ISSN: 1449-1907International Journal of Medical Sciences

Int J Med Sci 2018; 15(10):1062-1071. doi:10.7150/ijms.25527 This issue Cite
Research Paper
Rac1 promotes the survival of H9c2 cells during serum deficiency targeting JNK/c-JUN/Cyclin-D1 and AKT2/MCL1 pathways
Jinlong Zhao, Qiqiang Jie, Gang Li, Yong Li, Baoxin Liu, Hongqiang Li, Jiachen Luo, Xiaoming Qin, Zhiqiang Li, Yidong Wei ![]()
Department of Cardiology, Shanghai Tenth People's Hospital, Tongji University School of Medicine, Shanghai, China
Abstract
Rac1, known as a “molecular switch”, plays a crucial role in plenty of cellular processes. Rac1 aggravates the damage of myocardial cells in the process of myocardial ischemia-reperfusion during myocardial infarction through activating the NADPH oxidase and bringing about the reactive oxygen species(ROS) generation. Myocardial ischemia and hypoxia are the basic pathogenesis of myocardial infarction and the underlying mechanisms are intricate and varied. Moreover, the regulatory effect of Rac1 on myocardial cells in the condition of serum starvation and the potential mechanisms are still incompletely undefined. Therefore, heart-derived H9c2 cells cultured in 0% serum were used to mimic ischemic myocardial cells and to clarify the role of Rac1 in H9c2 cells and the underlying mechanisms during serum deficiency. After Rac1 was knocked down using specific siRNA, cell apoptosis was assessed by flow cytometry assay and cell proliferation was detected by CCK-8 assay and EdU assay. In addition, the expression and activation of protein in related signaling pathway were detected by Western blot and siRNAs was used to testify the signaling pathways. Our results indicated that Rac1 inhibited apoptosis, promoted proliferation and cell cycle progression of H9c2 cells during serum deficiency. We concluded that Rac1 inhibited apoptosis in an AKT2/MCL1 dependent way and promoted cell proliferation through JNK/c-JUN/Cyclin-D1.
Keywords: Rac1, apoptosis, cell proliferation, AKT2, JNK
Introduction
Rac1, a member of the small GTPase proteins including more than 150 members, is divided based on structural and functional similarities into 5 main subfamilies: Ras (Rap Ras and Ral), Rho (Rac, Cdc42 and RhoA), Arf, Rab and Ran [1]. The previous study found that Rac1 could be activated through binding to GTP and catalytically devitalized when binding to GDP [2-4]. Both processes require three groups of specific proteins, GTPase activating proteins (GAPs), guanine nucleotide exchange factors (GEFs) and guanine nucleotide dissociation inhibitors (GDIs) [5, 6].
Rac1 is a pivotal regulator of plenty of cellular processes. The activated Rac1 could facilitate actin polymerization at the cell periphery to stimulate the formation of the lamellipodia and membrane ruffles [2, 7-10]. Furthermore, Rac1 promotes cell migration, cycle progression, transformation and cell survival [11-13]. In the cardiovascular system, Rac1 is also an important regulatory factor. Rac1 might induce cardiomyocyte hypertrophy, regulate the proliferation and migration of vascular smooth muscle cell (VSMC) and the realignment of endothelial cells (ECs) [14-17]. Hypertrophy induced by angiotensin II (AngII) infusion in mouse that cardiac-specific deficiency of Rac1 could be alleviated through decreasing activities of the NADPH oxidase and reducing the generation of reactive oxygen species (ROS). In addition, the pro-hypertrophic effect of Rac1 was related to transcription factor NF-κB and signaling kinase-1 [18]. Overexpression of constitutively active Rac1 in mouse could result in atrial fibrillation at 16 months of age [19].
Myocardial infarction (MI) is one of the main causes of morbidity and mortality in coronary heart diseases (CHD) patients worldwide [20]. At the early phase of MI, myocardial cells undergo ischemia and effective myocardial reperfusion could reduce the area of myocardial infarction and alleviate myocardial injury. The process of ischemia-reperfusion can lead to the further death of cardiomyocytes is a phenomenon called ischemia-reperfusion injury (IR). During the process of IR, reactive oxygen species (ROS) generation and superabundant ROS production in the reperfusion phase are harmful to cardiac tissue, because cellular components could be attacked by ROS and then lead to apoptosis and necrosis [21, 22]. The difference between Rac1 and the other small GTPase proteins is that Rac1 is essential in the reactive oxygen species (ROS) generation through regulating NADPH oxidase activity. One of the crucial components of the NADPH oxidase complex, p67phox, would interact with Rac1 which is indispensable for the assembling of the multimeric enzyme and the catalytic activity [23]. Hence, Rac1 is a pronounced regulator in the myocardial ischemia-reperfusion injury (IR) during MI. Myocardial ischemia and hypoxia are the basic pathogenesis of myocardial infarction and the mechanisms of ischemia-reperfusion (IR) injury are intricate and varied. Most the previous studies focused on the role of Rac1 in the myocardial ischemia-reperfusion injury (IR). However, the regulatory effect of Rac1 on myocardial cells in the condition of serum starvation and the potential mechanism is not completely explicit. Therefore, in this study, heart-derived H9c2 cells were cultured in 0% serum to mimic the ischemic circumstance and to investigate the effect of Rac1 on the cardiomyocytes in the condition of serum starvation and explore the underlying mechanism.
Methods
Antibodies
The following antibodies were all purchased from Cell Signaling Technology (CST, MA, USA): anti-JNK, anti-phospho-JNK, anti-c-JUN, anti-phospho-c-JUN, anti-Cyclin-D1, anti-AKT2, anti-phospho-AKT2, anti-cleaved PARP89, anti-mouse lgG HRP-linked antibody, anti-rabbit lgG HRP-linked antibody. β-actin, mouse mAb was purchased from YCASEN (YCASEN, Shanghai, China). Anti-Rac1 antibody, anti-MCL1 antibody and anti-PARP antibody [E51] were purchased from Abcam (Cambridge, MA, USA).
Cell culture and si-RNAs transfection
H9c2 cells were purchased from ATCC and cultured in DMEM (Hyclone, USA) containing 10% fetal bovine serum (FBS), 1% penicillin/streptomycin and 4.5% sodium pyruvate at 37℃ and 5% CO2. According to our previous study, siRNAs transfection was operated using lipofectamine 2000 (Invitrogen, USA). H9c2 cells were seeded in 6-well plates 24h before transfection, incubated at 37℃ and 5% CO2 overnight, and then transfected at 50-70% confluence with 100nM siRNAs [24]. The siRNAs used in this study were purchased from Sigma (Table 1). The sequence of negative control used in this study was as follows: 5'→3': UUCUCCGAACGUGUCACGUTT; 3'→5': ACGUGACACGUUCGGAGAATT.
Article number (Art. No) and siRNA ID of siRNAs used in this study
| siRNAs | Article number (Art. No) | siRNA ID |
|---|---|---|
| Rac1 | NM_134366 | SASI_Rn01_00118554 |
| JNK | XM_341399 | SASI_Rn02_00392868 |
| AKT2 | NM_017093 | SASI_Rn01_00047688 |
| MCL1 | NM_021846 | SASI_Rn01_00262861 |
Western blot
After transfection with siRNAs for 48h, H9c2 cells were cultured in 10% and 0% serum conditions for 48h. Washed with cold PBS twice, lysed with 1XCell lysis Buffer (CST, MA, USA) containing 1× protease inhibitor cocktail (Roche, USA) and 1X phosphatase inhibitor cocktail (Roche, USA), total proteins were collected by centrifuging at 12000 rpm for 15 min at 4°C. Protein was loaded, separated and transferred. Incubated with primary antibodies overnight at 4°C and horseradish peroxidase (HRP)-conjugated secondary antibodies for 1h at room temperature. Hybridizing bands were visualized with High-sig ECL Western Blotting Substrate (Tanon, Shanghai, China) by Amersham Imager 600 ECL system (GE Healthcare, USA)
Cell proliferation assay
The cell proliferation assay was performed according to the manufacturer's instruction. H9c2 cells were seeded in 96-well plates in medium containing 10% serum and cultured overnight. After transfection with si-Rac1, cells were then cultured in 10% and 0% serum conditions. We added 10μl of Cell Counting Kit-8 (Dojindo, Japan) reagent to each well, incubated at 37°C for 2 h. OD value on 450nm was measure by SpectraMax i3 (MD, USA). The formula, [(OD experiment - OD blank) / (OD 1st day - OD blank)] x 100%, was calculated to represent cell proliferation rate. All experiments were performed in triplicate and repeated three times.
EdU assay
Click-iT EdU Imaging Kits (Invitrogen, USA) was used to detect cell cycle progression. Cells were plated on 6-well plate, label with 1X working solution of EdU (10 mM), incubated for 16h, fixed with 3.7% formaldehyde, permeated with 0.5% Triton X-100. Then, Click-iT reaction cocktail was used to detect EdU, 1XHoechst (1:2000 in PBS) was used to stain DNA. Finally, EdU (+) cells were detected by Olympus IX83 microscope (OLYMPUS, Japan).
Apoptosis assay
FITC Annexin V Apoptosis Detection KitⅠ(BD pharmingen, USA) was used to detect the Apoptosis of H9c2 cells according to manufacturer's protocol. H9c2 cells were seeded into 6-well plates. After transfection with si-Rac1, cells were cultured in 10% and 0% serum conditions for 48h and washed twice with cold PBS and resuspended in 1x binding buffer at a concentration of 1x106 cells/ml. 100μl of the solution was transferred to a 5 ml culture tube. Cells in each tube were stained using 5μl of FITC Annexin V and 5μl Propidium Iodide solution (PI) for 15 min at room temperature in the dark. 400 μl of 1x binding buffer was added to each tube and the cells were analyzed via flow cytometry.
Statistical analysis
Data are presented with means ± S.D. Statistical significance was determined by Student's t-tests. P < 0.05 was considered a statistically significant difference.
Results
Rac1 promoted the proliferation and cell cycle progression of H9c2 cells during serum deficiency
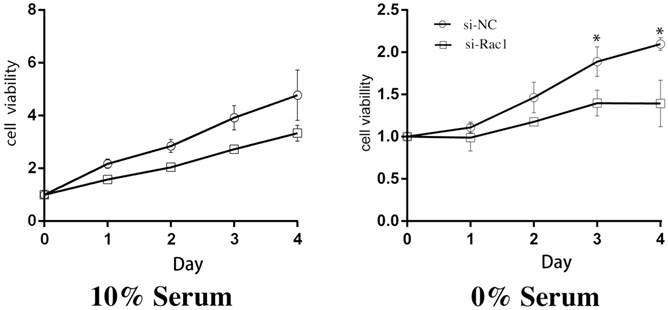
After aforementioned transfections of si-Rac1, we cultured H9c2 cells in medium containing 10% and 0% serum and then CCK-8 assay was performed daily as a cell viability indicator for 4 days. In 10% serum, compared with si-NC group, the viability of the si-Rac1 group was mildly decreased but has no significant difference (Figure 1). While in 0% serum, the cell viability of si-Rac1 group was significantly reduced compared with si-NC group (p < 0.05) (Figure 1). These results showed that Rac1 promoted cell proliferation in 0% serum condition rather than 10% serum condition.
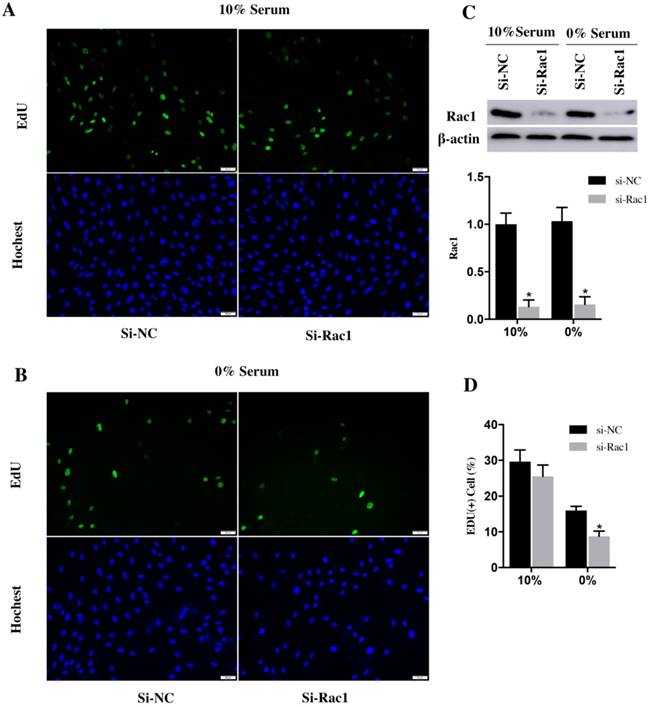
EdU assay was used to analyze the cell cycle progression. After transfection with si-Rac1 for 48h, H9c2 cells were cultured in medium containing 10% and 0% serum for 48h. In the medium of 10% serum, the si-NC and si-Rac1 group showed the similar percentage of EdU (+) cells (29.62±1.89% vs 25.53±1.83%) (Figure 2A and 2D). However, the percentage of EdU (+) H9c2 cells in si-Rac1 group (8.75±0.87%) was significantly decreased than in si-NC group (16.01±0.66%) in 0% serum condition (Figure 2B and 2D), which indicated that Rac1 promoted the cell cycle progression during serum-deficiency. The inhibitory effects of si-Rac1 on Rac1 expression were confirmed by Western Bolt after transfection for 48h (Figure 2C). The expression of Rac1 was significantly decreased (Figure 2C).
Together, all of these results demonstrated that Rac1 promoted cell proliferation and cell cycle progression of H9c2 cells during serum deficiency.
Rac1 promoted the proliferation of H9c2 cells during serum deficiency. The CCK-8 assay was performed to detect the viabilities of H9c2 cells and the cells were cultured in 10% and 0% serum conditions as indicated for 0-4 days after transfection for 48h. OD values standardized to day0 were considered to present the results. *P < 0.05 vs si-NC group.
Rac1 promoted the cycle progression of H9c2 cells during serum deficiency. A and B. Representative images of EdU assay for cell cycle progression in 10% and 0% serum conditions. The blue was the nuclei stained by Hochest (lower panel) and the green was EdU (+) cells (upper panel). Bars represent 50 μm. C. Western Bolt results of inhibitory effects of si-Rac1 on Rac1 expression after transfection for 48h. Diagram of inhibitory effects of si-Rac1. Densities were first normalized to β-actin and then to si-NC group. N=3. *P < 0.05 vs si-NC group. D. Diagram of EdU (+) cells of si-NC and si-Rac1 group in 10% and 0% serum conditions. *P < 0.05 vs si-NC group.
Rac1 inhibited apoptosis of H9c2 cells induced by serum deficiency
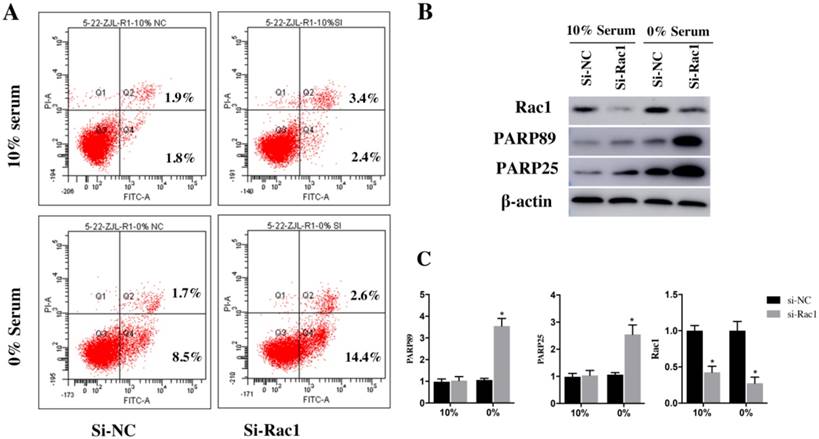
After being transfected with si-Rac1 for 48h, H9c2 cells were cultured in medium containing 10% serum and 0% serum for 48h. Two recognized apoptosis markers, the cleaved PARP89 and cleaved PARP25, were detected by Western blot. Compared with si-NC group, both cleaved PARP89 and PARP25 were significantly increased in si-Rac1 group cultured in 0% serum (Figure 3B and 3C). However, the expression of the cleaved PARP25 and PARP89 in H9c2 cells cultured in 10% serum condition was weakly detected (Figure 3B and 3C).
The Annexin-V assay was used to detect the apoptosis of H9c2 cells. The results demonstrated that apoptotic cells (sum of quadrant-2 and -4 on flow cytometry assay) have no significant difference between the si-NC group (3.23±0.89%) and si-Rac1 group (4.81±0.68%) at the condition of 10% serum. (Figure 3A). While in 0% serum condition, compared with si-NC group (9.2±0.96%) (Figure 3A), si-Rac1 group showed prominently enhanced apoptosis (16.5±0.79%) (p < 0.05). It showed that Rac1 inhibited apoptosis in 0% serum but not in 10% serum. The inhibitory effects of si-Rac1 on Rac1 expression were detected by Western Blot after transfection for 48h. (Figure 3B and 3C). The expression of Rac1 was significantly decreased (Figure 3B and 3C).
These data indicated that Rac1 inhibited apoptosis of H9c2 cells induced by serum deficiency effectively.
Rac1 inhibited apoptosis of H9c2 cells induced by serum deficiency. A. Flow cytometry was used to analyze the apoptosis of H9c2 cells cultured in 10% and 0% serum conditions for 48h after transfection with si-Rac1 for 48h. Q-2 presents the percentage of late apoptosis cells, Q-4 presents the percentage of early apoptosis cells and Q-3 presents live cells. B. Western Bolt results of inhibitory effects of si-Rac1 on Rac1 expression after transfection for 48h and the changes of the cleaved PARP25 and PARP89 in si-NC and si-Rac1 group in 10% and 0% serum. C. Diagram of inhibitory effects of si-Rac1 and the cleaved PARP25 and PARP89. Densities were first standardized to β-actin and then to si-NC group. N=3. *P < 0.05 vs si-NC group.
Rac1 promoted the proliferation and cell cycle progression of H9c2 cells through the JNK/c-JUN/Cyclin-D1 pathway
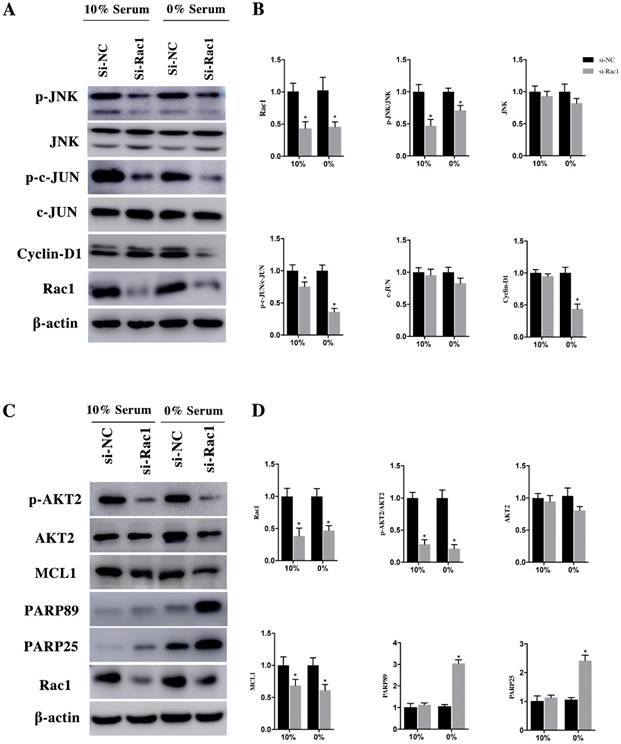
After being phosphorylated by JNK, the downstream effector of Rac1, c-JUN could interact with c-fos to form transcription factor AP1 to stimulate Cyclin-D1 expression. After transfection with si-Rac1 for 48h, H9c2 cells were cultured in medium having 10% and 0% serum for 48h. Compared with si-NC group, the Rac1 level of si-Rac1 group of 10% and 0% serum condition obviously reduced 62±10% and 47±6%, respectively (Figure 4A and 4B). The expression of p-JNK, downstream of Rac1, markedly decreased 47±8% and 29±6% in si-Rac1 group compared with si-NC group (Figure 4A and 4B). No significant diffidence was detected between si-Rac1 and si-NC group in the expression of JNK (Figure 4A and 4B). We also detected expression of c-JUN and p-c-JUN, the downstream of JNK. In both 10% and 0% serum conditions, c-JUN was slightly decreased but it showed no significant difference between si-NC group and si-Rac1 group, while the expression of p-c-JUN in si-Rac1 group reduced 25±6% and 64±4%, respectively, after Rac1 was knocked down (Figure 4A and 4B). As the downstream of c-JUN, Cyclin-D1 regulated G1/S-phase progression. In 10% serum condition, there was no significant difference between the expression of Cyclin-D1 in si-Rac1 group and si-NC group, however, in 0% serum condition, Cyclin-D1 significantly decreased 57±6% in si-Rac1 group (Figure 4A and 4B).
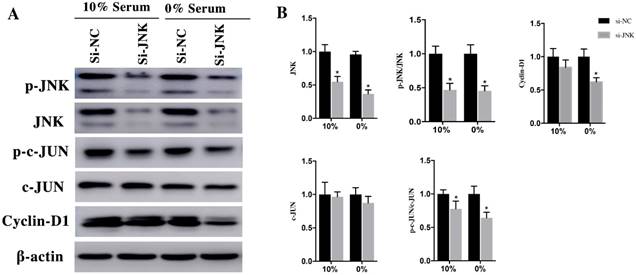
To verify the role of JNK/c-JNU/Cyclin-D1 pathway in Rac1 medicated pro-proliferation effects, we attenuated JNK expression using si-RNAs. After transfection with si-JNK for 48h, H9c2 cells were cultured in medium containing 10% and 0% serum conditions for 48h. Then the expression of JNK, p-JNK, c-JUN, p-c-JUN and Cyclin-D1 was detected using Western Blot. The results showed that both in 10% and 0% serum conditions, the expression of JNK decreased 45±7% and 63±5%, respectively, in si-JNK group and si-NC group. The p-JNK decreased 53±8% and 55±6%, respectively, in si-JNK group and si-NC group. The downstream of JNK, c-JUN remained unchanged in both 10% and 0% serum, while the expression of p-c-JUN was dramatically decrease 22±9% and 36±7% in si-JNK group compared with si-NC group (Figure 5A and 5B). The expression of Cyclin-D1, which remained unchanged in 10% serum condition. While in 0% serum the expression level decreased 35±10% in si-JNK group compared with si-NC group (Figure 5A and 5B).
These results indicated that Rac1 promotes H9c2 cell proliferation and cell cycle progression through the JNK/c-JUN/Cyclin-D1 pathway.
Rac1 promoted the proliferation and cell cycle progression of H9c2 cells through the JNK/c-JUN/Cyclin-D1 pathway and inhibits the apoptosis of H9c2 cells by AKT2/MCL1 pathway. A and C. Representative results of Western Blot detecting the expression of Rac1, JNK, p-JNK, c-JUN, p-c-JUN, Cyclin-D1, AKT2, p-AKT2, MCL1, PARP25 and PARP89 in si-NC and si-Rac1 group in 10% and 0% serum condition after transfection with si-Rac1 for 48h. β-actin was used as the control. B and D. Diagrams of A and C. Densities in A and C were first standardized to β-actin and then to si-NC group. N=3. *P < 0.05 vs si-NC group.
H9c2 cells were transfected with si-JNK for 48h and then cultured in medium with 10% and 0% serum for 48h. A. Changes of JNK, p-JNK, c-JUN, p-c-JUN, Cyclin-D1 in si-NC and si-JNK cells. β-actin was used as the control. B. Diagrams of A. Densities in A were first standardized to β-actin and then to si-NC group. N=3. *P < 0.05 vs si-NC group.
Rac1 inhibited the apoptosis of H9c2 cells by AKT2/MCL1 pathway
Previous studies reported that MCL1 was downstream of AKT which could inhibit cell apoptosis and promote the cell survival [25], but whether MCL1 is related to Rac1 is indistinct. Therefore, we hypothesized that Rac1 inhibits apoptosis though AKT2/MCL1 pathway.
In the same way, H9c2 cells were infected with si-Rac1 for 48h and cultured in medium containing 10% and 0% serum for 48h. In both 10% and 0% serum conditions, AKT2 was both mildly decreased in si-Rac1 group compared with si-NC group but has no significant difference (Figure 4C and 4D). However, to compare with si-NC group in 10% and 0% serum conditions, the p-AKT2 of si-Rac1 group decreased 72±6% and 79±5% (Figure 4C and 4D). In both 10% and 0% serum, MCL1 significantly reduced in si-Rac1 group compared with si-NC group (Figure 4C and 4D). The expression of the cleaved PARP89 and PARP25 were increased by 3.12±0.26 Fold and 2.58±0.16 Fold in si-Rac1 group with the condition of 0% serum. But the expression of the cleaved PARP89 and PARP25 in 10% serum condition was weakly detected (Figure 4C and 4D).
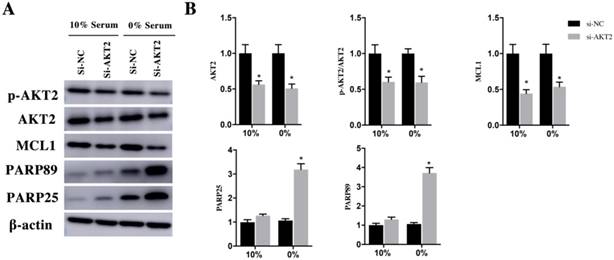
In order to verify the AKT2/MCL1 pathway, we attenuated AKT2 expression using si- AKT2 for 48h and cultured them in 10% and 0% serum for 48h. In both 10% and 0% serum conditions, AKT2 was both significantly decrease in si-AKT2 group compared with si-NC group (Figure 6A and 6B). Similarly, p-AKT2 was also decrease in si-AKT2 group compared with si-NC group (Figure 6A and 6B). The expression of MCL1 reduced 52±5% and 56±5% in si-AKT2 group compared with si-NC group in the 10% and 0% serum condition (Figure 6A and 6B). The cleaved PARP89 and PARP25 in 0% serum was significantly increased by 3.54±0.29 Fold and 3.14±0.49 Fold in si-AKT2 group compared with si-NC group. The expression of the cleaved PARP89 and PARP25 was weakly detected in 10% serum condition (Figure 6A and 6B).
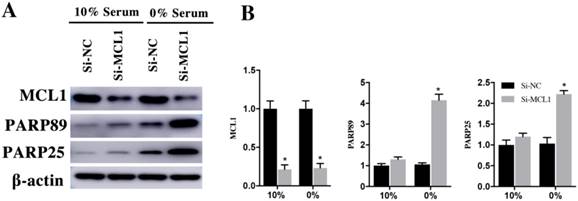
H9c2 cells were transfected with si-MCL1 for 48h and cultured them in 10% and 0% serum for 48h. Compared with si-NC group, the expression of MCL1 in si-MCL1 group decreased 79±5% and 77±5% in 10% and 0% serum group (Figure 7A and 7B). The expression of cleaved PARP89 and PARP25 was increased by 4.26±0.49 Fold and 2.15±0.39 Fold in si-MCL1 group at the 0% serum condition. Meanwhile, the expression of cleaved PRAP89 and PARP25 in 10% serum was weakly detected (Figure 7A and 7B). These results indicated that Rac1 inhibits the apoptosis of H9c2 cells by AKT2/MCL1 pathway.
H9c2 cells were transfected with si-AKT2 for 48h and then cultured in medium containing 10% and 0% serum for 48h. A. Changes of AKT2, p-AKT2, MCL1, cleaved PARP25 and PARP89 in si-NC and si-AKT2 cells. β-actin was used as the control. B. Diagrams of A. Densities in A were first standardized to β-actin and then to si-NC group. N=3. *P < 0.05 vs si-NC group.
H9c2 cells were transfected with si-MCL1 for 48h and then cultured in medium having 10% and 0% serum for 48h. A. Changes of MCL1, cleaved PARP25 and PARP89 in si-NC and si-MCL1 cells. β-actin was used as the control. B. Diagrams of A. Densities in A were first standardized to β-actin and then to si-NC group. N=3. *P < 0.05 vs si-NC group.
Discussion
In this study, we found: 1). Rac1 inhibited the apoptosis of H9c2 cells during serum deficiency; 2). Rac1 promoted H9c2 cells proliferation and cell cycle progression in the time of serum deficiency. 3). Rac1 promoted cell proliferation and cell cycle progression through JNK/c-JUN/Cyclin-D1 pathway. 4). The anti-apoptotic effect of Rac1 was AKT2/MCL1 dependent. Rac1 inhibited apoptosis of fibroblast induced by Ras over-activation, and its mechanism is dependent on the activation of NF-κB induced by Rac1 [26]. Shen et al. found that Rac1 played a crucial role in high glucose-induced myocardial apoptosis by regulating the activation of NADPH oxidase and increasing production of ROS. They also proved Rac1 inhibitor NSC23766 reduced the activity of NADPH oxidase and the apoptosis of myocardial cells, and mildly improved the function of myocardial cells [27]. In tumor cells, Rac1-knockdown could block the generation of Bcl-2-induced intracellular ROS and induce apoptosis [28]. Previous study indicated that in the breast cancer cells survived HFR (hyper-fractionated radiation) treatment, Rac1 protein expression increased compared to parental cells and Rac1 enhanced the survival of breast cancer cells through activating ERK1/2/NF-κB signaling pathways and then activating the anti-apoptotic protein Bcl-xL and Mcl-1 [29]. All these previous studies showed that Rac1 has varied anti-apoptotic effects in different cells and conditions. And we found that Rac1 inhibited the apoptosis of H9c2 cells during the serum starvation. Apoptosis, the autonomous death of cells controlled by genes, is critical for maintaining stable homeostasis in various cells. There are several types of cellular stress stimuli have been shown to induce apoptosis, including DNA damaging agents, oxidative stress, alimentary deficiency and ER stress. Serum deprivation (removal of nutrients) is also one of the stress to induce apoptosis. The process of apoptosis is regulated by various proteins including pro-apoptotic proteins and anti-apoptotic proteins. The most common anti-apoptotic family of proteins are FLICE-inhibitory proteins (FLIPs), the Bcl-2 family members McL-1 and Bcl-xL as well as inhibitors of Apoptosis Proteins (IAPs). Furthermore, Faim3, Erk1/2, AKT, Heat Shock Proteins (HSPs) and some enzymes like sphingosine kinase-1 are also the common anti-apoptotic proteins [30, 31]. Rac1 played an important role in anti-apoptotic signaling by stimulating phosphatidylinositol-3 kinase (PI3K) and AKT serine/threonine kinase. Rac1 also increased the expression of Cyclin-D1 and proteins associated with anti-apoptosis by activating the NADPH oxidase complex and then generating the production of reactive oxygen species (ROS) [32-35]. Previous studies showed the downstream of AKT, Bad, MDM2, Bax and Bak were pro-apoptotic factors. However, the member of Bcl- 2 family, MCL1, Bcl2 and Bcl-xL are anti-apoptotic [25, 36-40]. Our results indicated Rac1 inhibit the apoptosis of H9c2 cells by AKT2/MCL1 pathway.
Rac1 can activate nuclear factor kB (NF-kB), p21-activated kinase (PAK), Jun NH2-terminal kinase (JNK), p38 mitogen-activated protein kinase (MAPK) and these regulatory factors could activate AP1 transcription factor which could activate the expression of Cyclin D1 and c-JUN to promote cell cycle progression[25, 41-43]. In our study, the results showed that the expression of JNK decreased after attenuating the Rac1 expression (Figure 4A and 4C). Interference of JNK could decrease the expression of p-c-JUN and Cyclin-D1 in si-JNK cells. These results further indicated that Rac1 activated JNK and c-JUN, which promoted the Cyclin-D1 expression (Figure 5A and 5B).
Previous studies reported that rac1 played a key role in IR-induced injury [44, 45]. It is unequivocal that ROS generated in IR could cause organ damage and Rac1 is essential in the reactive oxygen species (ROS) generation through regulation of NADPH oxidase activity. Numerous researchers have found that the damages of tissue would be eliminated by inhibiting ROS production during ischemia/reperfusion. In the phase of reperfusion, the production of ROS could lead to various cellular events and eventually induce inflammation, necrosis, and/or apoptosis [46-48]. Inhibition of Rac1 protected IR-induced injury by decreasing the ROS production during reperfusion [44, 45]. Xiang et al reported that conditional cardiac-specific overexpression of RhoA in mice protected the heart against IR-induced injury [49]. Similarly, Bao et al has also found that inhibition of Rho-kinase protected the mouse heart against IR-induced injury [50]. All these researches indicated that small GTPase proteins including Rac1 aggravate the damage of oranges during IR-induced injury. However, in our study, we found that Rac1 has a positive and protective role in H9c2 cells through anti-apoptotic effect in an AKT2/MCL1 dependent way and pro-proliferative effect through JNK/c-JUN/Cyclin-D1 during the serum starvation. We suppose that there are three main reasons that led to the difference between our experimental results and previous studies. Firstly, it is the experimental condition. Myocardial ischemia and hypoxia is the basic pathological process in myocardial infarction and the mechanisms of ischemia-reperfusion (IR) injury are intricate and varied. And in our study, we discussed the effect of Rac1 on the cardiomyocytes in the condition of serum starvation, however, the previous studies mainly focused on the effect of Rac1 on the ischemia-reperfusion (IR) injury. Secondly, the experimental condition of our study was serum starvation and might only reflect nutrition deficiency aspect of myocardial ischemia. Other studies were focused on hypoxia and ROS production aspect. Thirdly, all of our findings were based on experiments in vitro and these were also the limitation of our study. H9c2 cells are heart-derived cell lines with permanent splitting capacity and have the characteristics of the cardiomyocyte, which could be utilized to stimulate various cardiac pathological processes [51]. Therefore, H9c2 cells were introduced as the substitute of isolated rat cardiomyocytes in our study.
In summary, we demonstrated that Rac1 inhibited apoptosis, promoted proliferation and cell cycle progression of H9c2 cells during serum deficiency. Rac1 inhibited apoptosis in an AKT2/MCL1 dependent way and promoted cell proliferation through JNK/c-JUN/Cyclin-D1.
Acknowledgments and fund resources
This research was supported by the Natural Science Foundation of Shanghai, Grant No. 18ZR1429700 (Y-D. W) and Natural Science Foundation of China, Grant No.81270193 (Y-D. W).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Etienne-Manneville S, Hall A. Rho GTPases in cell biology. Nature. 2002;420:629-35
2. Burridge K, Wennerberg K. Rho and Rac take center stage. Cell. 2004;116:167-79
3. Jaffe AB, Hall A. Rho GTPases: biochemistry and biology. Annu Rev Cell Dev Biol. 2005;21:247-69
4. Hall A. Rho family GTPases. Biochem Soc Trans. 2012;40:1378-82
5. Moon SY, Zheng Y. Rho GTPase-activating proteins in cell regulation. Trends Cell Biol. 2003;13:13-22
6. Heasman SJ, Ridley AJ. Mammalian Rho GTPases: new insights into their functions from in vivo studies. Nat Rev Mol Cell Biol. 2008;9:690-701
7. Ridley AJ, Hall A. The small GTP-binding protein rho regulates the assembly of focal adhesions and actin stress fibers in response to growth factors. Cell. 1992;70:389-99
8. Ridley AJ, Paterson HF, Johnston CL. et al. The small GTP-binding protein rac regulates growth factor-induced membrane ruffling. Cell. 1992;70:401-10
9. Ridley AJ. Rho GTPase signalling in cell migration. Curr Opin Cell Biol. 2015;36:103-12
10. Kaibuchi K, Kuroda S, Amano M. Regulation of the cytoskeleton and cell adhesion by the Rho family GTPases in mammalian cells. Annu Rev Biochem. 1999;68:459-86
11. Van Aelst L, D'Souza-Schorey C. Rho GTPases and signaling networks. Genes Dev. 1997;11:2295-322
12. Westwick JK, Lambert QT, Clark GJ. et al. Rac regulation of transformation, gene expression, and actin organization by multiple, PAK-independent pathways. Mol Cell Biol. 1997;17:1324-35
13. Karlsson R, Pedersen ED, Wang Z. et al. Rho GTPase function in tumorigenesis. Biochim Biophys Acta. 2009;1796:91-8
14. Lezoualc'h F, Metrich M, Hmitou I. et al. Small GTP-binding proteins and their regulators in cardiac hypertrophy. J Mol Cell Cardiol. 2008;44:623-32
15. Hordijk PL. Regulation of NADPH oxidases: the role of Rac proteins. Circ Res. 2006;98:453-62
16. Tzima E. Role of small GTPases in endothelial cytoskeletal dynamics and the shear stress response. Circ Res. 2006;98:176-85
17. Gregg D, Rauscher FM, Goldschmidt-Clermont PJ. Rac regulates cardiovascular superoxide through diverse molecular interactions: more than a binary GTP switch. Am J Physiol Cell Physiol. 2003;285:C723-34
18. Hingtgen SD, Tian X, Yang J. et al. Nox2-containing NADPH oxidase and Akt activation play a key role in angiotensin II-induced cardiomyocyte hypertrophy. Physiol Genomics. 2006;26:180-91
19. Adam O, Frost G, Custodis F. et al. Role of Rac1 GTPase activation in atrial fibrillation. J Am Coll Cardiol. 2007;50:359-67
20. Mozaffarian D, Benjamin EJ, Go AS. et al. Executive Summary: Heart Disease and Stroke Statistics-2016 Update: A Report From the American Heart Association. Circulation. 2016;133:447-54
21. McCord JM, Roy RS, Schaffer SW. Free radicals and myocardial ischemia. The role of xanthine oxidase. Adv Myocardiol. 1985;5:183-9
22. Murphy E, Steenbergen C. Mechanisms underlying acute protection from cardiac ischemia-reperfusion injury. Physiol Rev. 2008;88:581-609
23. Diekmann D, Abo A, Johnston C. et al. Interaction of Rac with p67phox and regulation of phagocytic NADPH oxidase activity. Science. 1994;265:531-3
24. Li G, Ying L, Wang H. et al. Rac1b enhances cell survival through activation of the JNK2/c-JUN/Cyclin-D1 and AKT2/MCL1 pathways. Oncotarget. 2016;7:17970-85
25. Mojsa B, Lassot I, Desagher S. Mcl-1 ubiquitination: unique regulation of an essential survival protein. Cells. 2014;3:418-37
26. Joneson T, Bar-Sagi D. Suppression of Ras-induced apoptosis by the Rac GTPase. Mol Cell Biol. 1999;19:5892-901
27. Shen E, Li Y, Li Y. et al. Rac1 is required for cardiomyocyte apoptosis during hyperglycemia. Diabetes. 2009;58:2386-95
28. Velaithan R, Kang J, Hirpara JL. et al. The small GTPase Rac1 is a novel binding partner of Bcl-2 and stabilizes its antiapoptotic activity. Blood. 2011;117:6214-26
29. Hein AL, Post CM, Sheinin YM. et al. RAC1 GTPase promotes the survival of breast cancer cells in response to hyper-fractionated radiation treatment. Oncogene. 2016;35:6319-6329
30. Portt L, Norman G, Clapp C. et al. Anti-apoptosis and cell survival: a review. Biochim Biophys Acta. 2011;1813:238-59
31. Fulda S, Gorman AM, Hori O. et al. Cellular stress responses: cell survival and cell death. Int J Cell Biol. 2010;2010:214074
32. Keely PJ, Westwick JK, Whitehead IP. et al. Cdc42 and Rac1 induce integrin-mediated cell motility and invasiveness through PI(3)K. Nature. 1997;390:632-6
33. Coniglio SJ, Jou TS, Symons M. Rac1 protects epithelial cells against anoikis. J Biol Chem. 2001;276:28113-20
34. Wennerberg K, Ellerbroek SM, Liu RY. et al. RhoG signals in parallel with Rac1 and Cdc42. J Biol Chem. 2002;277:47810-7
35. Ueyama T, Geiszt M, Leto TL. Involvement of Rac1 in activation of multicomponent Nox1- and Nox3-based NADPH oxidases. Mol Cell Biol. 2006;26:2160-74
36. Sarbassov DD, Guertin DA, Ali SM. et al. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005;307:1098-101
37. Yuan TL, Cantley LC. PI3K pathway alterations in cancer: variations on a theme. Oncogene. 2008;27:5497-510
38. Ji H, Ding Z, Hawke D. et al. AKT-dependent phosphorylation of Niban regulates nucleophosmin- and MDM2-mediated p53 stability and cell apoptosis. EMBO Rep. 2012;13:554-60
39. Porta C, Paglino C, Mosca A. Targeting PI3K/Akt/mTOR Signaling in Cancer. Front Oncol. 2014;4:64
40. Zhang J, Yu XH, Yan YG. et al. PI3K/Akt signaling in osteosarcoma. Clin Chim Acta. 2015;444:182-92
41. Bar-Sagi D, Hall A. Ras and Rho GTPases: a family reunion. Cell. 2000;103:227-38
42. Minden A, Lin A, Claret FX. et al. Selective activation of the JNK signaling cascade and c-JUN transcriptional activity by the small GTPases Rac and Cdc42Hs. Cell. 1995;81:1147-57
43. Chiariello M, Marinissen MJ, Gutkind JS. Regulation of c-myc expression by PDGF through Rho GTPases. Nat Cell Biol. 2001;3:580-6
44. Kim KS, Takeda K, Sethi R. et al. Protection from reoxygenation injury by inhibition of rac1. J Clin Invest. 1998;101:1821-6
45. Ozaki M, Deshpande SS, Angkeow P. et al. Inhibition of the Rac1 GTPase protects against nonlethal ischemia/reperfusion-induced necrosis and apoptosis in vivo. Faseb j. 2000;14:418-29
46. Atalla SL, Toledo-Pereyra LH, MacKenzie GH. et al. Influence of oxygen-derived free radical scavengers on ischemic livers. Transplantation. 1985;40:584-90
47. Gottlieb RA, Burleson KO, Kloner RA. et al. Reperfusion injury induces apoptosis in rabbit cardiomyocytes. J Clin Invest. 1994;94:1621-8
48. Cursio R, Gugenheim J, Ricci JE. et al. A caspase inhibitor fully protects rats against lethal normothermic liver ischemia by inhibition of liver apoptosis. Faseb j. 1999;13:253-61
49. Xiang SY, Vanhoutte D, Del Re DP. et al. RhoA protects the mouse heart against ischemia/reperfusion injury. J Clin Invest. 2011;121:3269-76
50. Bao W, Hu E, Tao L. et al. Inhibition of Rho-kinase protects the heart against ischemia/reperfusion injury. Cardiovasc Res. 2004;61:548-58
51. Pelloux S, Robillard J, Ferrera R. et al. Non-beating HL-1 cells for confocal microscopy: application to mitochondrial functions during cardiac preconditioning. Prog Biophys Mol Biol. 2006;90:270-98
Author contact
![]() Corresponding author: Yidong Wei, MD, Ph.D., Department of Cardiology, Shanghai Tenth People's Hospital, Tongji University School of Medicine, No. 301 Middle Yanchang Road, Shanghai, 200072, China. Telephone number: +862166306945; E-mail: yweiedu.cn
Corresponding author: Yidong Wei, MD, Ph.D., Department of Cardiology, Shanghai Tenth People's Hospital, Tongji University School of Medicine, No. 301 Middle Yanchang Road, Shanghai, 200072, China. Telephone number: +862166306945; E-mail: yweiedu.cn
Received 2018-2-12
Accepted 2018-6-7
Published 2018-6-23