ISSN: 1449-1907International Journal of Medical Sciences

Int J Med Sci 2019; 16(6):800-812. doi:10.7150/ijms.34172 This issue Cite
Research Paper
Identification of Key Genes and Pathways in Cervical Cancer by Bioinformatics Analysis
Xuan Wu1,2, Li Peng3, Yaqin Zhang1,2, Shilian Chen1,2, Qian Lei1,2, Guancheng Li1,2 ![]() , Chaoyang Zhang1,4
, Chaoyang Zhang1,4 ![]()
1. Key Laboratory of Carcinogenesis of the Chinese Ministry of Health and the Key Laboratory of Carcinogenesis and Cancer Invasion of Chinese Ministry of Education, Xiangya Hospital, Central South University, Changsha 410078, P.R. China;
2. Cancer Research Institute, Central South University, Changsha, P.R. China;
3. Guangdong Province Key Laboratory of Malignant Tumor Epigenetics and Gene Regulation, Research Center of Medicine, Sun Yat-Sen Memorial Hospital, Sun Yat-Sen University, Guangzhou, 510120, China;
4. Division of Functional Genome Analysis, German Cancer Research Centre (DKFZ), Heidelberg, Germany.
Abstract
Cervical cancer is a common malignant tumour of the female reproductive system that seriously threatens the health of women. The aims of this study were to identify key genes and pathways and to illuminate new molecular mechanisms underlying cervical cancer. Altogether, 1829 DEGs were identified, including 794 significantly down-regulated DEGs and 1035 significantly up-regulated DEGs. GO analysis suggested that the up-regulated DEGs were mainly enriched in mitotic cell cycle processes, including DNA replication, organelle fission, chromosome segregation and cell cycle phase transition, and that the down-regulated DEGs were primarily enriched in development and differentiation processes, such as tissue development, epidermis development, skin development, keratinocyte differentiation, epidermal cell differentiation and epithelial cell differentiation. KEGG pathway analysis showed that the DEGs were significantly enriched in cell cycle, DNA replication, the p53 signalling pathway, pathways in cancer and oocyte meiosis. The top 9 hub genes with a high degree of connectivity (over 72 in the PPI network) were down-regulated TSPO, CCND1, and FOS and up-regulated CDK1, TOP2A, CCNB1, PCNA, BIRC5 and MAD2L1. Module analysis indicated that the top 3 modules were significantly enriched in mitotic cell cycle, DNA replication and regulation of cell cycle (P < 0.01). The heat map based on TCGA database preliminarily demonstrated the expression change of the key genes in cervical cancer. GSEA results were basically coincident with the front enrichment analysis results. By comprehensive analysis, we confirmed that cell cycle was a key biological process and a critical driver in cervical cancer. In conclusion, this study identified DEGs and screened the key genes and pathways closely related to cervical cancer by bioinformatics analysis, simultaneously deepening our understanding of the molecular mechanisms underlying the occurrence and progression of cervical cancer. These results might hold promise for finding potential therapeutic targets of cervical cancer.
Keywords: Cervical cancer, Microarray, Bioinformatics analysis, Differentially expressed gene
Background
Cervical cancer is the fourth most common malignancy in women worldwide with an estimated 527,600 new cases and 265,700 deaths worldwide in 2012, displaying higher incidence and mortality in less developed countries [1]. The risk factors of cervical cancer chiefly include human papillomavirus (HPV) infection, high numbers of sexual partners, high parity, smoking cigarettes, long-term consumption of oral contraceptives and having the first sexual behaviour at a young age [2-8]. At present, surgery, radiotherapy and chemotherapy have been frequently used as treatment approaches to improve overall survival, disease-free survival, progression-free survival and decreased recurrence rate in cervical cancer patients [9, 10]. However, the 5-year survival rate for advanced cervical cancer patients, especially for metastatic cervical cancer patients, whose survival rates range from 5% to 15%, is still low [11]. Consequently much more effort is needed to further clarify the molecular mechanisms involved in tumour initiation and progression, which might help find better molecular targets for the treatment of cervical cancer.
As an innovative and high-throughput research method, gene expression profiling analysis based on microarray technology enables the simultaneous investigation of expression changes in thousands of genes in limited tumour samples and contributes to new drug target discovery, molecular diagnosis and prognosis [12]. In recent years, there have been many studies on the molecular mechanisms of cervical cancer occurrence and development by finding and analysing differentially expressed genes with microarray technologies. Scotto et al. examined the role of gains in the long arm of chromosome 20 (20q) and found a copy number increase in 20q in >50% of invasive cervical cancers by means of gene expression profiling [13]. Zhai et al. confirmed HOXC10 as a critical mediator of invasion by gene expression analysis of preinvasive and invasive cervical squamous cell carcinomas [14]. Medina-Martinez et al. investigated the impact of gene dosage on gene expression, biological processes and survival in the carcinogenesis of cervical cancer via microarray technology and data analysis [15]. However, there are no reliable molecular expression profiling that discriminates cancer tissue from normal cervix tissue identified at the clinic. Currently, a combination of gene expression profiling and bioinformatics analysis allows us to comprehensively detect mRNA expression changes in cervical cancer and subsequently identify key genes and pathways that exist in the interaction network of differentially expressed genes (DEGs).
In the current study, cervical cancer-associated gene expression dataset GSE7803 was downloaded from Gene Expression Omnibus (GEO, http://www.ncbi.nlm.nih.gov/geo/), which is a public functional genomics data repository with array- and sequence-based data. DEGs were identified by the comparison of cancerous and normal cervix tissue based on R software and Bioconductor. GO analysis, KEGG pathway analysis, PPI network analysis and GSEA helped find critical genes and pathways in cervical cancer.
Materials and Methods
Acquisition of microarray data. The targeted gene expression dataset GSE7803 was downloaded from the GEO database. GSE7803, submitted by Rork Kuick et al., included 10 normal cervix samples and 21 cervical cancer samples and was based on the GPL96 platform ([HG-U133A] Affymetrix Human Genome U133A Array) [14].
Identification of differentially expressed genes. The raw data were subjected to significance analysis with several packages of R statistical software (version 3.3.2, https://www.r-project.org/). First, we detected microarray quality by a quality control overview diagram based on the "simpleaffy" package, weights and residuals plot, relative log expression (RLE) box plot and normalized unscaled standard errors (NUSE) box plot based on the "affyPLM" and "RColorBrewer" packages, RNA degradation curve based on the "affy" package and clustering analysis diagram based on the "gcrma", "graph" and "affycoretools" packages to remove the unqualified samples. Afterwards, an optimal integrative algorithm was selected to perform pre-processing of the raw microarray data. Then, an empirical Bayes method was used for significance analysis of DEGs, which included six key steps: construction of a gene expression matrix, construction of an experimental design matrix, construction of a contrast matrix, fitting of a linear model, Bayes test, and generation of a results report. We defined a P value < 0.05 to be statistically significant. Finally, all the DEGs were annotated by the "annotate" package.
Gene Ontology and KEGG pathway enrichment analysis of DEGs. GO term enrichment analysis was conducted by the "GOstats" package of Bioconductor to excavate DEG function and biological significance. KEGG pathway enrichment analysis was performed to find pathways that were closely associated with cervical cancer with the "GeneAnswers" package of Bioconductor. A P value < 0.05 was considered statistically significant.
Module analysis of protein-protein interaction (PPI) network. Search Tool for the Retrieval of Interacting Genes (STRING) database (http://www.string-db.org/) was used to acquire PPI information for the DEGs. Cytoscape software was applied to visualize the PPI network according to PPI information. The top DEGs with a high degree of connectivity in the PPI network were selected to discuss their function and effect on cervical cancer. Then, we successively performed module analysis and GO analysis to identify the biological processes that the module genes were significantly enriched in by the plug-ins Molecular Complex Detection (MCODE) and Biological Network Gene Ontology tool (BiNGO) in Cytoscape. Finally, the expression level of these pivotal DEGs was verified through cervical cancer-associated transcription profiling in The Cancer Genome Atlas (TCGA) database (https://cancergenome.nih.gov/), which contains gene expression information for various cancers. A P value < 0.05 was considered to be statistically significant.
GSEA of DEGs on the whole gene expression level. GSEA is a powerful analytical method for interpreting genome-wide expression profiles, including three key elements: calculation of enrichment score (ES), estimation of significance level of ES and adjustment for multiple hypothesis testing [16]. We first created a chip expression profile file and a sample data file and then imported them into GSEA software. After choosing gene sets database and corresponding chip platform, setting other parameters as default, we could run GSEA and acquired pathway enrichment results on the total gene expression level.
Results
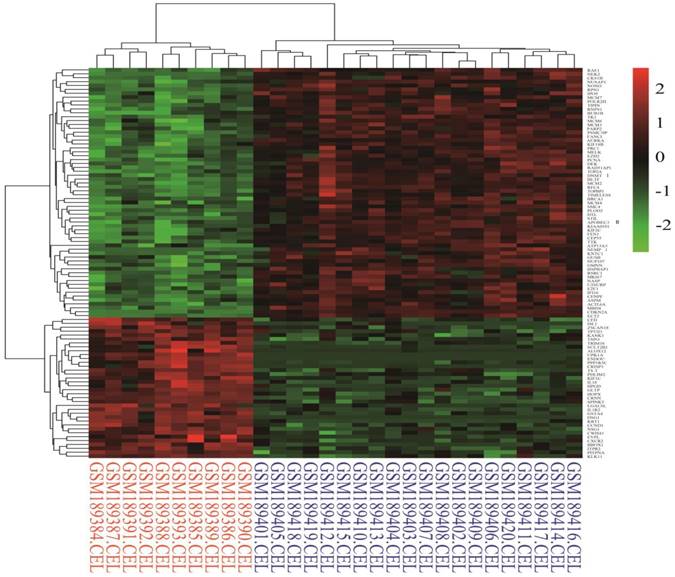
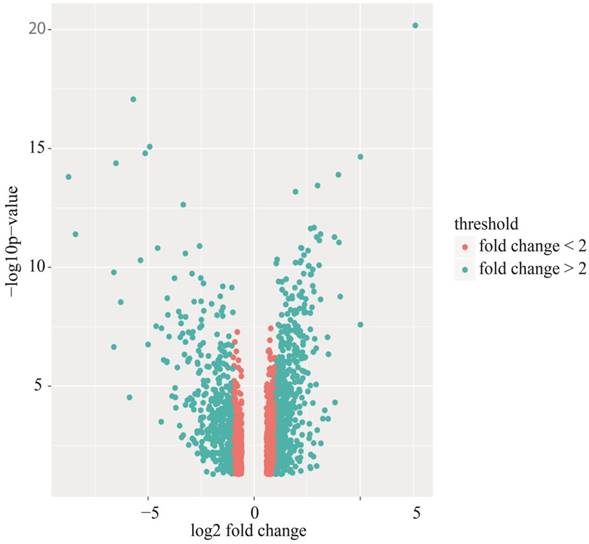
Identification of differentially expressed genes. Cervical cancer-associated gene expression dataset GSE7803 included 10 normal cervix samples and 21 cervical cancer samples. The cervical cancer sample GSM189421 was an outlier, and it was removed based on the cluster analysis results of the microarray data. The integrative algorithm gcRMA was selected for the pre-processing of microarray data. Significance analysis found that there were 1829 DEGs in cervical cancer samples compared with normal samples, among which 794 DEGs were significantly down-regulated and 1035 DGEs were significantly up-regulated. A heat map showed expression profiling of the top 100 DEGs in the analysis result (Figure 1). The top 100 DEGs, including 36 significantly down-regulated genes and 64 significantly up-regulated genes, could effectively distinguish cervical cancer samples from normal cervix samples. Some of these DEGs might have relatively high potential to function as diagnostic biomarkers and therapeutic targets of cervical cancer. Table 1 shows detailed information on the top 10 DEGs in the significance analysis result, including gene symbols, gene names, average expression values, expression fold changes, and statistics, such as t values and P values. We chose 887 DEGs with fold changes over 2, including 469 up-regulated DEGs and 418 down-regulated DEGs, to carry out the subsequent bioinformatics analysis, as displayed in the volcano plot (Figure 2).
Heat map of the top 100 differentially expressed genes in the analysis result (36 down-regulated genes and 64 up-regulated genes). Red: up-regulation; Green: down-regulation.
Volcano plot of differentially expressed genes (DEGs). Red: DEGs with fold changes less than 2; Green: DEGs with fold changes over 2.
Detailed information on the top 10 differentially expressed genes in the analysis result, including gene symbol, gene name, average expression value, expression fold change, and statistics, such as t value and P value.
| Symbol | Name | AveExpr | t | P.Value | adj.P.Val | B | logFC |
|---|---|---|---|---|---|---|---|
| CDKN2A | cyclin dependent kinase inhibitor 2A | 8.208 | 25.038 | 6.68E-21 | 1.49E-16 | 37.806 | 7.602 |
| UPK1A | uroplakin 1A | 4.159 | -19.185 | 8.51E-18 | 9.48E-14 | 30.665 | -5.699 |
| ENDOU | endonuclease, poly(U) specific | 3.927 | -16.091 | 8.33E-16 | 6.19E-12 | 26.039 | -4.927 |
| IL1R2 | interleukin 1 receptor type 2 | 5.210 | -15.689 | 1.59E-15 | 8.88E-12 | 25.383 | -5.140 |
| ECT2 | epithelial cell transforming 2 | 7.054 | 15.478 | 2.25E-15 | 1.00E-11 | 25.033 | 5.005 |
| DSG1 | desmoglein 1 | 4.430 | -15.109 | 4.16E-15 | 1.55E-11 | 24.410 | -6.513 |
| MCM2 | minichromosome maintenance complex component 2 | 7.032 | 14.451 | 1.28E-14 | 4.07E-11 | 23.271 | 3.969 |
| KRT1 | keratin 1 | 5.976 | -14.333 | 1.57E-14 | 4.38E-11 | 23.063 | -8.747 |
| RFC4 | replication factor C subunit 4 | 7.824 | 13.858 | 3.64E-14 | 9.02E-11 | 22.209 | 2.990 |
| MBD4 | methyl-CpG binding domain 4, DNA glycosylase | 9.357 | 13.528 | 6.63E-14 | 1.48E-10 | 21.601 | 1.944 |
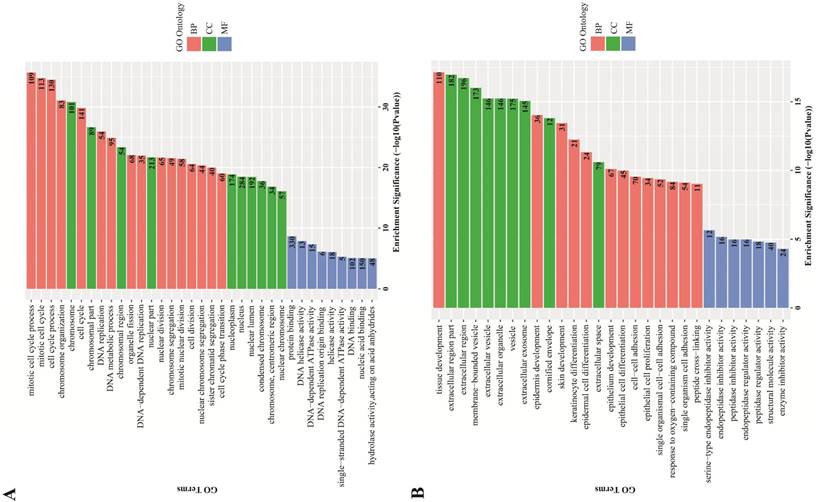
GO term enrichment analysis. GO term enrichment analysis found an obvious quantity variance and significance level difference among the 887 DEGs that were enriched in biological processes, molecular functions and cellular components. For biological processes, the up-regulated DEGs were mainly enriched in mitotic cell cycle processes, including DNA replication, organelle fission, chromosome segregation and cell cycle phase transition, suggesting that these DEGs could act as oncogenes to promote Human cervical carcinoma by accelerating cell cycle phase. The down-regulated DEGs were significantly enriched in development and differentiation processes, such as tissue development, epidermis development, skin development, keratinocyte differentiation, epidermal cell differentiation and epithelial cell differentiation, suggesting that cell differentiation-associated DEGs might play a negative role in the malignant proliferation of Human cervical carcinoma cells. Regarding cellular components, the up-regulated DEGs were significantly enriched in the chromosomal region and the nuclear part, indicating that the cellular localization of DEGs was consistent with the abovementioned biological functions, and the down-regulated DEGs were primarily enriched in the extracellular region part, such as extracellular vesicles, extracellular organelles and extracellular exosomes. For molecular functions, the up-regulated DEGs were significantly enriched in protein binding, DNA helicase activity and DNA-dependent ATPase activity, which participated in and facilitated DNA replication to promote cell cycle, and the down-regulated DEGs were significantly enriched in endopeptidase inhibitor activity, peptidase regulator activity and enzyme inhibitor activity. All the detailed GO term enrichment analysis results are displayed in Figure 3.
KEGG pathway enrichment analysis. KEGG pathway analysis found that DEGs were significantly enriched in 5 pathways as shown in Figures 4-5. To be specific, 33 up-regulated DEGs and 3 down-regulated DEGs were significantly enriched in cell cycle-associated pathways. Thirteen up-regulated DEGs were significantly enriched in DNA replication-associated pathways. Ten up-regulated DEGs and eight down-regulated DEGs were significantly enriched in p53 signalling pathways. Eleven down-regulated DEGs and 26 up-regulated DEGs were significantly enriched in pathways in cancer. Thirteen up-regulated DEGs and five down-regulated DEGs were significantly enriched in oocyte meiosis. The detailed analysis results of the top 5 pathways with high enrichment significance levels are displayed in Table 2; these results indicated that cell cycle-associated pathways had the highest fold over-expression.
GO terms enrichment analysis of DEGs. Red: biological process; Green: cellular component; Blue: molecular function. Number in the bar plot: Count of DEGs enriched in corresponding GO classification. (A) GO analysis results of up-regulated DEGs, which were significantly enriched in cell cycle, cell nucleus and DNA replication-associated enzyme activity; (B) GO analysis results of down-regulated DEGs, which were significantly enriched in tissue development and cell differentiation, extracellular region and peptidase inhibitor activity.
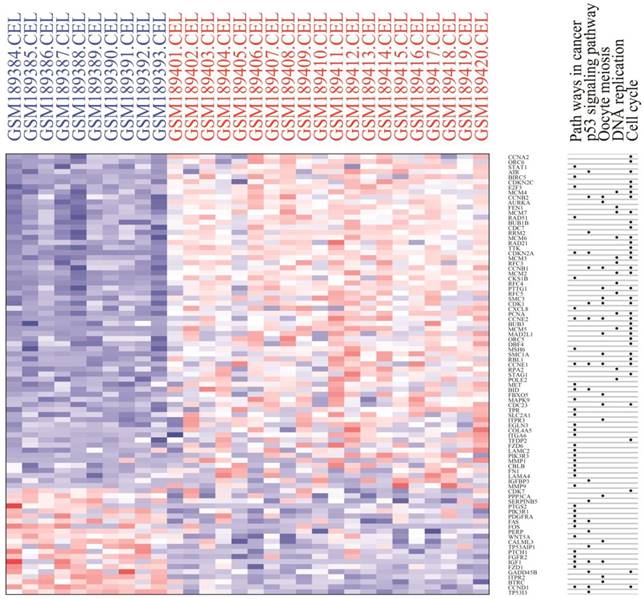
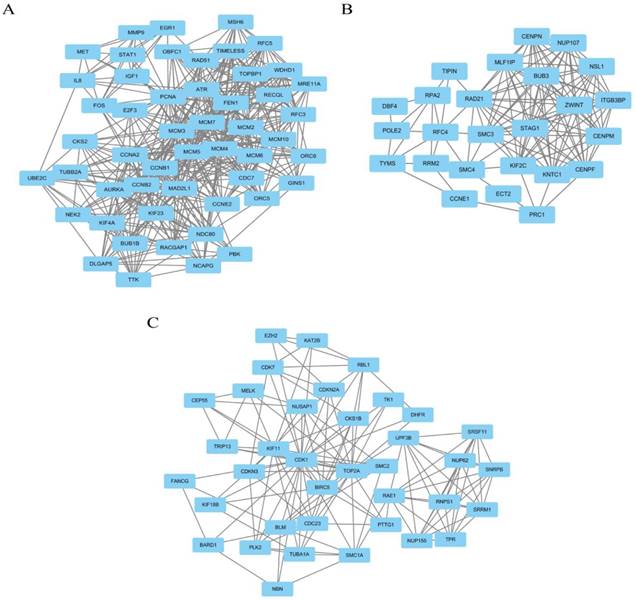
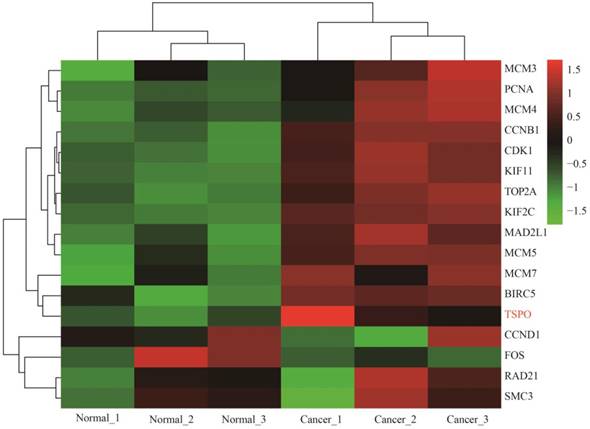
Module analysis of PPI network. PPI information was acquired from the STRING database. A PPI network graph was conducted by Cytoscape software, and based on this graph, we selected the top 9 DEGs with a degree of connectivity larger than 72 as hub genes that were closely associated with cervical cancer. These hub genes were down-regulated translocator protein (TSPO), cyclin D1 (CCND1), Fos proto-oncogene, and AP-1 transcription factor subunit (FOS) and up-regulated cyclin B1 (CCNB1), topoisomerase (DNA) II alpha (TOP2A), proliferating cell nuclear antigen (PCNA), cyclin dependent kinase 1 (CDK1), MAD2 mitotic arrest deficient-like 1 (MAD2L1), and baculoviral IAP repeat containing 5 (BIRC5). Among these DEGs, TSPO displayed the highest degree of connectivity which was 149. The plug-ins MCODE and BiNGO were used for PPI module screening and corresponding GO term enrichment analysis in Cytoscape. The top 3 modules with high scores were selected for display and were significantly enriched in mitotic cell cycle, DNA replication and the regulation of cell cycle (Table 3-5, P < 0.01). Specifically, module 1 contained 51 nodes (genes) and 477 edges (interaction), among which PCNA, MCM5, MCM3, MCM4, MCM7 and CCNB1 had a degree of connectivity larger than 30 and could function as hub module genes in DNA replication processes (Figure 6A and Table 3). Relevant studies have corroborated this speculation. Up-regulated PCNA induced by long noncoding RNA CCHE1 could promote the cell proliferation of cervical cancer [17]. MCM2-7 could strengthen DNA helicase activity to accelerate the cell cycle [18,19]. CCNB1, one of the highly conserved cyclin family members, is involved in regulating cell cycle at the G2/M transition by forming maturation-promoting factor (MPF) with p34, suggesting that its over-expression can promote the progression of cancers [20,21]. Module 2 contained 25 nodes and 120 edges, among which RAD21, SMC3 and KIF2C had a degree of connectivity larger than 14 and could act as critical module genes in cell cycle processes (Figure 6B and Table 4). RAD21, involved in the repair of DNA double-strand breaks, is essential for supporting normal DNA replication in the cell cycle [22]. Dysregulated SMC3 could contribute to the maintenance of sister chromatid cohesion by acetylation in cell cycle processes [23]. As a tumour antigen, NY-CO-58/KIF2C is overexpressed in various solid tumours and its expression level correlates with the proliferative activity of cancer cells [24]. Module 3 contained 37 nodes and 141 edges, among which KIF11, CDK1, BIRC5 and TOP2A had a degree of connectivity larger than 10 and could be considered to be key module genes in cell cycle-associated pathways (Figure 6C and Table 5). KIF11 is an evolutionarily conserved chromosome instability gene, and aberrant expression of KIF11 may promote the pathogenesis of cancer by inducing chromosome instability [25]. The up-regulated DEG CDK1 is a key signalling molecule in the regulation of cell cycle and holds promise as a drug target in various tumours, although there are not many studies on CDK1 in cervical cancer [26]. BIRC5, known as survivin, can contribute to cell growth, proliferation, migration and metastasis in cervical cancer [27-31]. Up-regulated TOP2A participates in controlling DNA topological structure, chromosome segregation and cell cycle progression by encoding a 170-kDa nuclear enzyme [32-34]. Cervical cancer-associated transcription profiling in the TCGA database further validated the expression changes of these key DEGs (Figure 7). TSPO was detected as up-regulated, but this might be due to a few paracancerous tissue samples of cervical cancer in the TCGA database.
Heat map of KEGG pathways in which differentially expressed genes were significantly enriched. There were 5 pathways in the heat map, including cell cycle, DNA replication, p53 signalling pathway, pathways in cancer and oocyte meiosis. Red: up-regulation; Blue: down-regulation.
Relation graph of KEGG pathways in which differentially expressed genes were significantly enriched. There were 5 pathways in the relation graph, including cell cycle, DNA replication, p53 signalling pathway, pathways in cancer and oocyte meiosis. Red: up-regulation; Blue: down-regulation.
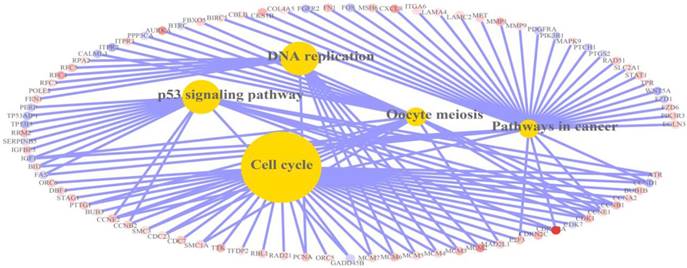
The detailed information of the top 5 significantly enriched KEGG pathways.
| KEGG ID | KEGG name | genes in Category | percent in the observed List | percent in the genome | fold of overrepresents | odds ratio | p value |
|---|---|---|---|---|---|---|---|
| 4110 | Cell cycle | 36 | 0.099 | 0.021 | 4.681 | 6.756 | 9.50E-16 |
| 4115 | p53 signaling pathway | 18 | 0.049 | 0.012 | 4.268 | 5.676 | 8.81E-08 |
| 3030 | DNA replication | 13 | 0.036 | 0.006 | 5.822 | 8.828 | 1.02E-07 |
| 4114 | Oocyte meiosis | 18 | 0.049 | 0.019 | 2.591 | 2.995 | 0.000157 |
| 5200 | Pathways in cancer | 37 | 0.102 | 0.056 | 1.830 | 2.042 | 0.000207 |
The top 5 significantly enriched GO terms and corresponding gene information in module 1.
| GO-ID | p-value | corr p-value | N | Description | Genes in test set |
|---|---|---|---|---|---|
| 6260 | 3.82E-27 | 3.70E-24 | 21 | DNA replication | RFC5|GINS1|FEN1|RFC3|PCNA|MCM7|RECQL|MRE11A|MCM10|CDC7|IGF1| |
| ORC5|ORC6|RAD51|CCNE2|MCM3|MCM4|MCM5|MCM6|MCM2|ATR | |||||
| 7049 | 5.23E-24 | 2.53E-21 | 29 | cell cycle | MCM7|BUB1B|NCAPG|TTK|AURKA|CCNB2|CCNB1|RACGAP1|PBK|NEK2 |
| |E2F3|DLGAP5|UBE2C|MRE11A|KIF23|CDC7|NDC80|MSH6|CCNA2|TUBB2A | |||||
| |RAD51|IL8|CKS2|MCM3|TIMELESS|MCM6|MCM2|ATR|MAD2L1 | |||||
| 6259 | 2.76E-23 | 8.91E-21 | 25 | DNA metabolic process | FEN1|PCNA|MCM7|MCM10|ORC5|ORC6|OBFC1|TOPBP1|RFC5|GINS1|RFC3|RECQL| |
| MRE11A|CDC7|FOS|IGF1|MSH6|RAD51|CCNE2|MCM3|MCM4|MCM5|MCM6|MCM2|ATR | |||||
| 279 | 8.85E-20 | 2.14E-17 | 20 | M phase | UBE2C|BUB1B|NCAPG|MRE11A|KIF23|TTK|NDC80|AURKA|MSH6|CCNA2|CCNB2| |
| CCNB1|TUBB2A|RAD51|PBK|CKS2|TIMELESS|NEK2|DLGAP5|MAD2L1 | |||||
| 22403 | 2.59E-19 | 4.25E-17 | 21 | cell cycle phase | UBE2C|BUB1B|NCAPG|MRE11A|KIF23|TTK|CDC7|NDC80|AURKA|MSH6|CCNA2| |
| CCNB2|CCNB1|TUBB2A|RAD51|PBK|CKS2|TIMELESS|NEK2|DLGAP5|MAD2L1 |
The top 5 significantly enriched GO terms and corresponding gene information in module 2.
| GO-ID | p-value | corr p-value | N | Description | Genes in test set |
|---|---|---|---|---|---|
| 278 | 4.53E-17 | 1.94E-14 | 14 | mitotic cell cycle | TIPIN|SMC3|SMC4|NSL1|ZWINT|STAG1|CENPF| |
| DBF4|PRC1|CCNE1|RAD21|KNTC1|KIF2C|BUB3 | |||||
| 51301 | 2.02E-16 | 4.29E-14 | 13 | cell division | TIPIN|SMC3|SMC4|NSL1|ZWINT|STAG1|CENPF| |
| PRC1|CCNE1|RAD21|KNTC1|KIF2C|BUB3 | |||||
| 22403 | 3.00E-16 | 4.29E-14 | 14 | cell cycle phase | TIPIN|SMC3|SMC4|NSL1|ZWINT|STAG1|CENPF| |
| DBF4|PRC1|CCNE1|RAD21|KNTC1|KIF2C|BUB3 | |||||
| 22402 | 1.70E-14 | 1.31E-12 | 14 | cell cycle process | TIPIN|SMC3|SMC4|NSL1|ZWINT|STAG1|CENPF|DBF4| |
| PRC1|CCNE1|RAD21|KNTC1|KIF2C|BUB3 | |||||
| 280 | 1.84E-14 | 1.31E-12 | 11 | nuclear division | TIPIN|STAG1|CENPF|RAD21|KNTC1| |
| KIF2C|BUB3|SMC3|SMC4|NSL1|ZWINT |
The top 5 significantly enriched GO terms and corresponding gene information in module 3.
| GO-ID | p-value | corr p-value | N | Description | Genes in test set |
|---|---|---|---|---|---|
| 7049 | 3.96E-15 | 3.07E-12 | 19 | cell cycle | BARD1|BLM|CDKN2A|PLK2|KIF11|SMC1A|CKS1B|SMC2|KAT2B|CDC23| |
| RBL1|PTTG1|NUSAP1|CDK1|BIRC5|NBN|TRIP13|CEP55|CDKN3 | |||||
| 51726 | 6.19E-14 | 2.40E-11 | 15 | regulation of cell cycle | BARD1|BLM|CDKN2A|SMC1A|FANCG|CKS1B|KAT2B|CDK7| |
| CDC23|TPR|NUSAP1|CDK1|BIRC5|NBN|CDKN3 | |||||
| 22402 | 1.58E-13 | 4.08E-11 | 16 | cell cycle process | BARD1|BLM|CDKN2A|KIF11|SMC1A|SMC2|KAT2B|CDC23| |
| PTTG1|NUSAP1|CDK1|BIRC5|NBN|TRIP13|CEP55|CDKN3 | |||||
| 22403 | 9.69E-13 | 1.88E-10 | 14 | cell cycle phase | BLM|CDKN2A|KIF11|SMC1A|SMC2|CDC23|PTTG1| |
| NUSAP1|CDK1|BIRC5|NBN|TRIP13|CEP55|CDKN3 | |||||
| 278 | 3.56E-12 | 5.52E-10 | 13 | mitotic cell cycle | BLM|CDKN2A|PLK2|KIF11|SMC1A|SMC2|CDC23| |
| PTTG1|NUSAP1|CDK1|BIRC5|CEP55|CDKN3 |
The top 3 modules with relatively high scores selected from the protein-protein interaction network. (A) module 1 with 51 nodes and 477 edges was significantly enriched in DNA replication, cell cycle, DNA metabolic process, M phase and cell cycle phase; (B) module 2 with 25 nodes and 120 edges was significantly enriched in mitotic cell cycle, cell division, cell cycle phase, cell cycle process and nuclear division; and (C) module 3 with 37 nodes and 141 edges was significantly enriched in cell cycle, regulation of cell cycle, cell cycle process, cell cycle phase and mitotic cell cycle.
Heat map of expression profiling of several key genes from the protein-protein interaction network analysis results. Four genes were down-regulated and thirteen genes were up-regulated, which preliminarily validated the bioinformatics analysis results of the microarray data.
The significant enriched KEGG pathways from GSEA results (P < 0.01, FDR < 0.25).
| NAME | SIZE | ES | NES | NOM p-val | FDR q-val | FWER p-val | RANK AT MAX |
|---|---|---|---|---|---|---|---|
| DNA_REPLICATION_INDEPENDENT_NUCLEOSOME_ORGANIZATION | 38 | -0.764 | -1.547 | 0.0000 | 0.159 | 0.998 | 328 |
| CELL_CYCLE_CHECKPOINT | 155 | -0.617 | -1.547 | 0.0000 | 0.162 | 0.998 | 2191 |
| MEIOTIC_CHROMOSOME_SEGREGATION | 34 | -0.783 | -1.549 | 0.0000 | 0.165 | 0.998 | 234 |
| NEGATIVE_REGULATION_OF_ORGANELLE_ORGANIZATION | 281 | -0.526 | -1.531 | 0.0000 | 0.169 | 0.999 | 2145 |
| NEGATIVE_REGULATION_OF_CYTOSKELETON_ORGANIZATION | 160 | -0.577 | -1.615 | 0.0000 | 0.170 | 0.940 | 1517 |
| NEGATIVE_REGULATION_OF_CELLULAR_CATABOLIC_PROCESS | 120 | -0.532 | -1.529 | 0.0000 | 0.170 | 0.999 | 2145 |
| MICROTUBULE_BASED_MOVEMENT | 132 | -0.495 | -1.562 | 0.0000 | 0.170 | 0.996 | 1792 |
| NUCLEAR_TRANSPORT | 263 | -0.479 | -1.566 | 0.0000 | 0.173 | 0.996 | 2615 |
| REGULATION_OF_MICROTUBULE_BASED_PROCESS | 170 | -0.611 | -1.616 | 0.0000 | 0.175 | 0.939 | 1539 |
| DNA_REPLICATION | 150 | -0.707 | -1.566 | 0.0000 | 0.176 | 0.996 | 2187 |
| PROTEIN_SUMOYLATION | 98 | -0.686 | -1.617 | 0.0000 | 0.179 | 0.935 | 2304 |
| MISMATCH_REPAIR | 26 | -0.725 | -1.568 | 0.0000 | 0.180 | 0.996 | 3098 |
| CHROMATIN_ORGANIZATION | 456 | -0.525 | -1.575 | 0.0000 | 0.180 | 0.992 | 2632 |
| SISTER_CHROMATID_COHESION | 74 | -0.760 | -1.585 | 0.0000 | 0.183 | 0.981 | 1747 |
| PEPTIDYL_LYSINE_MODIFICATION | 226 | -0.581 | -1.617 | 0.0000 | 0.185 | 0.932 | 2529 |
| COVALENT_CHROMATIN_MODIFICATION | 238 | -0.510 | -1.587 | 0.0000 | 0.185 | 0.979 | 2609 |
| MICROTUBULE_ORGANIZING_CENTER_ORGANIZATION | 59 | -0.706 | -1.619 | 0.0000 | 0.189 | 0.929 | 1370 |
| DNA_CONFORMATION_CHANGE | 200 | -0.689 | -1.635 | 0.0000 | 0.194 | 0.897 | 1965 |
| REGULATION_OF_CYTOSKELETON_ORGANIZATION | 369 | -0.432 | -1.470 | 0.0000 | 0.201 | 1.000 | 1539 |
| DNA_REPAIR | 346 | -0.604 | -1.619 | 0.0000 | 0.204 | 0.927 | 2161 |
| DNA_GEOMETRIC_CHANGE | 70 | -0.718 | -1.645 | 0.0000 | 0.217 | 0.862 | 1309 |
| NEGATIVE_REGULATION_OF_CHROMOSOME_ORGANIZATION | 77 | -0.637 | -1.646 | 0.0000 | 0.234 | 0.861 | 2502 |
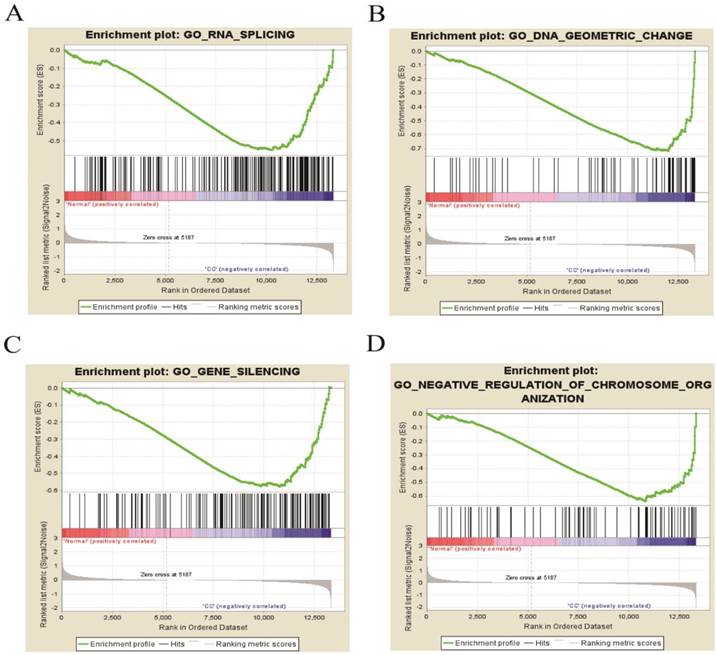
GSEA of DEGs on the whole gene expression level. We further explored and verified KEGG pathway which DEGs involved on the whole gene expression level by GSEA. The results, as shown in Table 6, identified 97 significantly enriched pathways such as DNA replication, meiotic cell cycle and signal transduction by p53 class mediator (P < 0.01, FDR < 0.25), which were basically consistent with the above analysis results. Figure 8 displayed four pathways including RNA splicing, gene silencing, DNA geometric change and negative regulation of chromosome organization, illuminating new molecular mechanisms that DEGs participated in cervical occurrence and progression.
Discussion
Cervical cancer is a common female malignancy that involves multiple factors, multiple gene variations, and a multi-step and multi-stage progression. It is of critical importance to understand the molecular mechanisms of cervical cancer occurrence and development for clinical therapy and early diagnosis. As a high-throughput platform, microarray technology has been widely used to measure gene expression profiles in various tumours to find pivotal genes and pathways as potential molecular targets. In our previous study, we identified several critical DEGs and pathways, such as EGFR and immune response, through bioinformatics analysis of microarray data [35]. In the present study, we acquired the original cervical cancer microarray dataset GSE7803 and identified 1035 significantly up-regulated DEGs and 794 significantly down-regulated DEGs between normal and cancerous cervix samples. Then, we selected 887 DEGs with fold changes over 2 for subsequent bioinformatics analysis.
GO term enrichment analysis showed that the up-regulated DGEs were significantly enriched in cell cycle process, chromosomes, and nuclear and DNA helicase activity, suggesting that some of these DEGs could locate in the nucleus and be involved in cell cycle processes to promote cell proliferation by enhancing DNA helicase activity in cervical cancer. At present, several studies have demonstrated that the up-regulated DEGs CCNE1, CDKN3, SOX4, CDK1 and MDC1 could function as oncogenes involved in the cell cycle during the progression of cervical cancer [36-40]. In contrast, the down-regulated DEGs were significantly enriched in development and differentiation processes, extracellular regions and enzyme inhibitor activity, indicating that some of these DEGs could localize to the extracellular region, inhibit peptidase activity and be involved in cell differentiation, which could play a suppressive role in cervical cancer. For example, the significantly down-regulated DEG serpin peptidase inhibitor (SERPINB5) can function as a tumour suppressor to repress the invasion and metastasis of cancer cells [41]. In addition, the down-regulated DEG Cystatin M (CST6) can act as an inhibitor of lysosomal cysteine proteases, which can contribute to cancer cell invasion by degrading the extracellular matrix [42-46]. Therefore, some of the down-regulated DEGs could be promising candidate genes of antitumour drugs. KEGG pathway enrichment analysis found that some DEGs were significantly enriched in cell cycle, DNA replication, the p53 signalling pathway, pathways in cancer and oocyte meiosis. Mine KL et al. demonstrated that cell cycle and antiviral genes could function as major drivers of cervical cancer [47]. Furthermore, some studies found that multiple abnormally expressed genes, such as microRNAs, CYB5D2 and oral cancer overexpressed 1 (ORAOV1), could regulate cell cycle to influence cervical cancer progression, which highlighted the biological significance of the cell cycle in cervical cancer [48-51]. MCM2-7, which was significantly enriched in DNA replication (as shown in Figure 5), played a key role in cell cycle progression by licensing DNA replication only once per cell cycle, and it was highly expressed to promote the malignant proliferation of cervical cancer cells [52]. P53, a pivotal tumour suppressor gene, and its signalling pathways were involved in the proliferation and apoptosis of cervical cancer cells and displayed relatively high prognostic value [53-55]. In brief, the enriched GO terms and KEGG pathways explained the specific molecular mechanisms of cervical cancer to some extent.
The four enrichment plots from the GSEA results, CC: Cervical Cancer. (A) RNA splicing (P = 0.002, FDR = 0.186); (B) DNA geometric change (P < 0.01, FDR = 0.217); (C) gene silencing (P = 0.008, FDR = 0.195); (D) negative regulation of chromosome organization (P < 0.01, FDR = 0.234).
We then constructed a PPI network with DEGs to screen the top 9 hub genes with a relatively high degree of connectivity, including down-regulated TSPO, CCND1, and FOS and up-regulated CDK1, TOP2A, CCNB1, PCNA, BIRC5 and MAD2 L1. These hub genes exerted a considerable influence on cervical cancer initiation and progression from different sides. TSPO had the highest degree of connectivity; it is an 18-kD protein that plays an important role in various cellular processes, such as inducing cell apoptosis and cell cycle arrest [56-58]. Therefore, the down-regulation of TSPO could contribute to the anti-apoptosis and proliferation of cervical cells. CCND1 was significantly down-regulated according to our analysis, and this result was consistent with the studies performed by Skomedal H and Bae DS [59, 60]. However, three microRNAs, including microRNA-2861, microRNA-195 and microRNA-202, have been demonstrated to inhibit cell proliferation and progression in cervical cancer by directly targeting CCND1, suggesting that CCND1 was a tumour promoter [61-63]. Consequently, more investigation is needed to determine the function of down-regulated CCND1 in cervical cancer. The significantly down-regulated FOS gene has been shown to have tumour suppressor activity and could inhibit cell cycle progression and tumour formation [64]. Nevertheless, on the contrary, other studies have demonstrated that FOS could function as a proto-oncogene to facilitate tumourigenesis, showing over-expression in various cancers, including invasive cervical cancer [65-67]. Therefore, additional efforts are needed to further confirm the molecular mechanism of FOS in cervical cancer. As a mitotic checkpoint protein, MAD2L1 goes hand-in-hand with cell cycle in various cancers [68-71]. In conclusion, the top 9 hub DEGs, in different aspects, deeply influence the progression of cervical cancer, including cell growth, proliferation, invasion and metastasis, suggesting that some up-regulated DEGs have the potential to be effective molecular targets. Module analysis of the PPI network further indicated that the progression of cervical cancer was significantly associated with cell cycle and DNA replication, which was basically consistent with what we have discussed in the above text. Cervical cancer-associated transcription profiling data in the TCGA database validated the expression changes of key DEGs, including the top 9 DEGs with a high degree of connectivity in PPI network and 8 module hub genes, further demonstrating the high performance of DEG significance analysis based on R (Figure 7).
GSEA results further confirmed the identified pathways and simultaneously found some new biological processes DEGs significantly enriched such as RNA splicing and Gene silencing [72, 73]. Mole S, et al demonstrated that HPV16 E2 as a transcription factor could regulate the expression of RNA splicing factors SF2/ASF, SRp20 and SC35 to facilitate cervical cancer progression [74]. Gene silencing of oncogenes such as UHRF1 and MAPK1 could inhibit cell proliferation and metastasis [75, 76].
These new biological processes identified by GSEA helped us gain a better knowledge of molecular mechanism existing in cervical cancer. Finally, based on GO term enrichment analysis, KEGG pathway analysis, PPI network module analysis and GSEA results, we found that some obvious similarities existed in the different analysis results. By comprehensive analysis, we confirmed that cell cycle, including DNA replication, chromosome segregation, organelle fission, cell cycle phase transition and so on, was a key biological process and a critical driver in cervical cancer initiation and progression.
As high-throughput technologies develops fast, the cost of microarray and sequencing are reducing rapidly. As a result, there are more and bigger high-throughput data sets from various chips or sequencers such as Affymetrix and Agilent chips in cervical cancer research field. In this article, we could not find more new molecular targets for cervical cancer due to the limitation of the bioinformatics analysis on single data set. However, we performed a comprehensive analysis and presented more details about our finding, which have been partially confirmed by some other research groups [77-79]. In future, the integrated analysis on multiple data sets could help a lot on the identification of more key targets.
Conclusion
In conclusion, we conducted a comprehensive bioinformatics analysis on microarray data of cervical cancer. Pivotal DEGs and pathways were identified and screened to provide a theoretical basis for potential drug target discovery and the molecular mechanisms of cervical cancer. Nevertheless, additional relevant studies are needed to further confirm the identified DEGs and pathways in cervical cancer.
Acknowledgements
This work was supported by the Open Sharing Fund for the Large-scale Instruments And Equipments of Central South University, Hunan Provincial Innovation Foundation for Postgraduates, and Fundamental Research Funds for the Central Universities of Central South University.
Authors' contributions
Xuan Wu, Chaoyang Zhang and Guancheng Li conceived the study, manuscript, and stastics analysis; Li Peng, Yaqin Zhang, Shilian Chen and Qian Lei assistance revisions of the manuscript; All authors read and approved the final manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Torre LA, Bray F, Siegel RL. et al. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65:87-108
2. Appleby P, Beral V, Franceschi S. et al. Cervical carcinoma and reproductive factors: collaborative reanalysis of individual data on 16,563 women with cervical carcinoma and 33,542 women without cervical carcinoma from 25 epidemiological studies. Int J Cancer. 2006;119:1108-1124
3. Appleby P, Beral V, Berrington de Gonzalez A. et al. Cervical cancer and hormonal contraceptives: collaborative reanalysis of individual data for 16,573 women with cervical cancer and 35,509 women without cervical cancer from 24 epidemiological studies. Lancet. 2007;370:1609-1621
4. Moreno V, Bosch FX, Munoz N. et al. Effect of oral contraceptives on risk of cervical cancer in women with human papillomavirus infection: the IARC multicentric case-control study. Lancet. 2002;359:1085-1092
5. Munoz N, Franceschi S, Bosetti C. et al. Role of parity and human papillomavirus in cervical cancer: the IARC multicentric case-control study. Lancet. 2002;359:1093-1101
6. Plummer M, Herrero R, Franceschi S. et al. Smoking and cervical cancer: pooled analysis of the IARC multi-centric case-control study. Cancer Causes Control. 2003;14:805-814
7. Burk RD, Chen Z, Saller C. et al. Cancer Genome Atlas Research Network. Integrated genomic and molecular characterization of cervical cancer. Nature. 2017;543:378-384
8. van Dam PA, van Dam PJ, Rolfo C. et al. In silico pathway analysis in cervical carcinoma reveals potential new targets for treatment. Oncotarget. 2016;7:2780-95
9. de Freitas AC, Gomes-Leitao MC, Coimbra EC. Prospects of molecularly-targeted therapies for cervical cancer treatment. Curr Drug Targets. 2015;16:77-91
10. Duenas-Gonzalez A, Serrano-Olvera A, Cetina L, Coronel J. New molecular targets against cervical cancer. Int J Womens Health. 2014;6:1023-1031
11. Martin-Hirsch PL, Wood NJ. Cervical cancer. BMJ Clin Evid. 2011;2011:0818
12. Nannini M, Pantaleo MA, Maleddu A. et al. Gene expression profiling in colorectal cancer using microarray technologies: results and perspectives. Cancer Treat Rev. 2009;35:201-209
13. Scotto L, Narayan G, Nandula SV. et al. Identification of copy number gain and overexpressed genes on chromosome arm 20q by an integrative genomic approach in cervical cancer: potential role in progression. Genes Chromosomes Cancer. 2008;47:755-765
14. Zhai Y, Kuick R, Nan B. et al. Gene expression analysis of preinvasive and invasive cervical squamous cell carcinomas identifies HOXC10 as a key mediator of invasion. Cancer Res. 2007;67:10163-10172
15. Medina-Martinez I, Barron V, Roman-Bassaure E. et al. Impact of gene dosage on gene expression, biological processes and survival in cervical cancer: a genome-wide follow-up study. PLoS One. 2014;9:e97842
16. Subramanian A, Tamayo P, Mootha VK. et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102:15545-15550
17. Yang M, Zhai X, Xia B, Wang Y, Lou G. Long noncoding RNA CCHE1 promotes cervical cancer cell proliferation via upregulating PCNA. Tumour Biol. 2015;36:7615-7622
18. Simon NE, Schwacha A. The Mcm2-7 replicative helicase: a promising chemotherapeutic target. 2014. 2014:549719
19. You Z, Komamura Y, Ishimi Y. Biochemical analysis of the intrinsic Mcm4-Mcm6-mcm7 DNA helicase activity. Mol Cell Biol. 1999;19:8003-8015
20. Miyazaki T, Arai S. Two distinct controls of mitotic cdk1/cyclin B1 activity requisite for cell growth prior to cell division. Cell Cycle. 2007;6:1419-1425
21. Takizawa CG, Morgan DO. Control of mitosis by changes in the subcellular location of cyclin-B1-Cdk1 and Cdc25C. Curr Opin Cell Biol. 2000;12:658-665
22. Yamamoto G, Irie T, Aida T. et al. Correlation of invasion and metastasis of cancer cells, and expression of the RAD21 gene in oral squamous cell carcinoma. Virchows Arch. 2006;448:435-441
23. Beckouet F, Hu B, Roig MB. et al. An Smc3 acetylation cycle is essential for establishment of sister chromatid cohesion. Mol Cell. 2010;39:689-699
24. Gnjatic S, Cao Y, Reichelt U. et al. NY-CO-58/KIF2C is overexpressed in a variety of solid tumors and induces frequent T cell responses in patients with colorectal cancer. Int J Cancer. 2010;127:381-393
25. Asbaghi Y, Thompson LL, Lichtensztejn Z, McManus KJ. KIF11 silencing and inhibition induces chromosome instability that may contribute to cancer. Genes Chromosomes Cancer. 2017;56:668-80
26. Zhang S, Bao Y, Ju X. et al. BA-j as a novel CDK1 inhibitor selectively induces apoptosis in cancer cells by regulating ROS. Sci Rep. 2015;5:13626
27. Chen ZY, Liang K, Lin Y, Yang F. Study of the UTMD-based delivery system to induce cervical cancer cell apoptosis and inhibit proliferation with shRNA targeting Survivin. Int J Mol Sci. 2013;14:1763-1777
28. Kogo R, How C, Chaudary N. et al. The microRNA-218~Survivin axis regulates migration, invasion, and lymph node metastasis in cervical cancer. Oncotarget. 2015;6:1090-1100
29. Li QX, Zhao J, Liu JY. et al. Survivin stable knockdown by siRNA inhibits tumor cell growth and angiogenesis in breast and cervical cancers. Cancer Biol Ther. 2006;5:860-866
30. Wu SF, Zhang JW, Qian WY. et al. Altered expression of survivin, Fas and FasL contributed to cervical cancer development and metastasis. Eur Rev Med Pharmacol Sci. 2012;16:2044-2050
31. Zhou XL, Wang M. Expression levels of survivin, Bcl-2, and KAI1 proteins in cervical cancer and their correlation with metastasis. Genet Mol Res. 2015;14:17059-17067
32. Isaacs RJ, Davies SL, Sandri MI. et al. Physiological regulation of eukaryotic topoisomerase II. Biochim Biophys Acta. 1998;1400:121-137
33. Isaacs RJ, Harris AL, Hickson ID. Regulation of the human topoisomerase IIalpha gene promoter in confluence-arrested cells. J Biol Chem. 1996;271:16741-16747
34. Watt PM, Hickson ID. Structure and function of type II DNA topoisomerases. Biochem J. 1994;303:681-695
35. Zhang C, Peng L, Zhang Y. et al. The identification of key genes and pathways in hepatocellular carcinoma by bioinformatics analysis of high-throughput data. Med Oncol. 2017;34:101
36. Berumen J, Espinosa AM, Medina I. Targeting CDKN3 in cervical cancer. Expert Opin Ther Targets. 2014;18:1149-1162
37. Luo Y, Wu Y, Peng Y. et al. Systematic analysis to identify a key role of CDK1 in mediating gene interaction networks in cervical cancer development. Ir J Med Sci. 2016;185:231-239
38. Sun R, Jiang B, Qi H. et al. SOX4 contributes to the progression of cervical cancer and the resistance to the chemotherapeutic drug through ABCG2. Cell Death Dis. 2015;6:e1990
39. Yuan C, Bu Y, Wang C. et al. NFBD1/MDC1 is a protein of oncogenic potential in human cervical cancer. Mol Cell Biochem. 2012;359:333-346
40. Zubillaga-Guerrero MI, Alarcon-Romero Ldel C, Illades-Aguiar B. et al. MicroRNA miR-16-1 regulates CCNE1 (cyclin E1) gene expression in human cervical cancer cells. Int J Clin Exp Med. 2015;8:15999-16006
41. Sheng S, Carey J, Seftor EA. et al. Maspin acts at the cell membrane to inhibit invasion and motility of mammary and prostatic cancer cells. Proc Natl Acad Sci U S A. 1996;93:11669-11674
42. Buck MR, Karustis DG, Day NA, Honn KV, Sloane BF. Degradation of extracellular-matrix proteins by human cathepsin B from normal and tumour tissues. Biochem J. 1992;282( Pt 1):273-278
43. Maciewicz RA, Wotton SF, Etherington DJ, Duance VC. Susceptibility of the cartilage collagens types II, IX and XI to degradation by the cysteine proteinases, cathepsins B and L. FEBS Lett. 1990;269:189-193
44. Mai J, Sameni M, Mikkelsen T, Sloane BF. Degradation of extracellular matrix protein tenascin-C by cathepsin B: an interaction involved in the progression of gliomas. Biol Chem. 2002;383:1407-1413
45. Ni J, Abrahamson M, Zhang M. et al. Cystatin E is a novel human cysteine proteinase inhibitor with structural resemblance to family 2 cystatins. J Biol Chem. 1997;272:10853-10858
46. Sotiropoulou G, Anisowicz A, Sager R. Identification, cloning, and characterization of cystatin M, a novel cysteine proteinase inhibitor, down-regulated in breast cancer. J Biol Chem. 1997;272:903-910
47. Mine KL, Shulzhenko N, Yambartsev A. et al. Gene network reconstruction reveals cell cycle and antiviral genes as major drivers of cervical cancer. Nat Commun. 2013;4:1806
48. Jiang L, Zeng X, Wang Z. et al. Oral cancer overexpressed 1 (ORAOV1) regulates cell cycle and apoptosis in cervical cancer HeLa cells. Mol Cancer. 2010;9:20
49. Mou Z, Xu X, Dong M, Xu J. MicroRNA-148b Acts as a Tumor Suppressor in Cervical Cancer by Inducing G1/S-Phase Cell Cycle Arrest and Apoptosis in a Caspase-3-Dependent Manner. Med Sci Monit. 2016;22:2809-2815
50. Xie Y, Shen YT, Kapoor A. et al. Dataset on the effects of CYB5D2 on the distribution of HeLa cervical cancer cell cycle. Data Brief. 2016;6:811-816
51. Zhao JL, Zhang L, Guo X. et al. miR-212/132 downregulates SMAD2 expression to suppress the G1/S phase transition of the cell cycle and the epithelial to mesenchymal transition in cervical cancer cells. IUBMB Life. 2015;67:380-394
52. Das M, Prasad SB, Yadav SS. et al. HPV-type-specific response of cervical cancer cells to cisplatin after silencing replication licensing factor MCM4. Tumour Biol. 2015;36:9987-9994
53. Li Q, Li X, Wang C. Uc.206 regulates cell proliferation and apoptosis by targeting P53 in cervical cancer cells. Neoplasma. 2016;63:411-418
54. Sharma G, Rana NK, Singh P. et al. p53 dependent apoptosis and cell cycle delay induced by heteroleptic complexes in human cervical cancer cells. Biomed Pharmacother. 2017;88:218-231
55. Zhou R, Wei C, Liu J, Luo Y, Tang W. The prognostic value of p53 expression for patients with cervical cancer: a meta analysis. Eur J Obstet Gynecol Reprod Biol. 2015;195:210-213
56. Decaudin D. Peripheral benzodiazepine receptor and its clinical targeting. Anticancer Drugs. 2004;15:737-745
57. Papadopoulos V, Baraldi M, Guilarte TR. et al. Translocator protein (18kDa): new nomenclature for the peripheral-type benzodiazepine receptor based on its structure and molecular function. Trends Pharmacol Sci. 2006;27:402-409
58. Sutter AP, Maaser K, Hopfner M. et al. Specific ligands of the peripheral benzodiazepine receptor induce apoptosis and cell cycle arrest in human esophageal cancer cells. Int J Cancer. 2002;102:318-327
59. Bae DS, Cho SB, Kim YJ. et al. Aberrant expression of cyclin D1 is associated with poor prognosis in early stage cervical cancer of the uterus. Gynecol Oncol. 2001;81:341-347
60. Skomedal H, Forus A, Holm R. Deregulation of D-type cyclins in uterine cancers. Cyclin D1/D3 is differentially expressed in cervical cancer. Anticancer Res. 2003;23:3929-3935
61. Li Z, Wang H, Wang Z, Cai H. MiR-195 inhibits the proliferation of human cervical cancer cells by directly targeting cyclin D1. Tumour Biol. 2016;37:6457-6463
62. Xu J, Wan X, Chen X. et al. miR-2861 acts as a tumor suppressor via targeting EGFR/AKT2/CCND1 pathway in cervical cancer induced by human papillomavirus virus 16 E6. Sci Rep. 2016;6:28968
63. Yi Y, Li H, Lv Q. et al. miR-202 inhibits the progression of human cervical cancer through inhibition of cyclin D1. Oncotarget. 2016;7:72067-72075
64. Mikula M, Gotzmann J, Fischer AN. et al. The proto-oncoprotein c-Fos negatively regulates hepatocellular tumorigenesis. Oncogene. 2003;22:6725-6738
65. Cheung TH, Leung JO, Chung TK. et al. c-fos overexpression is associated with the pathoneogenesis of invasive cervical cancer. Gynecol Obstet Invest. 1997;43:200-203
66. Chiu R, Boyle WJ, Meek J. et al. The c-Fos protein interacts with c-Jun/AP-1 to stimulate transcription of AP-1 responsive genes. Cell. 1988;54:541-552
67. Fan Q, He M, Deng X. et al. Derepression of c-Fos caused by microRNA-139 down-regulation contributes to the metastasis of human hepatocellular carcinoma. Cell Biochem Funct. 2013;31:319-324
68. Liang X, Liu Y, Zeng L. et al. miR-101 inhibits the G1-to-S phase transition of cervical cancer cells by targeting Fos. Int J Gynecol Cancer. 2014;24:1165-1172
69. Ko YH, Roh JH, Son YI. et al. Expression of mitotic checkpoint proteins BUB1B and MAD2L1 in salivary duct carcinomas. J Oral Pathol Med. 2010;39:349-355
70. Percy MJ, Myrie KA, Neeley CK. et al. Expression and mutational analyses of the human MAD2L1 gene in breast cancer cells. Genes Chromosomes Cancer. 2000;29:356-362
71. Scintu M, Vitale R, Prencipe M. et al. Genomic instability and increased expression of BUB1B and MAD2L1 genes in ductal breast carcinoma. Cancer Lett. 2007;254:298-307
72. Zhou C, Shen L, Mao L. et al. miR-92a is upregulated in cervical cancer and promotes cell proliferation and invasion by targeting FBXW7. Biochem Biophys Res Commun. 2015Feb27;458(1):63-9
73. Huang J, Liou YL, Kang YN. et al. Real-time colorimetric detection of DNA methylation of the PAX1 gene in cervical scrapings for cervical cancer screening with thiol-labeled PCR primers and gold nanoparticles. Int J Nanomedicine. 2016Oct12;11:5335-5347
74. Mole S, McFarlane M, Chuen-Im T. et al. RNA splicing factors regulated by HPV16 during cervical tumour progression. J Pathol. 2009;219:383-391
75. Ge TT, Yang M, Chen Z. et al. UHRF1 gene silencing inhibits cell proliferation and promotes cell apoptosis in human cervical squamous cell carcinoma CaSki cells. J Ovarian Res. 2016;9:42
76. Li XW, Tuergan M, Abulizi G. Expression of MAPK1 in cervical cancer and effect of MAPK1 gene silencing on epithelial-mesenchymal transition, invasion and metastasis. Asian Pac J Trop Med. 2015;8:937-943
77. Wu K, Yi Y, Liu F. et al. Identification of key pathways and genes in the progression of cervical cancer using bioinformatics analysis. Oncology Letters. 2018;16:1003-1009
78. Ge Y, Zhang C, Xiao S. et al. Identification of differentially expressed genes in cervical cancer by bioinformatics analysis. Oncology Letters. 2018;16:2549-2558
79. Wang S, Chen X. Identification of potential biomarkers in cervical cancer with combined public mRNA and miRNA expression microarray data analysis. Oncology Letters. 2018;16:5200-5208
Author contact
![]() Corresponding authors: Guancheng Li: ligc61edu.cn and Chaoyang Zhang: chaoyang.zhangde
Corresponding authors: Guancheng Li: ligc61edu.cn and Chaoyang Zhang: chaoyang.zhangde
Received 2019-2-17
Accepted 2019-5-6
Published 2019-6-2