding author: Bai Ji, MD, Department of Hepatobiliary and Pancreatic Surgery, the First Hospital of Jilin University, Changchun 130021, China (Tel: 86-431-81875160; Email:jirulincom)