ISSN: 1449-1907International Journal of Medical Sciences
- Current issue
- Volume 21; 2024
- Volume 20; 2023
- Volume 19; 2022
- Volume 18; 2021
- Volume 17; 2020
- Archive
- Cover images
- Index & coverage
- Cover suggestion
- Special issues
Introduction
Materials and Methods
Results
Discussion
Conclusions
Acknowledgements
References
Author Biography

Int J Med Sci 2019; 16(1):135-144. doi:10.7150/ijms.29134 This issue Cite
Research Paper
Bilirubin Attenuates ER Stress-Mediated Inflammation, Escalates Apoptosis and Reduces Proliferation in the LS174T Colonic Epithelial Cell Line
Rohit Gundamaraju1 ![]() , Ravichandra Vemuri1, Wai Chin Chong1, Andrew Cameron Bulmer2, Rajaraman Eri1
, Ravichandra Vemuri1, Wai Chin Chong1, Andrew Cameron Bulmer2, Rajaraman Eri1
1. School of Health Sciences, University of Tasmania, Launceston, Tasmania, Australia
2. School of Medical Science and Menzies Health Institute Queensland, Griffith University, Gold Coast, Qld, Australia
Abstract

Mildly elevated serum unconjugated bilirubin (UCB) concentrations are associated with protection against disease conditions underpinned by cellular and metabolic stress. To determine the potential therapeutic efficacy of UCB we tested it in an in vitro model of gut inflammation. Tunicamycin TUN (10 µg/mL) was used to induce endoplasmic reticular stress (ERS) affecting N-glycosylation in LS174T cells. Cultured cells were investigated with addition of UCB at doses 0.1, 1 and 10µM (resulting in bilirubin:albumin ratios of 0.325-0.003)against ER stress-mediated effects including inflammation, cell survival (determined by apoptosis) and proliferation. Gene expression of ER stress markers (Grp78, Perk, XBP1 and ATF6) were evaluated in addition to cytokine concentrations in media after six hours of treatment. We then verified the potential role of UCB in executing programmed cell death via PARP, Caspase3 and Annexin V assays and further explored cell proliferation using the Click-iT EdU assay. A dose of 10µM UCB most potently reduced tunicamycin-mediated effects on enhanced UPR markers, inflammatory cytokines and proliferation; however all the doses (i.e.0.1-10µM) reduced the expression of ER stress and inflammatory markers Grp78, NLRP3, IL1-b, XBP1, PERK and ATF6. Furthermore, media concentrations of pro-inflammatory cytokines IL-8, IL-4 and TNFα decreased and the anti-inflammatory cytokine IL-10 increased (P<0.05). A dose of 10µM UCB initiated intrinsic apoptosis via Caspase 3 and in addition reduced cellular proliferation. Collectively, these data indicate that co treatment with UCB resulted in reducing ER stress response to TUN in gastrointestinal epithelial cells, reduced the subsequent inflammatory response, induced cancer cell death and decreased cellular proliferation. These data suggest that mildly elevated circulating or enteric UCB might protect against gastrointestinal inflammatory disorders.
Keywords: ER stress, colon cancer, inflammation, cell proliferation, apoptosis
Introduction
The prime function of ER stress is to activate specific enzymes and transcription factors in order to maintain homeostasis within the endoplasmic reticulum (ER). However, initiation of inflammatory signalling or conditions typified by increased programmed cell death (apoptosis) occurs if ER stress becomes chronic [1]. Disturbances in ER function trigger the unfolded protein response (UPR), a tightly orchestrated collection of intracellular signal transduction reactions designed to restore protein homeostasis. A critical initiator of the UPR is Grp78. Grp78 binds to three major molecules, namely IRE1α, PERK and ATF6. IRE1α-mediated signalling orchestrates cell-fate decisions during stress, i.e. it signals the intensity of cellular stress thereby clearly showing that ER stress has a role in cell survival [2]. Translation of proteins occurs in the ER and there is increased protein folding and transport during conditions such as carcinogenesis. The ER stress response, which is cytoprotective, is involved in supporting tumour growth and adaptation [3]. Cancerous cells adapt to prevent ER stress-mediated apoptosis in order to survive by expression of inhibition of apoptosis (IAP) proteins [4]. Novel therapeutics against ER stress are required in order to arrest cancer growth by increasing apoptosis and decreasing cellular proliferation. Some initial targets included inhibition of reactive oxygen species (ROS) generation and reducing ER stress [5]. Cells deficient in PERK have comparatively less ROS compared to those with PERK, demonstrating that loss of PERK has an impact on ROS-induced ER stress, leading to apoptosis. Oxidative stress in this scenario directed PERK-mediated ER stress signaling [5]. Antioxidants, including polyphenols, induce cancer cell death via various pathways including NF-kB/p53, suppression of MMP-2 expression, ERK and c-Jun N-terminal kinase (JNK). They can also target angiogenesis- related pathways including PI3K/Akt/Fork head box O (FoxO) [6]. As ROS inhibition influences ER stress pathways, PERK, which is one of the regulators of the growth of cancer cells, was chosen as a target. PERK inhibition via DNA damage checkpoint prevented mammary carcinoma cells from forming solid tumours in vivo, denoting its role in tumour initiation and redox homeostasis [3], and suggesting that compounds with antioxidant capacity might represent effective treatments to prevent ER stress and cellular proliferation. Benzodiazepines [7] were used to attenuate ER stress via Grp78 reduction in neural stem cells. Kifunensine mannosidase inhibitors [8, 9] were employed to decrease the ER stress via impeding CHOP expression in cervical cancer cells. Stocker and colleagues demonstrated that the endogenous heme catabolite unconjugated bilirubin (UCB) exhibited potent antioxidant effects and inhibited lipid oxidation in vitro [10, 11]. UCB also scavenges oxidants (i.e. hydrogen peroxide and other peroxides) and multiple radical species [12] demonstrating broad specificity in its ROS-neutralising effects [13]. Bilirubin's antioxidant capacity and ability to inhibit lipid peroxidation are supported in vivo, with serum bilirubin positively correlating with total anti-oxidant capacity in plasma [14, 15] and negatively correlating with susceptibility to copper induced lipid oxidation [16].
Endogenously elevated UCB is also associated with protection from circulating oxidative stress in an animal model of adenine-induced renal failure [17]. Biliveridin (BV), which is chemically reduced to bilirubin in vivo, also protects against vascular injury, ischemia reperfusion injury [18] and inhibits Toll-like receptor4 (TLR4) activation in mouse macrophages [19, 20]. BV also ameliorates complement-mediated inflammation and reduces pro-inflammatory cytokine expression including TNF-α and IL-6 [19]. A strong evidence suggests that mild hyperbilirubinemia may exhibit protection against diabetic vascular complications and also impede oxidative stress [21]. A study by Barateiro et al., has demonstrated the interrelation between UCB and ERS and associated cascade of events [22]. Despite these findings which describe antioxidant and anti-inflammatory effects, very little is known regarding bilirubin's impact on gut health. Therefore, we aimed to determine whether bilirubin, which is produced during stress conditions and possesses antioxidant activity, might prevent ER stress, inhibit inflammatory responses and encourage apoptosis in LS174T cells.
Materials and Methods
Unconjugated bilirubin
UCB was obtained from Frontier Scientific (Utah, USA) and was added to media without further purification. All UCB solutions were protected from light using foil and replaced daily in cell culture. UCB was dissolved in 0.1% DMSO. UCB is carried by albumin with the bilirubin: albumin concentration dictating its toxicity in vivo. Therefore, we aimed to test UCBs efficacy at bilirubin to albumin ratios of <0.5.
The concentration of albumin in FBS was calculated using an album in specific kit (ALB2) and a Cobas Integra 400+ clinical chemistry analyzer (Roche Diagnostics, North Ryde, NSW, Australia). Given that the FBS concentration in media was 10%v/v, FBS albumin concentrations were divided by 10 to obtain the bilirubin: albumin ratio in culture. To achieve this, the albumin concentration was converted to a molar concentration, assuming a molecular mass for albumin of 66.5 kDa. The albumin concentration in FBS equaled 20.5 g/L (308 µM), and therefore equaled 30.8 µM in media. This resulted in bilirubin: albumin ratios of 0.325, 0.032 and 0.003 for 10,1 and 0.1 µM UCB concentrations in media.
Cell culture
Human colorectal adenocarcinoma cells LS174T (ATCC) were cultured in RPMI (1640medium with L-glutamine (Life Technologies),supplemented with 10% fetal bovine serum (Gibco, AUS); penicillin (1000 U/mL) and streptomycin (1000 ug/L) (Gibco BRL, AUS).Cells were grown till they reached 100 % confluency at 370C5%CO2, In humidified conditions. Media was replaced every 2 days. Cells were then harvested by 0.25% TrypLe express (Life Technologies, AUS) and detached cells were washed twice in cell culture media and cell number and viability assessed by Countess® cell counter (Life Technologies, AUS).
ERS
Tunicamycin (TUN) was procured from Sigma, AUS in a solution form (DMSO solution) at a concentration of 5mg/mL. The concentration was finalized to 10µg/mL before adding to the cells.
RNA Extraction and cDNA synthesis
RNA was extracted from LS174T cells with a cell population of 3x105using RNeasy Mini Kits (Qiagen, AUS) with gDNA removed using the RNase-Free DNase set (Qiagen, AUS). RNA (quantitative and qualitative) analysis was performed using an Experion automated electrophoresis system (Bio-Rad Laboratories, AUS) and RNA samples with an RQI > 7.0 were considered suitable for expression analysis. An iScript cDNA synthesis kit (Bio-Rad Laboratories, AUS) was used to transcribe one microgram of total RNA to cDNA as per the manufacturer's protocol.
RT-PCR
The RT-PCR was performed using Taqman ® probes (Life Technologies, AUS) for GAPDH (Hs03929097_gl), ATF6 (Hs00232586_m1), XBP1 (Hs00231936_m1), GRP78 (Hs0060719_gH), CHOP (Hs00358796_g1), NLRP3 (Hs00918082_m1), IL1β (Hs01555410_m1) and PERK (Hs00984006_m1). The RT-reaction mixture consisted of 40ng of cDNA, Taqman Fast Advanced Master mix (Life Technologies, AUS), 1µL of gene specific probe/ total volume of 20µL. The reactions were run in duplicate on an RT-PCR machine (StepOne Plus-Life Technologies).Thermo cycling conditions included: 90oC for 20s, 40 cycles at 95oC for 1 s and 60oC for 20s. Gene expression was quantified using the comparative (ΔΔCT) method where the threshold cycle (CT) for each gene was normalized to reference gene GAPDH.
Cytokine quantification by Bio-Plex
LS174T cells were used for the quantification of IL-8, IL-4, TNF-α and IL-10 cytokine levels. Cells were subjected to the respective treatments (tunicamycin and bilirubin) and were cultured in 12-well culture plates (Greiner, AUS). Wells were seeded at a density of 3.0 × 105 cells in 2.0 mL of medium and incubated overnight at 37oC/5% CO2 to allow the cells to adhere. Medium was replaced the next day Tunicamycin (10µg/mL in DMSO) and 4PBA (40µg/mL) were incubated for 6 hours at 37oC/5% CO2. The treatment groups then consisted of: no-treatment (LS174T cells with media), tunicamycin only, tunicamycin+ UCB 10µM and UCB 10µM only. After 6 hr of treatment, the media from each well was collected and used for quantification of cytokines using Bio-Plex® Pro human cytokine assay kits (Bio-Rad®), according to the manufacturer's protocol. Briefly, 50 µL of cytokine beads were added to the 96-well plate and incubated for 30 min before washing twice with wash buffer. Then, 50 µL of each standard, blank and samples were added to the respective wells and incubated at room temperature on a shaker at 850 rpm for 30 min. After incubation, the wells were washed thrice and 25 µL of detection antibody was added to each well and incubated at room temperature on a shaker at 850 rpm for 30 min. Later, 50 µL of streptavidin-PE was added to each well and incubated at room temperature in a shaker at 850 rpm for 10 min. After three washes, 125 μL assay buffer was added to each well and incubated at room temperature for 30 sec. After incubation, the plates were read on the Bio-Plex® 200 system and data was analyzed in Bio-Plex Data ProTM Software. All the experiments were performed in triplicate.
Apoptosis assays
Caspase- 3 assay
The caspase-3 fluorometric assay was performed on cell lysates which were collected after the 6 hour treatment according to assay kit instructions and published research protocols [23]. This enzyme assay works based upon the hydrolysis of the caspase-3 peptide substrate (acetyl-Asp-Glu-Val-Asp or AC-DEVD) conjugated to a fluorochrome at the C-terminal Asp, resulting in the release of the fluorescent moiety. After the fluorometric treatment, the fluorescence (absolute units) was measured using the CytoFluor Multi-Well Plate Reader Series 4000 spectro-fluorometer from PerSeptive Biosystems (Framingham, MA, USA).
Annexin V assay
The Annexin-V-Fluos assay (cat. no. 11828681001, Roche Diagnostics, Zug Switzerland) was utilized to measure apoptotic (annexin V) cell populations. After the 6 hour treatment cells were incubated in incubation buffer ((10 mM HEPES at pH 7.4, 140 mMNaCL, 2.5 mM CaCl2) supplemented with annexin V-PI mix for 15 min at RT and the cells were analyzed by confocal microscopy with DAPI for nuclear staining and FITC channel (fluorescein) for visualizing apoptotic cells (Nikon AR1MP) [24].
Toxicity Assay Lactate Dehydrogenase (LDH)
After incubation with the respective treatments, the supernatants were collected for the determination of cytotoxicity by using the lactate dehydrogenase (LDH) assay. The cellular cytotoxicity was assessed by the LDH in-vitro cytotoxicity assay (TOX7, Sigma-Aldrich, St. Louis, MO, USA). The culture supernatants were centrifuged at 250×g for 4 min. An aliquot containing 50 µL of either blank (complete medium) or control (cells only) and cells with DMSO supernatants (various concentrations obtained after the respective time point incubations was mixed with 100 µL of a solution containing the LDH assay mixture (LDH substrate, LDH dye, and LDH cofactor). The mixture was then incubated at room temperature for 20 to 30 min and the reaction was quenched by the addition of 1N hydrochloric acid (15 μL). The absorbance was measured spectro-photo-metrically by using a plate reader (Spectra Max M2 microplate reader, Sunnyvale, CA, USA) at a wavelength of 490 nm. The cellular viability was examined by a Trypan Blue exclusion staining assay using a Countess Automated Cell Counter (Thermo-Fisher, Waltham, MA, USA).
Western blot of PARP assay
Total cell protein was extracted from 1 × 106 treated (6 hrs) LS174T cells by firstly washing the cells with HBSS followed by homogenization in 2 mL of RIPA buffer/10% of protease inhibitor (Sigma-Aldrich, AUS). Cell supernatant was generated by centrifugation at 12000 rpm for 20 min at 4 °C. Thirty micrograms of protein from each sample was denatured in Laemmli loading buffer (Bio-Rad Laboratories, AUS; 1:1 v/v) and separated on precast 12% SDS-PAGE gels (Bio-Rad Laboratories, AUS) followed by overnight transfer onto PVDF membranes (Millipore, AUS) at 30 mV at 4 °C. The blot was blocked with 5% non-fat milk, before being incubated with anti-GADPH (#14C10, 1:3000, Novus Biologicals, AUS), poly(ADP-ribose)polymerase (PARP), cleaved PARP (Sigma-Aldrich, Australia) overnight at 4 °C in blocking buffer. The blot was washed in Phosphate-buffered saline (PBST) and incubated with appropriate species monoclonal horseradish peroxidase- conjugated anti-IgG secondary antibodies (1:5000) for 1 h at 20 °C. Bands were visualized using Supersignal West Pico chemiluminesce kit (Thermo Scientific, AUS), digitized and band intensities determined using a Fuji LAS-3000 Imager (Fuji Life Sciences, Japan). Samples from all groups were included in individual blots to ensure accurate quantification across multiple blots.
Proliferation assay
The proliferation assay 5-ethynyl-2′-deoxyuridine (EdU)) Invitrogen, Australia) was performed as per manufacturer instruction and published protocols [25].In brief, Click-iT™ EdU Flow Cytometry Assay Kit, Invitrogen™ was added at a 50 μM final concentration. For the Click reaction, cells were collected into 3 ml of PBS containing 1% BSA, centrifuged and fixed with 100 μl of 4% para formaldehyde for 15 min. Cells were visualized using confocal microscopy (Nikon AR1MP) with DAPI as nuclear staining.
Statistical analysis
Statistical significance of the differences between groups among repeated experiments was determined by one-y and two way ANOVA and Fisher's LSD-tests using GraphPad Prism 4 software (GraphPad Software Ltd, La Jolla, CA, USA). The results are expressed as the mean values ± standard deviation. In all statistical tests, a P-value <0.05 was considered statistically significant.
Results
Effect of DMSO on LS174T cells
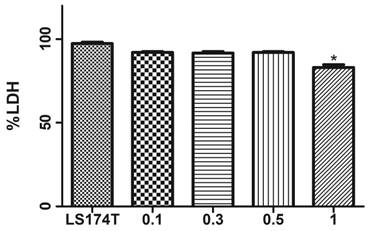
Initially to assess the toxic effects of DMSO (if any exists) against the LS174T cells, we have carried a LDH assay to verify any cell death due to toxicity. Compared to the cells alone group which has shown 99 % viability, 0.1%, 0.3% and 0.5% concentrations have shown a very minimal toxicity. 1% DMSO has exhibited about 80% toxicity. We have for this reason employed 0.5 % DMSO in our cell culture media either to dissolve TUN or UCB in our study.
Toxicity effects of DMSO on LS174T cells. Data represented in the above figure demonstrates cells alone (without DMSO) and cells in media with DMSO concentrations 0.1 %, 0.3 %, 0.5 % and 1%) Data are shown as the mean fold change ± SEM (vs. vehicle: *, p < 0.05)
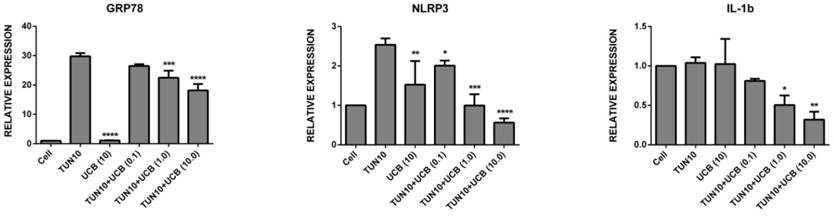
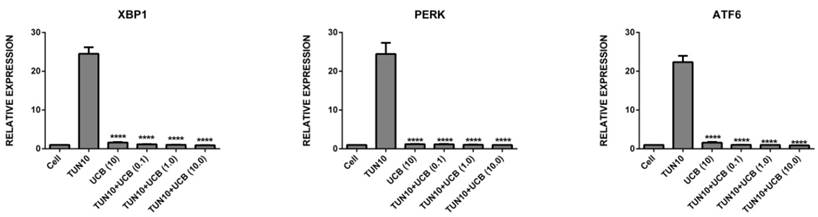
UCBreduces ER stress in LS174T colon cancer cells
First of all, we investigated the efficacy of UCB in attenuating ER stress in LS174T cells. The addition of tunicamycin 10µg/mL for six hours was used to increase mRNA expression of all the ER stress and inflammatory markers (Figure 2), namely Grp78 (29.8±2.0 fold; p<0.05), NLRP3 (2.5±0.15 fold; p<0.05), IL1-β (1.04±0.5 fold, p<0.05), XBP1 (24.5±2.0 fold; p<0.05), PERK (24.5±2.0 fold; p<0.05) and ATF6 (22.4±1.2 fold; p<0.05) (mean ± SEM; n=3) corresponding to ER stress induction. Co-treatment with UCB for six hours significantly reduced ER stress marker mRNA expression. The mRNA expression levels of the UCB alone and UCB+tunicamycin groups were as follows: Grp78 (UCB alone (1.13±3.0) UCB+tunicamycin 10µM (18.2±2.0), where there was a surprising marked decrease in the ER stress markers; NLRP3 (UCB alone (1.5±0.4) UCB+tunicamycin (0.9± 0.3) UCB+tunicamycin 10µM (0.56±0.9); IL1-β (UCB alone (no difference) UCB+tunicamycin (0.50±0.08) UCB+tunicamycin (0.32±0.07); XBP1 (UCBalone (1.6± 0.10) UCB+tunicamycin 0.1mM (1.15±0.05) UCB+tunicamycin 1mM (0.035±0.02) UCB+tunicamycin 10mM (0.92±0.03). All the bilirubin treatments exhibited significance, with PERK (UCB alone (1.21±0.4) UCB+tunicamycin 0.1 mM (1.16±0.07) UCB+tunicamycin 1mM (1.05±0.05) UCB+tunicamycin 10mM (1.0±0.01), and ATF6 (bilirubin alone (1.58±0.14) UCB+tunicamycin 0.1mM (1.04±0.015) UCB+tuncamycin 1mM (1.0±0.01) UCB+tunicamycin 10mM (0.90±0.02) (P< 0.05).
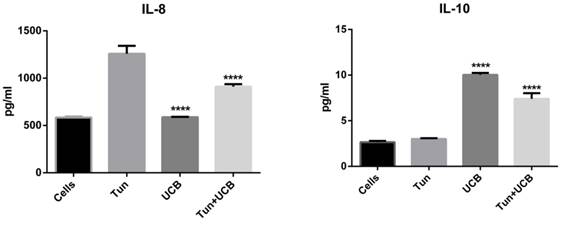
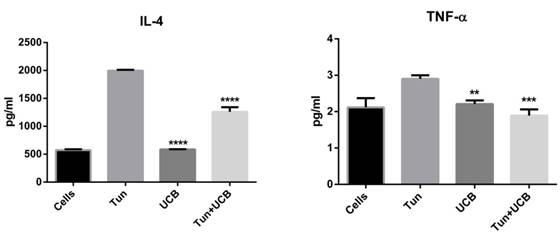
UCB ameliorates tunicamycin-mediated inflammatory responses
When LS174T cells were treated with tunicamycin, media IL-8, IL-4 and TNFα concentrations increased to 1258±90, 1997±13 and 2.9±0.1 pg/mL respectively (Figure 3). During co-treatment with UCB 10µMalone and UCB 10µM+tunicamycin, concentrations of IL-8, IL-4 and TNFα decreased significantly (p<0.05) compared to the tunicamycin group. For the IL-8 group, the levels were downregulated by 587.51±3.39 pg/mL for bilirubin 10mM and 910.273± 29.85 pg/mL for bilirubin 10mM+tunicamycin 10µg/ mL. IL-4 levels were down to 585.85±4.33 pg/mL for bilirubin 10mM and 1258.39±8.6pg/mL for bilirubin 10µM+tunicamycin 10µg/mL. TNFα was reduced to 2.20±0.1 pg/mL for bilirubin 10mM and 1.89±0.11 pg/mL for bilirubin 10mM+tunicamycin 10µg/mL. IL-10 concentrations increased from 3.0±0.10 pg/mL to 7.40±0.71 pg/mL for UCB 10µM+tunicamycin 10µg/mL.
Bilirubin increases apoptosis in the LS174T cancer cell line
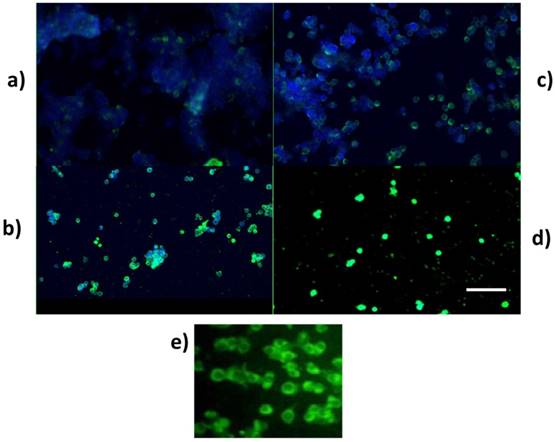
To verify the induction of apoptosis, cells after treatments were stained with Annexin V (FITC) (Figure 4). Greater apoptosis occurred in cells treated with UCB (10µM) (Figure 4d) alone compared to the UCB (10µM) and tunicamycin treated group (Figure 4b). The control group cells that were treated with the solvent control only did not show increased Annexin V positive staining. Cells treated with UCB alone (10µM) also demonstrated a change in their morphology such as condensed nucleus and accumulation of Annexin V in cytoplasm, suggesting enhanced permeability when compared to the solvent control group and tunicamycin group shown under higher magnification (Figure 4e).
mRNA expression of ER stress markers [relative mRNA expression levels are vs. control and normalized to GAPDH (n = 3). Data are shown as the mean fold change ± SEM (vs. vehicle: *, **, ***; p < 0.05].
Cytokine concentrations in the media of LS174T cell line supernatant. Groups designated with different letters are significantly different (p < 0.05) *.
Annexin V assay on LS174T cell line. a. Solvent control; b. TUN+UCB; c.TUN; d. UCB (10µM) alone; e. High magnification of UCB 10µM alone depicting disoriented nucleus. Annexin staining is denoted by green fluorescence and DAPI (nuclear staining) by blue fluorescence. Scale bar represents 200µm
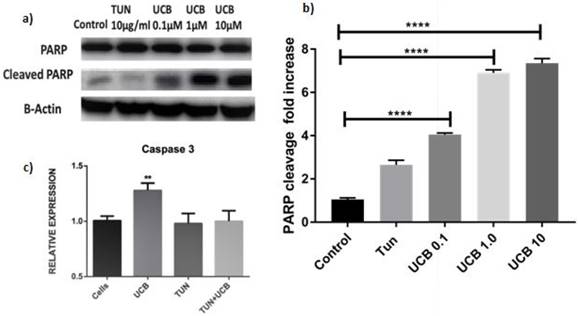
For investigating PARP activity, protein from the cells (LS174T) was obtained after treatment and processed for protein expression (Figure 5). Cleaved PARP, which is a caspase substrate activator, was detected in the UCB-treated groups, suggesting that UCB initiated apoptosis. These results were supported by Caspase 3 analysis, which showed a significant increase in Caspase 3 expression in the UCB alone group compared to tunicamycin and tunicamycin+UCB.
Bilirubin reduces ERS mediated cellular proliferation
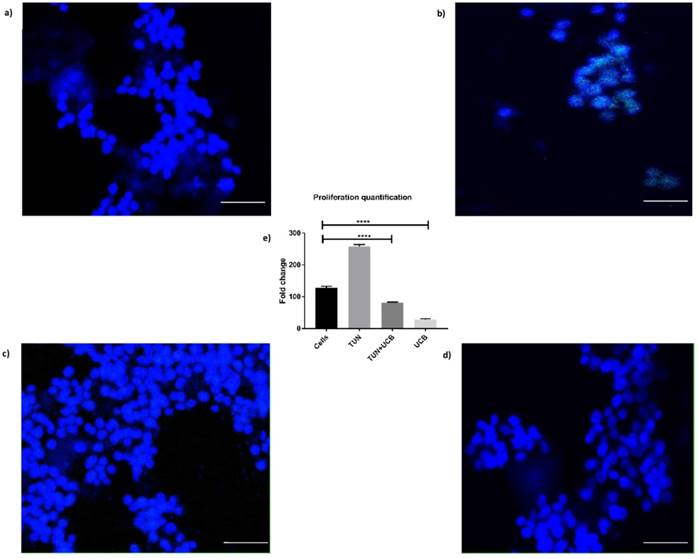
Considering the results of the apoptosis assay, we then sought to determine whether differences in cellular viability would influence proliferation. The EdU assay for in vitro proliferation was applied and assessed through DNA-synthesis and detected the incorporation of the alkyne-modified nucleoside EdU (5-Ethynyl-2′-deoxyuridine) into DNA using copper-catalysed azide-alkyne click chemistry to attach fluorescent probes. We have here included only the highest concentration of bilirubin (10µM) as it was the most significant dose observed in the previous assays. The results indicated reduced cell proliferation was highest in the TUN group (Figure 6b) showing that ERS induction in a cancer cell line leads to increased proliferation. The proliferation was reduced in UCB+ TUN treated groups, compared to the TUN only group and the cells alone (Figure 6a). The lowest proliferation was observed in the UCB alone treated group (Figure 6d). The proliferation rate was quantified in Figure 6e.
Quantification of PARP and Caspase 3 expression. a.Western blot of PARP cleavage of LS174T cells treated with TUN and UCB (0.1, 1 and 10 µM) b. PARP cleavage quantification c. Caspase 3/7 fluoremetric assay
Proliferation assay of LS174T cells. a) Cells alone; b) Proliferation in cells with TUN treatment; c) TUN+UCB; d) UCB only e) Quantification of proliferation by Image J®. Scale bar represents 100µm.
Discussion
In the present study, we have shown that a co-treatment with UCB reduces the mRNA expression of ERS and inflammation induced by TUN, increases apoptosis and reduces cellular proliferation in the tumour-derived LS174T cell line. These data are novel and suggest that bile pigments present in the gut may inhibit inflammation in the gut and related effects. The concentrations tested can also be correlated with clinical conditions of Gilbert's syndrome (GS). The serum bilirubin concentration in GS ranges between 20 and 50 µM [26], and approximates a bilirubin: albumin ratio of 0.04-0.1. The 1µM dose (bilirubin: albumin 0.032) in this study therefore most closely replicates the bilirubin:albumin ratio in GS.
The crucial role of the unfolded protein response (UPR) in numerous cancers and cancer development is well accepted and documented [27]. The UPR, which is a key player in the signalling of ERS, has various effectors including XBP1, PERK and ATF6 which are increased in many neoplasms including brain and pancreatic lesions. Furthermore, Grp78 is over-expressed in a number of cancer cell lines. The UPR is also linked to the presence of cell dormancy, secretory switch mechanisms, epithelial -to-mesenchymal transition (EMT), tumour angiogenesis and tumour autophagy which are associated with ATF6, PERK and IRE1 activation. These phenomena are characteristic of the role of ERS in colitis and colon cancer. XBP1 has been correlated to hypoxia inducible factor-1α activation thereby facilitating tumour survival, activating SNAIL (snail-related protein) thereby promoting metastasis via EMT and glucose uptake [28]. Moreover, long-standing ERS activation is related to metastasis and drug resistance and thus targeting ATF6, PERK and other ER stress responses holds potential for anti-cancer therapy [29]. We employed a similar strategy in this study whereby we initially showed that UCB inhibits tunicamycin- induced ER stress in LS174T cells. Bilirubin at a dose of 10µM reduced the expression of UPR genes Grp78, XBP1, PERK, ATF6 and inflammatory mediators such asNLRP3 and IL-1β. Previous effective therapeutic strategies are also harnessed by bitter melon extracts against ER stress in the treatment of tunicamycin- induced ERS [30].
With the present experiment results, we aimed to demonstrate the link between ER stress and inflammation induced by TUN by assessing concentrations of pro- and anti-inflammatory cytokines after six hours of co-incubation. The activation of pathways within UPR is interconnected to inflammation through mechanisms including ROS, calcium release from ER and the acute phase response [31]. For example, interleukin-8 (IL-8) may exert tumourigenic effects demonstrated in IL-8 silencing studies which reversed the tumour-like characteristics and drug resistance of HCT116 and Caco2 cells [32]. Bilirubin alone (10µM) significantly decreased the concentration of IL-8 and furthermore demonstrated similar effects in combination with tunicamycin. Targeting IL-8 and its receptor CXCR2 represents a strategic mechanism for targeting cancers and also chemosensitising tumours [33]. In the colon, IL-8 transfectants demonstrate increased proliferation and cell migration with its silencing reversing the effect [32]. Many of the pro-inflammatory cytokines are implicated in the intestinal microenvironment, including IL-4. IL-4 induces colitis and its over-expression induces acute and fatal colitis [34]. Self-renewing cancers formed from stem cells (CSC) can be resistant to chemical therapies. Blockade of IL-4 leads to initiation of apoptotic signalling in CSCs, suggesting that IL-4 could be used as one of the many potential therapeutic targets [35]. Bilirubin at a dose of 10µM decreased IL-4 concentrations in the conditioned media of LS174T cells when compared to the TUN treatment. Furthermore, IL-4 expression was accompanied by increased TNF-α expression, suggesting its active role in intestinal diseases and cancer [34, 36]. TNF-α alone could be used as a diagnostic marker for colorectal cancer and represents a promising therapeutic target [37]. Similar to IL-4, UCB reduced TNF-α secretion compared to the TUN group, where it was increased due to ER stress. These conclusions are supported by a study by Zins et al. where the human TNF-alpha gene silencing decreased mouse macrophage TNF-alpha, CSF-1, MMP-2, and VEGF-A mRNA expression when co-cultured with human cancer cells [38]. As much as reducing pro-inflammatory cytokines concentrations are important, it is also important to increase the anti-inflammatory cytokines in order to counter-regulate inflammatory responses. In the in vivo models, oral administration of IL-10 microparticles decreased polyposis in the ApcMin/+ model by suppressing the development of IL-17-producing Tregcells and inducing conventional, IL-17-negative Treg cells [39]. This finding helps to explain the therapeutic nature and role of IL-10 in colon cancer. IL-10 demonstrates immune-suppressant effects in cancer [40]. Our study could emphasise the potency of bilirubin in increasing the anti-inflammatory cytokine IL-10 in LS174T cells in accordance with a previous study where IL-10-deficient mice demonstrated increased pro-inflammatory cytokine production. In these mice, IL-10 treatment improved intestinal inflammation and aided in ameliorating disease progression [41].
Induction of apoptosis via activation of caspases is a critical feature associated with the effectiveness of potential cancer treatments [42, 43] including the use of tetrapyrrolic bile pigments in human cancer cells [44]. Apart from being used as a novel biomarker for nasopharyngeal carcinoma [45], UCB has the potential to cause cell cycle arrest at G0/G1 and exert pro-oxidant effects at high concentrations [46]. Human biliverdin reductase, which chemically reduces biliverdin to bilirubin, was recently implicated as a regulator in cancer development and maybe useful in designing biomarkers for cancer patients [47]. A similar study reported that lower serum bilirubin levels occur in colorectal cancer patients with a 1µM decrease in serum bilirubin related to a 7% rise in CRC risk [48]. Bilirubin possessed anti-tumour properties in vitro in HRT-18 cell lines and also BALB/c nude mice bearing HRT-18 colon cancer xenografts where it has shown that bilirubin defended against cancer by interfering with pro-carcinogenic pathways [49]. Bilirubin may also demonstrate synergistic anti-cancer effects inducing apoptosis in HeLa cells [50]. It was, however, necessary to consider the mode of cell death and determine whether bilirubin induced intrinsic cell death, and therefore we demonstrated that bilirubin induced apoptosis via PARP and Caspase 3. Caspase 3 and PARP are regarded as downstream activators of Caspase 9 [51], which may suggest the initiation of mitochondrial-dependent intrinsic cell death similar to previous studies showing D-limonene induced programmed cell death [51]. We support this conclusion by demonstrating increased AnnexinV staining to show that bilirubin initiates early apoptosis in LS174T cells. Annexin V is a 35-36 kDaCa2+-dependent phospholipid-binding protein with high affinity for PS, and binds to exposed apoptotic cell surface phospholipid phosphatidylserine [52, 53]. Our results clearly show the early cell death recorded in cells treated with 10µM bilirubin compared to that of the TUN group which evidently supports a conclusion that bilirubin exerts pro-apoptotic effects in cancer cell lines.
In parallel to the apoptosis assay, we assessed the proliferation rate via the EdU click non-radioactive method [54] which showed reduced proliferation in the bilirubin 10 µM group compared to the TUN group. In previous studies, bilirubin and biliverdin inhibited smooth muscle cell proliferation at the G1 phase and also phosphorylation of the retinoblastoma tumour suppressor protein inhibition in primary rat and mouse vascular smooth muscle cells (VSMCs) [55]. We have performed the in vitro proliferation assay in congruence with these results supporting the cell proliferation arresting ability of bilirubin in LS174T cells.
Conclusions
In conclusion, bilirubin despite of reducing ERS and ERS mediated inflammation, also increases cancer cell apoptosis and reduces proliferation. These data add further weight to the possibility that bilirubin can be used as a potential anti-inflammatory and anti-cancer agent in the colon. In the future we intend to translate this study to in-vivo and use unconjugated bilirubin as therapeutic agent in reducing the inflammatory mediated negative effects in a mouse carcinogenic model and assess the downstream effects.
Acknowledgements
The authors are thankful to the technical staff of School of Health Sciences (University of Tasmania).
Author Contributions
RG performed all the experiments with the help of WCC and RV. RG and ACB has drafted the manuscript with technical inputs and supervision from RE.
Funding
This work was supported by Takeda-IBD Research Grant (E0025316) allocated to Dr. Rajaraman Eri.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Hasnain SZ, Lourie R, Das I. et al. The interplay between endoplasmic reticulum stress and inflammation. Immunol Cell Biol. 2012;90:260-70
2. Sano R, Reed JC. ER stress-induced cell death mechanisms. Biochem Biophys Acta. 2013;1833:3460-70
3. Yadav RK, Chae S, Kim H. et al. Endoplasmic Reticulum Stress and Cancer. J Cancer Prev. 2014;19:75-88
4. Wang W, Groenendyk J, Michalak M. Endoplasmic reticulum stress associated responses in cancer. Biochim Biophys Acta. 2014;1843:2143-9
5. Farooqi AA, Li K, Fayyaz S. Anticancer drugs for the modulation of endoplasmic reticulum stress and oxidative stress. Tumor Biol. 2015;36:5743-52
6. Zhou YE, Zheng JG, Li Y. et al. Natural Polyphenols for Prevention and Treatment of Cancer. Nutrients. 2016:5
7. Park SW, Lee CH, Lee JG. et al. Protective effects of atypical antipsychotic drugs against mpp(+)-induced oxidative stress in pc12 cells. Neurosci Res. 2011;69:283-90
8. Singh M, Chaudhry P, Parent S. et al. Ubiquitin-proteasomaldegradation of cox-2 in tgf-beta stimulated human endometrialcells is mediated through endoplasmic reticulum mannosidasei. Endocrinology. 2012;153:426-37
9. Elfrink HL, Zwart R, Baas F. et al. Inhibition of endoplasmicreticulum associated degradation reduces endoplasmic reticulumstress and alters lysosomal morphology and distribution. Mol Cells. 2013;35:291-7
10. Stocker R, Yamamoto Y, McDonagh AF. et al. Bilirubin is an antioxidant of possible physiological importance. Science. 1987;235:1043-6
11. Stocker R, Peterhans E. Synergistic interaction between vitaminE and the bile pigments bilirubin and biliverdin. Biochim Biophys Acta. 1989;1002:238-44
12. Stocker R. Antioxidant activities of bile pigments. Antioxid Redox Signal. 2004;6:841-9
13. Tomaro ML, Batlle AM. Bilirubin: its role in cytoprotection against oxidative stress. Int J Biochem Cell Biol. 2002;34:216-20
14. Gopinathan V, Miller NJ, Milner AD. et al. Bilirubin and ascorbate antioxidant activity in neonatal plasma. FEBS Lett. 1994;349:197-200
15. Bellanger S, Lavole JC, Chessex P. Influence of bilirubin on the antioxidant capacity of plasma in newborn infants. Biol Neonate. 1997;71:233-8
16. Bulmer AC, Blanchfield JT, Toth I. et al. Improved resistance to serum oxidation in Gilbert's syndrome: a mechanism for cardiovascular protection. Atherosclerosis. 2008;199:390-6
17. Boon AC, Bulmar AC, Coombes JS. et al. Circulating bilirubin and defense against kidney disease and cardiovascular mortality: mechanisms contributing to protection in clinical investigations. Am J Physiol Renal Physiol. 2014;307:F123-F36
18. Bakrania B, Du Toit EF, Wagner KH. et al. Pre- or post-ischemic bilirubin ditaurate treatment reduces oxidative tissue damage and improves cardiac function. Int J Cardiol. 2016;202:27-33
19. Bisht K, Tampe J, Shing C. et al. Endogenous Tetrapyrroles Influence Leukocyte Responses to Lipopolysaccharide in Human Blood: Pre-Clinical Evidence Demonstrating the Anti-Inflammatory Potential of Biliverdin. J Clin Cell Immunol. 2014;5:1000218
20. Kinderlerer AR, I PG, Hamdulay SS. et al. Heme oxygenase-1expression enhances vascular endothelial resistance to complement-mediated injury through induction of decay-accelerating factor: a role for increased bilirubin and ferritin. Blood. 2009;113:1598-607
21. Inoguchi T, Sonoda N, Maeda Y. Bilirubin as an important physiological modulator of oxidative stress and chronic inflammation in metabolic syndrome and diabetes: a new aspect on old molecule. Diabetology International. 2016;7:338-41
22. Barateiro A, Vaz AR, Silva SL. et al. ER stress, mitochondrial dysfunction and calpain/JNK activation are involved in oligodendrocyte precursor cell death by unconjugated bilirubin. Neuromolecular Med. 2012;14:285-302
23. Ray R, Hauck S, Kramer R. A convenient fluorometric method to study sulfur mustard-induced apoptosis in human epidermal keratinocytes monolayer microplate culture. Drug Chem Toxicol. 2008;28:105-16
24. Fytianos K, Rodriguez-lorenzo L, Clift MJ. Uptake efficiency of surface modified gold nanoparticles does not correlate with functional changes and cytokine secretion in human dendritic cells in vitro. Nanomedicine. 2015;11:633-44
25. Yu Y, Arora A, Min W. EdU incorporation is an alternative non-radioactive assay to [(3)H]thymidine uptake for in vitro measurement of mice T-cell proliferations. J Immunol Methods. 2009;350:29-35
26. Hirschfield GM, Alexander GJ. Gilbert's syndrome: an overview for clinical biochemists. Ann Clin Biochem. 2008;43:340-3
27. Vandewynckel YP, Laukens D, Geerts A. The paradox of the unfolded protein response in cancer. Anticancer Res. 2013;33:4683-94
28. Avril T, Vauléon E, Chevet E. Endoplasmic reticulum stress signaling and chemotherapy resistance in solid cancers. Oncogenesis. 2017;6:e373
29. Cubillos-ruiz JR, Bettigole SE, Glimche LH. Tumorigenic and Immunosuppressive Effects of Endoplasmic Reticulum Stress in Cancer. Cell. 2017;168:692-706
30. Kunde DA, Chong WC, Nerurkar PV. Bitter melon protects against ER stress in LS174T colonic epithelial cells. BMC Complement Altern Med. 2017;17:2
31. Zhang K, Kaufman RJ. From endoplasmic-reticulum stress to the inflammatory response. Nature. 2008;454:455-62
32. Ning Y, Manegold PC, Hong YK. et al. Interleukin-8 is associated with proliferation, migration, angiogenesis and chemosensitivity in vitro and in vivo in colon cancer cell line models. Int J Cancer. 2011;128:2038-49
33. Ning Y, Lenz HJ. Targeting IL-8 in colorectal cancer. Expert Opin Ther Targets. 2012;16:491-7
34. Van Kampen C, Gauldie JP, Collins SM. Proinflammatory properties of IL-4 in the intestinal microenvironment. Am J PhysiolGastrointest Liver Physiol. 2005;288:G111-7
35. Francipane MG, Alea MP, Lombardo Y. et al. Crucial role of interleukin-4 in the survival of colon cancer stem cells. Cancer Res. 2008;68:4022-5
36. Stanilov N, Miteva L, Dobreva Z. et al. Colorectal cancer severity and survival in correlation with tumour necrosis factor-alpha. Biotechnol Biotechnol Equip. 2014;28:911-7
37. Al Obeed OA, Alkhayal KA, Al Sheikh A. et al. Increased expression of tumor necrosis factor-α is associated with advanced colorectal cancer stages. World J Gastroenterol. 2014;20:18390-6
38. Zins K, Abraham D, Sioud M. et al. Colon cancer cell-derived tumor necrosis factor-alpha mediates the tumor growth-promoting response in macrophages by up-regulating the colony-stimulating factor-1 pathway. Cancer Res. 2007;67:1038-45
39. Chung AY, Li Q, Blair SJ. et al. Oral interleukin-10 alleviates polyposis via neutralization of pathogenic T-regulatory cells. Cancer Res. 2014;74:5377-85
40. Dennis KL, Blatner NR, Gounari F. Current status of IL-10 and regulatory T-cells in cancer. Curr Opin Oncol. 2013;25:637-45
41. Berg DJ, Davidson N, Kühn R. et al. Enterocolitis and colon cancer in interleukin-10-deficient mice are associated with aberrant cytokine production and CD4(+) TH1-like responses. J Clin Invest. 1996;98:1010-20
42. Khan N, Adham VM, Mukhtar H. Apoptosis by dietary agents for prevention and treatment of cancer. Biochem Pharmacol. 2008;76:1333-9
43. Sun SY, Hail Jr, Lotan R. Apoptosis as a novel target for cancer chemoprevention. J Natl Cancer Inst. 2004;96:662-72
44. Mölzer C, Pfleger B, Putz E. et al. In vitro DNA-damaging effects of intestinal and related tetrapyrroles in human cancer cells. Exp Cell Res. 2013;319:536-45
45. Deng CC, Xu M, Li J. et al. Unconjugated Bilirubin Is a Novel Prognostic Biomarker for Nasopharyngeal Carcinoma and Inhibits Its Metastasis via Antioxidation Activity. Cancer Prev Res (Phila). 2016;9:180-8
46. Rao P, Suzuki R, Mizobuchi S. et al. Bilirubin exhibits a novel anti-cancer effect on human adenocarcinoma. BiochemBiophys Res Commun. 2006;342:1279-83
47. Zhang M, Xin W, Yi Z. et al. Human biliverdinreductase regulates the molecular mechanism underlying cancer development. J Cell Biochem. 2018;119:1337-45
48. Jirásková A, Novotný J, Novotný L. et al. Association of serum bilirubin and promoter variations in HMOX1 and UGT1A1 genes with sporadic colorectal cancer. Int J Cancer. 2012;131:1549-55
49. Ollinger R, Kogler P, Troppmair J. et al. Bilirubin inhibits tumor cell growth via activation of ERK. Cell Cycle. 2007;6:3078-85
50. Dwarka D, Thaver V, Naidu M. et al. In vitro chemo-protective activity of Strelitzia Nicolai aril extract containing bilirubin. Afr J Tradit Complement Altern Med. 2017;14:147-56
51. Jia S, Xi G, Zhang M. Induction of apoptosis by D-limonene is mediated by inactivation of Akt in LS174T human colon cancer cells. Oncol Rep. 2012;29:349-54
52. van Engeland M, Ramaekers FC, Schutte B. et al. A novel assay to measureloss of plasma membrane asymmetry during apoptosis of adherent cells in culture. Cytometry. 1996;24:131-9
53. Casciola-Rosen L, Rosen A, Petri M. et al. Surface blebs on apoptotic cells are sites ofenhancedprocoagulant activity: implications for coagulation events and antigenic spread insystemic lupus erythematosus. Proc Natl Acad Sci U S A. 1996;93:1624-9
54. Yu Y, Arora A, Min W. et al. EdU incorporation is an alternative non-radioactive assay to [(3)H] thymidine uptake for in vitro measurement of mice T-cell proliferations. J Immunol Methods. 2009;350:29-35
55. Ollinger R, Bilban M, Erat A. et al. Bilirubin: a natural inhibitor of vascular smooth muscle cell proliferation. Circulation. 2005;112:1030-9
Author Biography
Rohit Gundamaraju is a PhD student in School of Health Sciences under Dr. Raj Eri, recipient of International Post Graduate Research Scholarship at University of Tasmania. Rohit's research is primarily unraveling the relation between endoplasmic reticular stress and programmed cell death in colon cancer. He has published numerous papers in the field of gut physiology. He has been as an investigator in projects like obesity, dengue and cancer. His recent work was on understanding the effects of ER stress inhibition in attenuating inflammation and cancer.
![]() Corresponding author: rohit.gundamarajuedu.au
Corresponding author: rohit.gundamarajuedu.au
Received 2018-8-10
Accepted 2018-11-29
Published 2019-1-1