ISSN: 1449-1907International Journal of Medical Sciences

Int J Med Sci 2017; 14(3):275-283. doi:10.7150/ijms.17860 This issue Cite
Research Paper
Regulation of DMT1 on autophagy and apoptosis in osteoblast
Fei Liu, Wei-Lin Zhang, Hong-Zheng Meng, Zheng-Yu Cai, Mao-Wei Yang ![]()
Department of Orthopedics, the First Hospital of China Medical University, Shenyang, Liaoning, China.
Abstract
Iron overload has recently been associated with the changes in the bone microstructure that occur in osteoporosis. However, the effect of iron overload on osteoblasts is unclear. The purpose of this study was to explore the function of divalent metal transporter 1 (DMT1) in the pathological processes of osteoporosis. Osteoblast hFOB1.19 cells were cultured in medium supplemented with different concentrations (0, 50, 100, 200, 300, 400, 500 μmol/L) of ferric ammonium citrate (FAC) as a donor of ferric ions. We used western blotting and immunofluorescence to determine the levels of DMT1 after treatment with FAC. Apoptosis was evaluated by detecting the levels of cleaved caspase 3, BCL2, and BAX with western blotting. Autophagy was evaluated by detecting the levels of LC3 with western blotting and immunofluorescence. Beclin-1 expression was also assessed with western blotting. The autophagy inhibitor 3-methyladenine was used to determine whether autophagy affects the apoptosis induced by FAC. Our results show that FAC increased the levels of DMT1, upregulated the expression of BCL2, and downregulated the apoptosis-related proteins cleaved caspase 3 and BAX. Both LC3I/LC3II levels and beclin-1 were also increased, indicating that FAC increases the accumulation of autophagosomes in hFOB1.19 cells. FAC-induced autophagy was increased by the apoptosis inhibitor 3-MA but was reduced in DMT1 shRNA hFOB1.19 cells. These results suggest that the increased expression of DMT1 induces iron overload and iron overload induces osteoblast autophagy and apoptosis, thus affecting the pathological processes of osteoporosis. Clarifying the mechanisms underlying the effects of DMT1 will allow the identification of novel targets for the prevention and treatment of osteoporosis.
Keywords: divalent metal transporter 1, DMT1, osteoporosis, autophagy, apoptosis, osteoblast.
Introduction
Osteoporosis is the most common systemic metabolic bone disease in the elderly. It is characterized by microarchitectural deterioration, low bone mass, and an increased risk of fracture. This common disease occurs when bone breakdown occurs more rapidly than bone formation [1]. The various diverse complications of osteoporosis, including fractures, affect the patient's quality of life and health, and the economic burden of osteoporosis is markedly increasing as the world population ages [2].
Osteoporosis and iron overload are clearly related. Some researchers have shown that iron overload has an inhibitory effect on osteogenesis [3], whereas others have demonstrated a higher incidence of osteoporosis in patients with iron overload diseases, including hereditary hemochromatosis, thalassemia, and sickle-cell anemia [4-8]. Iron overload also reduces osteoblast function by inducing osteoblast apoptosis, which can cause osteoporosis [9]. However, in osteoporotic patients, the processes underlying the changes in the bone microstructure and the precise mechanism of osteoblast iron overload are unclear.
Divalent metal transporter 1 (DMT1) is a 12-transmembrane-domain protein found in a range of tissues, including the duodenum, kidney, and bone, that transports a number of divalent cations [10]. The cellular transport of Fe2+ is heavily dependent on DMT1 [11]. In a previous study, we found that osteoporosis was closely associated with DMT1 [12], and Chew et al. reported that the overexpression of DMT1 causes dramatic iron overload [13]. Therefore, an intimate relationship between DMT1 expression and iron overload has been suggested. Several studies have also reported that iron overload can induce either cell autophagy or apoptosis [14, 15]. However, no study has reported that the DMT1 expressed in osteoblasts affects cell autophagy by regulating the concentration of Fe2+ ions, thereby affecting apoptosis.
Autophagy is the primary metabolic process by which eukaryotic cells are degraded and damaged macromolecules and organelles are salvaged [16, 17]. It plays a housekeeping role in eliminating old organelles, misfolded proteins, and damaged molecules, and a role in recycling limited nutrients and oxygen [18]. Recent studies have shown that autophagy plays a complex and important role during osteogenesis [19]. Autophagy is closely associated with apoptosis and the functional relationship between apoptosis and autophagy is complex [20]. Autophagy sometimes constitutes a stress adaptation that suppresses apoptosis, whereas in other cellular situations, it constitutes an alternative cell-death pathway [18, 21-25]. However, how autophagy affects the pathology of iron overload in osteoblast cells remains unclear.
All the data discussed above suggest close associations between DMT1 expression, iron overload, and osteoporosis. However, how autophagy affects the pathology of iron overload caused by DMT1 in osteoblast cells remains unclear. The aims of this study were to determine the role of autophagy in osteoporosis-related fracture by examining the autophagy levels in osteoblasts exposed to iron overload; to determine whether autophagy acts through DMT1; and to explore the relationship between autophagy and apoptosis in terms of iron overload.
Materials and Methods
Cell culture and materials
The human fetal osteoblastic cell line hFOB 1.19, kindly provided by Dr. M. Subramaniam [26], was maintained in a 1:1 mixture of Ham's F12 Medium Dulbecco's Modified Eagle Medium without phenol red (Gibco, USA), supplemented with 10% fetal bovine serum (FBS) (HyClone, USA) and 0.3 g/L G418 (Sigma, USA), in a humidified 5% CO2 atmosphere at 33.5°C, and the medium was changed every other day. The cell was subcultured using trypsin-EDTA to replace the cells and begin the experiment. The hFOB 1.19 cells were plated at 104 cells/cm2 for 24 h before treatment.
The 3-(4,5-dimethyl-thiazol-2-yl)- 2,5-diphenyl-tetrazolium bromide (MTT) and FAC were obtained from Sigma. The DMT1-shRNA lentiviral was purchased from Genechem (China). Primary antibodies for LC3 was purchased from Cell Signaling Technology (CST, USA) and for DMT1, beclin-1, BAX, cleaved caspase 3 and BCL2 were purchased from Abcam (USA).
Cell viability and proliferation analysis
Cell viability was measured using MTT. Briefly, the cells were seeded onto 96-well plates (6000 cells/well) for 24 h, and the medium was the replaced with 10% serum medium. After treatment, culture media was changed for serumfree culture media. MTT dissolved in phosphate buffer saline (PBS) was added to each well and then incubated for 4 h. After this interval, the serum-free culture media containing MTT was discarded and dimethyl sulfoxide (DMSO) was added to each well dissolving the precipitate. The optical densities were measured at 490 nm spectral wavelengths using a microplate reader (Spectra Thermo, Switzerland). Cell proliferation was estimated using a BrdU kit (Roche, China) following the protocol of the manufacturer. Cell viability and proliferation results were expressed as percentages. The absorbency measured from untreated cells was taken to be 100%.
Cell apoptosis analysis
Cell apoptosis was determined by detecting phosphatidylserine exposure on the cellular plasma membranes using the fluorescent dye Annexin V-APC/7AAD apoptosis detection kit according to the manufacturer's protocols. In brief, the cells were harvested, washed twice in ice-cold phosphate buffer saline (PBS), resuspended in 500 μL of binding buffer, incubated with 5 μL of Annexin V-APC and 5 μL of 7AAD solution for 15 min at room temperature in the dark and then immediately analyzed by bivariate flow cytometry using a FACScan-LSR equipped with Cell Quest software. Approximately 5×105 cells were analyzed in each of the samples. The experiment was repeated three times.
Western blotting
After treatment, the cells were extracted with lysis buffer (150 mm NaCl, 1% NP-40, 0.1% SDS, 2 μg/mL aprotinin, 1 mm PMSF) for 30 min at 4°C. The supernatants were centrifuged at 12,000 g for 15 min at 4°C. The supernatant containing total protein was harvested. Aliquots containing 50 μg of proteins were separated by a 12% SDS-PAGE and transferred to PVDF membranes at 60 V or 40 V for 2 h at low temperature. The membranes were soaked in blocking buffer (5% skimmed milk) for 2 h. Subsequently, proteins were detected using primary antibodies at 1:500 or 1:1000 dilution for overnight at 4°C, then visualized using anti-goat or anti-rabbit IgG conjugated with peroxidase (HRP) at 1:6000 or 1:8000 dilution for 2 h at room temperature. The EC3 Imaging System (UVP Inc. Upland, CA, USA) was used to catch up the specific bands, and the optical density of each band was measured using an Image J software (NIH, Bethesda, MD, USA). The rate between interesting proteins and β-actin of the same sample was calculated as relative content and expressed graphically.
Transmission electron microscopy
The cells from each group were digested after 24 h of culture, followed by centrifugation, and the floating cells were collected. The cells were washed twice with cold PBS and fixed in 5% glutaraldehyde. Subsequently, the cells were conventionally dehydrated, embedded, sectioned, and stained, and the formation of autophagosomes was observed using transmission electron microscopy. The number of intracellular autophagosomes in every ten fields was counted.
Immunofluorescence
Cells were fixed with 4% paraformaldehyde at room temperature for 15 min. After washing with PBS, cells were permeabilized with 0.2% Triton X-100 for 5 min. After washing with PBS, secions were incubated in a blocking buffer containing 5% BSA for 30 min at room temperature, followed by incubation with anti-LC3 (1:200) and anti-DMT1(1:200)antibody overnight at 4 ̊C. Secondary antibodies labeled with fluorescein (1:500, Abcam, USA) were applied for 120 min. After incubating with 0.1% DAPI for 5 min and another washing step with PBS, coverslips were transferred onto glass slides. Images were captured on a wide-field fluorescent microscopy (Olympus, Japan). A CLSM was used to measure the green fluorescence when excited at 492 nm and emitted at 517 nm.
RNA interference
For the gene knockdown experiments, we obtained lentiviral shRNA purchased from Shanghai GenePharma. The shRNA was designed against DMT1 (sense, 5'-GAGCAGTGGCTGGATTTAAG-3'; antisense, 5'-CGGTGACATACTTCAGCAAG-3'). DMT1-shRNA-lentivirus was added into target hFOB 1.19 cells at multiplicity of infection [27] with ENi.S and 5 µg/mL polybrene to obtain stably- transfected DMT1-shRNA.
Statistical analyses
The experiments were repeated 3 times. The quantitative data are presented as means ± S.E.M. The Statistical Package for Social Science (SPSS) 17.0 software was used for analysis. Comparisons among multiple groups were performed using one-way analysis of variance (ANOVA). Pair-wise comparisons were performed using the t test. P<0.05 indicated that the observed difference was significant.
Results
Effects of FAC on osteoblastic hFOB1.19 cells
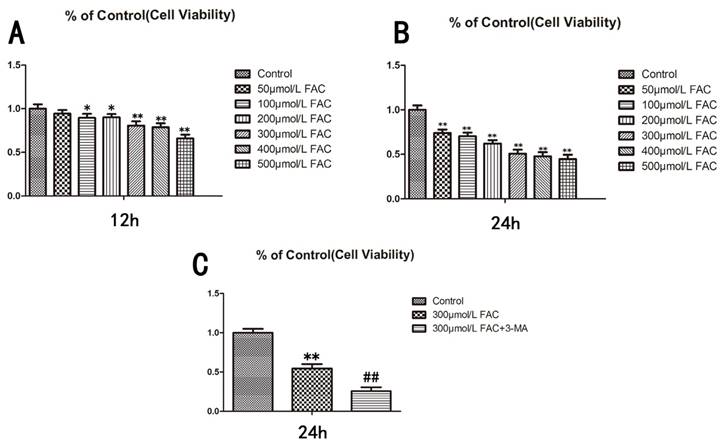
To determine the influence of FAC on the viability of osteoblasts, we treated human hFOB1.19 osteoblasts with different concentrations of FAC (0, 50, 100, 200, 300, 500 µmol/L) for different time periods (12, 24 h), and then assessed the viability of the treated cells with a 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay (Fig.1A,B). The viability of the cells treated with 300 µmol/L FAC for 24 h decreased by 50% relative to that of the control cells. Therefore, a FAC concentration of 300 µmol/L was used for subsequent experiments (P < 0.001 vs. control cells).
To investigate the effect of autophagy on cell viability, 3-methyladenine (3-MA), a classic inhibitor of autophagy, was used to inhibit cell autophagy. 3-MA significantly enhanced the effect of 300 µmol/L FAC in reducing the viability of hFOB1.19 cells (Fig.1C), suggesting that the effect of FAC in inhibiting cell viability is related to cell autophagy.
DMT1 is upregulated in FAC-treated hFOB1.19 cells
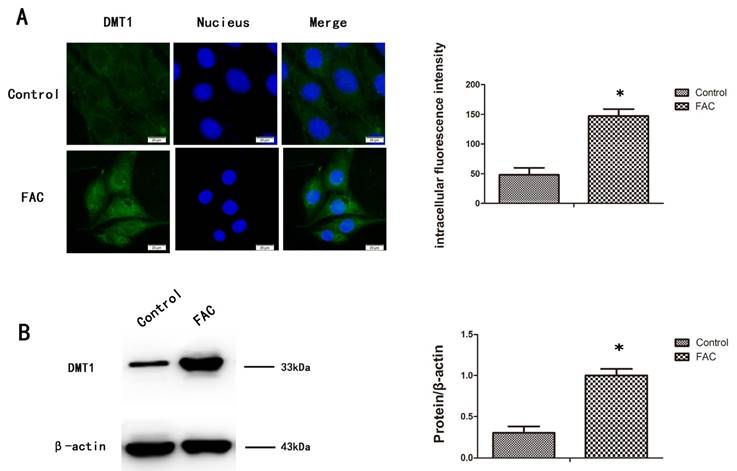
To measure the expression of DMT1 after treatment with 300 µmol/L FAC, we used a western blotting analysis and immunofluorescence to confirm that DMT1 was significantly upregulated in hFOB1.19 cells after exposure to FAC for 24 h (Fig.2A,B).
Transfection of hFOB1.19 cells with a DMT1-encoding small hairpin RNA (DMT1-shRNA) lentivirus
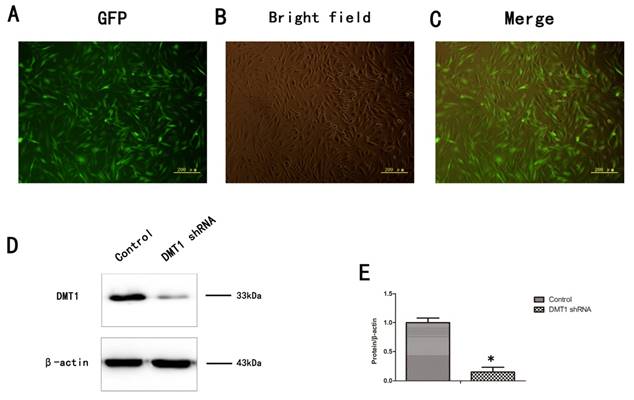
To verify the role of DMT1 in human osteoporosis, we altered the expression of DMT1 in hFOB1.19 cells. More than 80% of the cells expressed green fluorescent protein (GFP) (Fig.3A,B,C). We then used a western blotting analysis to detect the expression of DMT1 after the hFOB1.19 cells were transfected with the DMT1-shRNA lentivirus (Fig.3D). The results confirmed that the cells were successfully.
Viability of hFOB1.19 cells treated with increasing concentrations of FAC and autophagy is involved in the effect of FAC was tested via an MTT assay. A. Cells were treated with various concentrations of FAC for 12 h. B. Cells were treated with various concentrations of FAC for 24 h. C. Cells were then treated with or without 5 mmol/L 3-MA and incubated with 300 µmol/L FAC for 24 h. Values represent the mean ± S.E.M. of at least three independent experiments. *P < 0.05 vs. control, **P < 0.01 vs. control, #P<0.05 vs. FAC treatment, ##P<0.01 vs. FAC treatment.
Expression of DMT1 in hFOB1.19 cells after treatment with FAC. A. Fluorescence intensity of DMT1 (green fluorescence) in hFOB1.19 cells treated with 300 µmol/L FAC for 24 h. Histogram shows the fluorescence intensity of DMT1 in the different groups. B. Western blot shows the DMT1 protein levels in hFOB1.19 cells treated with 300 µmol/L FAC for 24 h. Histogram shows the expression of DMT1 in the different groups, analyzed with western blotting. Values represent the mean ± S.E.M. of at least three independent experiments. *P < 0.05 vs. control.
Expression of DMT1 in hFOB1.19 cells transfected with DMT1-shRNA-encoding lentivirus was analyzed using western blotting. A. Expression of green fluorescent protein (GFP)-labeled DMT1 in hFOB1.19 cells was assessed under an inverted fluorescence microscope. B. hFOB1.19 cells were observed with an optical microscope. C. Merging of A and B. D. Western blotting shows DMT1 protein levels in hFOB1.19 cells. Histogram shows the expression of DMT1 in the different groups, analyzed with western blotting. Values represent the mean ± S.E.M. of at least three independent experiments. *P < 0.05 vs. control.
Relationship between FAC and apoptosis
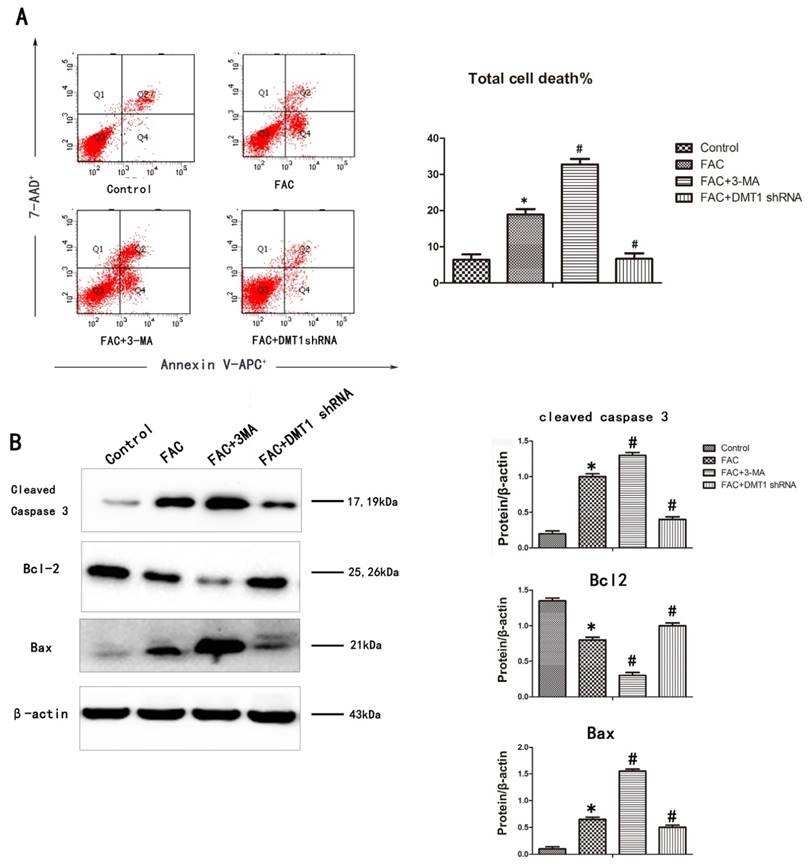
To determine whether the inhibitory effect of 300 µmol/L FAC for 24 h on osteoblast viability is related to cell apoptosis, an annexin V-APC/7AAD apoptosis detection kit was used. The DNA fragmentation ratio in the FAC-treated hFOB1.19 cells was higher than that in the DMT1-shRNA hFOB1.19 cells and the control group. In contrast, 3-MA markedly enhanced the FAC-induced inhibition of cell viability and promoted apoptotic cell death (Fig.4A).
A western blotting analysis was used to investigate the mechanisms responsible for DMT1-mediated cell protection. The expression of the proapoptotic proteins BAX and cleaved caspase 3 increased, whereas the expression of the antiapoptotic protein BCL2 deceased in the hFOB1.19 cells. The addition of 3-MA increased BAX and cleaved caspase 3 even further and also further reduced the expression BCL2. Compared with the past, this result was opposite that observed in the DMT1-shRNA hFOB1.19 cells (Fig.4B). These findings demonstrate that caspase-dependent FAC-induced apoptotic cell death was significantly exacerbated by the inhibition of autophagy, but was attenuated in the DMT1-shRNA hFOB1.19 cells.
Relationship between FAC and the accumulation of autophagosomes
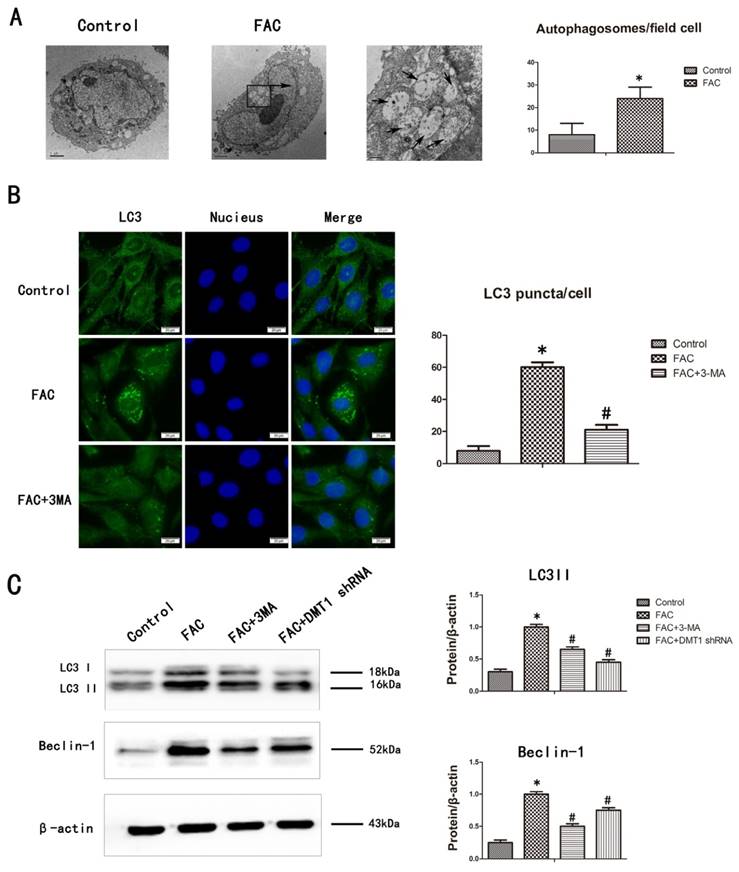
We confirmed that FAC induced cell autophagy by counting the number of autolysosomes present in the cells with transmission electron microscopy (TEM) and fluorescence microscopy. TEM is the gold standard technique for monitoring autophagy [28]. After treatment with 300 µmol/L FAC for 24 h, the number of autolysosomes in the hFOB1.19 cells increased significantly compared with those in the controls (Fig.5A). LC3 is mainly located on the surface of pre-autophagosomes and autophagosomes, and is known as a general biomarker of autophagy [28]. We used fluorescence microscopy to observe the punctate aggregation (autolysosomes) of internal LC3. The results were consistent with the results of TEM. The punctate aggregation of both proteins increased significantly after treatment with 300 µmol/L FAC for 24 h compared with the controls, and when 3-MA was added, the punctate aggregations decreased significantly compared with those in the FAC-treated cells without 3-MA(Fig.5B). These results suggest that FAC increases the accumulation of autophagosomes of hFOB1.19 cells.
The levels of LC3-II protein, which is formed by the lipidation of the cytosolic LC3-I protein, and beclin-1 correlate with the number of autophagosomes present in cells [28]. Their expression also correlates positively with the level of autophagy. To determine whether FAC affects the autophagy of hFOB1.19 cells, we examined the changes in the expression of autophagy-associated proteins, including LC3I/LC3II and beclin-1, with a western blotting analysis. Treatment with 300 µmol/L FAC for 24 h increased the expression of both LC3II and beclin-1 in hFOB1.19 cells to much higher levels than those in the DMT1-shRNA hFOB1.19 cells. 3-MA significantly inhibited the expression of both LC3II and beclin-1 induced by 300 µmol/L FAC in hFOB1.19 cells (Fig.5C). The concurrent increases in LC3II and beclin-1 in these cell types indicate that FAC causes an accumulation of autophagosomes [28], whereas DMT1 reduced the FAC-induced accumulation of autophagosomes in hFOB1.19 cells.
FAC-induced apoptosis in hFOB1.19 cells. A. Cells were collected, stained with annexin V-APC/7AAD, and examined with flow cytometry. Histogram shows the apoptosis rate in each group. B. Western blotting shows cleaved caspase 3, BCL2, and BAX levels in hFOB1.19 cells treated with or without 5 mmol/L 3-MA and incubated with 300 µmol/L FAC for 24 h. Histogram shows the expression of cleaved caspase 3, BCL2, and BAX in the different groups, analyzed with western blotting. Values represent the mean ± S.E.M. of at least three independent experiments. *P < 0.05 vs. control, #P<0.05 vs. FAC treatment.
FAC-induced accumulation of autophagosomes in hFOB1.19 cells is blocked by 3-MA. A. Transmission electron microscopy shows autophagosomes (bold arrows) in hFOB1.19 cells treated with 300 µmol/L FAC for 24 h. Quantitation of autophagosomes. B. Fluorescence microscopy shows endogenous LC3 (green fluorescence) and nuclei (blue fluorescence) in hFOB1.19 cells treated with or without 5 mmol/L 3-MA and incubated with 300 µmol/L FAC for 24 h. Quantitation of LC3 puncta. C. Western blotting shows LC3 and beclin-1 levels in hFOB1.19 cells treated with or without 5 mmol/L 3-MA and incubated with 300 µmol/L FAC for 24 h. Histogram shows the expression of LC3 and beclin-1 in the different groups, analyzed with western blotting. Values represent the mean ± S.E.M. of at least three independent experiments. *P < 0.05 vs. control, #P<0.05 vs. FAC treatment.
Discussion
Osteoporosis is an important complication of many diseases, including hemochromatosis, which is associated with iron overload [29, 30], and one study found that postmenopausal osteoporosis induces the accumulation of Fe2+ ions [31]. These data indicate that iron plays a crucial role in osteoporosis. Iron overload can induce apoptosis in osteoblasts and impair osteoblast differentiation and mineralization, as well as their bone-mineralizing capacity [32]. Some researchers have shown that iron overload alters the iron metabolism and suppresses the differentiation and functions of cells in the osteoblast lineage, which is associated with increased apoptosis [33]. Other researchers have reported that excessive iron can increase apoptosis and reduce the mRNA expression of collage-1 (COL-I) and osteocalcin (OCN) in hFOB1.19 cells [9]. Kanako Yamasaki suggested that iron overload gives rise to osteoporosis by inhibiting osteoblast proliferation and differentiation [29]. In summary, iron overload has been shown to reduce osteoblast function by inducing osteoblast apoptosis. In this study, we used the hFOB1.19 cell line as a cellular model because it is homologous to human osteoblasts and known to have a strong proliferative capacity [34]. We found that 300 µmol/L FAC caused the apoptosis of osteoblastic hFOB1.19 cells. FAC is considered a supplier of Fe2+ ions, allowing an iron overload environment to develop [32]. At the same concentration of FAC, we also found that iron overload induced the accumulation of autophagosomes. When 3-MA was added, the accumulation of autophagosomes decreased and the apoptosis increased. Therefore, we suggest that the iron-overload-induced accumulation of autophagosomes, which adversely affects osteoblasts, interferes with their apoptosis.
DMT1 is the major apical transporter responsible for intestinal Fe2+ absorption, and is also ubiquitously expressed in endosomal compartments, where it is responsible for Fe2+ export from the endosome during the transferrin cycle [10, 35]. Recently, a study found that the total iron content was significantly elevated in PD patients, with a corresponding increase in DMT1 transcripts in the same region [36]. In a previous study, we found that DMT1 is markedly increased in osteoporosis and the loss of DMT1 can reduce the iron content of bone [12]. This suggests a close association between DMT1 expression and iron overload. It has already been confirmed that iron overload promotes cell autophagy [13, 37]. Therefore, there is also a close relationship between DMT1 and autophagy. Consistent with previous studies, we found that cells treated with FAC expressed more DMT1 than untreated cells. Therefore, we used DMT1-shRNA hFOB1.19 cells to confirm that DMT1 affects the cellular accumulation of autophagosomes by regulating the Fe2+ concentration in osteoblasts, which can affect apoptosis. We found that the FAC-induced accumulation of autophagosomes was reduced in the DMT1-shRNA hFOB1.19 cells. We also found that apoptosis was also reduced in the DMT1-shRNA hFOB1.19 cells. Therefore, we hypothesized that DMT1 regulates the Fe2+ concentrations in osteoblasts, which in turn affect the cellular accumulation of autophagosomes. The accumulation of autophagosomes then affects apoptosis.
Autophagy and apoptosis may be triggered by common upstream signals, which sometimes results in both autophagy and apoptosis. Under other circumstances, the cell switches between the two responses in a mutually exclusive manner. Therefore, on the molecular level, the apoptotic and autophagic response mechanisms share common pathways, which either link or polarize the cellular responses [20]. Although the interaction between apoptosis and autophagy has not been studied comprehensively, it is currently a topical research issue. We found that autophagy can reduce the apoptosis of osteoblasts through the intervention of autophagy. This result is consistent with current related studies in osteoblasts [38, 39]. Many factors can affect both the autophagy and apoptosis of osteoblasts, including fluoride, high glucose, and paxilitaxel [40-42]. However, the effect of DMT1 on the apoptosis and autophagy of osteoblasts is first reported here. Iron overload can induce oxidative stress and increase reactive oxygen species (ROS), and it has been reported that ROS-induced apoptosis delays autophagy [43]. Therefore, we infer that DMT1 affects osteoblast apoptosis and autophagy through ROS.
In summary, iron overload had an adverse effect on the viability of hFOB1.19 cells in vitro. In this study, we have demonstrated that DMT1 is increased in osteoporosis and induces iron overload. Iron overload affects osteoblast autophagy and apoptosis, thus affecting the pathological processes of osteoporosis. Clarification of the mechanism of DMT1 will allow the identification of novel targets for the prevention and treatment of osteoporosis.
Acknowledgements
This study was supported by the National Natural Science Foundation of China (No.:81471094), the National Science Foundation of Liaoning Province (No.:2015020725), and the Foundation for Science of Shenyang City(No.:F16-205-1-30).
Author Contributions
FL, WLZ, HZM, ZYC, MWY conceived of the study, participated in the design of the study and performed the statistical analyses. All authors carried out the experiments. FL drafted the manuscript with the help of WLZ, HZM and YMW. All authors have read and approved the final manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. An J, Yang H, Zhang Q, Liu C, Zhao J, Zhang L. et al. Natural products for treatment of osteoporosis: The effects and mechanisms on promoting osteoblast-mediated bone formation. Life sciences. 2016;147:46-58
2. Leboime A, Confavreux CB, Mehsen N, Paccou J, David C, Roux C. Osteoporosis and mortality. Joint, bone, spine: revue du rhumatisme. 2010;77(Suppl 2):S107-12
3. Chen B, Yan YL, Liu C, Bo L, Li GF, Wang H. et al. Therapeutic effect of deferoxamine on iron overload-induced inhibition of osteogenesis in a zebrafish model. Calcified tissue international. 2014;94:353-60
4. Guggenbuhl P, Deugnier Y, Boisdet JF, Rolland Y, Perdriger A, Pawlotsky Y. et al. Bone mineral density in men with genetic hemochromatosis and HFE gene mutation. Osteoporosis international: a journal established as result of cooperation between the European Foundation for Osteoporosis and the National Osteoporosis Foundation of the USA. 2005;16:1809-14
5. Morabito N, Russo GT, Gaudio A, Lasco A, Catalano A, Morini E. et al. The "lively" cytokines network in beta-Thalassemia Major-related osteoporosis. Bone. 2007;40:1588-94
6. Sarrai M, Duroseau H, D'Augustine J, Moktan S, Bellevue R. Bone mass density in adults with sickle cell disease. British journal of haematology. 2007;136:666-72
7. Skordis N, Toumba M. Bone disease in thalassaemia major: recent advances in pathogenesis and clinical aspects. Pediatric endocrinology reviews: PER. 2011;8(Suppl 2):300-6
8. Valenti L, Varenna M, Fracanzani AL, Rossi V, Fargion S, Sinigaglia L. Association between iron overload and osteoporosis in patients with hereditary hemochromatosis. Osteoporosis international: a journal established as result of cooperation between the European Foundation for Osteoporosis and the National Osteoporosis Foundation of the USA. 2009;20:549-55
9. Zhao GY, Zhao LP, He YF, Li GF, Gao C, Li K. et al. A comparison of the biological activities of human osteoblast hFOB1.19 between iron excess and iron deficiency. Biological trace element research. 2012;150:487-95
10. Gunshin H, Mackenzie B, Berger UV, Gunshin Y, Romero MF, Boron WF. et al. Cloning and characterization of a mammalian proton-coupled metal-ion transporter. Nature. 1997;388:482-8
11. East E, Baker D, Pryce G, Lijnen HR, Cuzner ML, Gveric D. A role for the plasminogen activator system in inflammation and neurodegeneration in the central nervous system during experimental allergic encephalomyelitis. The American journal of pathology. 2005;167:545-54
12. Zhang WL, Meng HZ, Yang MW. Regulation of DMT1 on Bone Microstructure in Type 2 Diabetes. International journal of medical sciences. 2015;12:441-9
13. Chew KC, Ang ET, Tai YK, Tsang F, Lo SQ, Ong E. et al. Enhanced autophagy from chronic toxicity of iron and mutant A53T alpha-synuclein: implications for neuronal cell death in Parkinson disease. The Journal of biological chemistry. 2011;286:33380-9
14. Chen G, Jing CH, Liu PP, Ruan D, Wang L. Induction of autophagic cell death in the rat brain caused by iron. The American journal of the medical sciences. 2013;345:369-74
15. Li SW, Liu CM, Guo J, Marcondes AM, Deeg J, Li X. et al. Iron overload induced by ferric ammonium citrate triggers reactive oxygen species-mediated apoptosis via both extrinsic and intrinsic pathways in human hepatic cells. Human & experimental toxicology. 2016;35:598-607
16. Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132:27-42
17. Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature. 2008;451:1069-75
18. Hocking LJ, Whitehouse C, Helfrich MH. Autophagy: a new player in skeletal maintenance?. Journal of bone and mineral research: the official journal of the American Society for Bone and Mineral Research. 2012;27:1439-47
19. Carames B, Taniguchi N, Otsuki S, Blanco FJ, Lotz M. Autophagy is a protective mechanism in normal cartilage, and its aging-related loss is linked with cell death and osteoarthritis. Arthritis and rheumatism. 2010;62:791-801
20. Maiuri MC, Zalckvar E, Kimchi A, Kroemer G. Self-eating and self-killing: crosstalk between autophagy and apoptosis. Nature reviews Molecular cell biology. 2007;8:741-52
21. Levine B, Yuan J. Autophagy in cell death: an innocent convict?. The Journal of clinical investigation. 2005;115:2679-88
22. Kroemer G, Jaattela M. Lysosomes and autophagy in cell death control. Nature reviews Cancer. 2005;5:886-97
23. Kondo Y, Kanzawa T, Sawaya R, Kondo S. The role of autophagy in cancer development and response to therapy. Nature reviews Cancer. 2005;5:726-34
24. Gozuacik D, Kimchi A. Autophagy and cell death. Current topics in developmental biology. 2007;78:217-45
25. Lockshin RA, Zakeri Z. Programmed cell death and apoptosis: origins of the theory. Nature reviews Molecular cell biology. 2001;2:545-50
26. Subramaniam M, Jalal SM, Rickard DJ, Harris SA, Bolander ME, Spelsberg TC. Further characterization of human fetal osteoblastic hFOB 1.19 and hFOB/ER alpha cells: bone formation in vivo and karyotype analysis using multicolor fluorescent in situ hybridization. Journal of cellular biochemistry. 2002;87:9-15
27. Li L, Li L, Zhang Z, Jiang Z. Hypoxia promotes bone marrow-derived mesenchymal stem cell proliferation through apelin/APJ/autophagy pathway. Acta biochimica et biophysica Sinica. 2015;47:362-7
28. Klionsky DJ, Abdelmohsen K, Abe A, Abedin MJ, Abeliovich H, Acevedo Arozena A. et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy. 2016;12:1-222
29. Yamasaki K, Hagiwara H. Excess iron inhibits osteoblast metabolism. Toxicology letters. 2009;191:211-5
30. Doyard M, Fatih N, Monnier A, Island ML, Aubry M, Leroyer P. et al. Iron excess limits HHIPL-2 gene expression and decreases osteoblastic activity in human MG-63 cells. Osteoporosis international: a journal established as result of cooperation between the European Foundation for Osteoporosis and the National Osteoporosis Foundation of the USA. 2012;23:2435-45
31. Liu G, Men P, Kenner GH, Miller SC. Age-associated iron accumulation in bone: implications for postmenopausal osteoporosis and a new target for prevention and treatment by chelation. Biometals: an international journal on the role of metal ions in biology, biochemistry, and medicine. 2006;19:245-51
32. He YF, Ma Y, Gao C, Zhao GY, Zhang LL, Li GF. et al. Iron overload inhibits osteoblast biological activity through oxidative stress. Biological trace element research. 2013;152:292-6
33. Messer JG, Kilbarger AK, Erikson KM, Kipp DE. Iron overload alters iron-regulatory genes and proteins, down-regulates osteoblastic phenotype, and is associated with apoptosis in fetal rat calvaria cultures. Bone. 2009;45:972-9
34. Harris SA, Enger RJ, Riggs BL, Spelsberg TC. Development and characterization of a conditionally immortalized human fetal osteoblastic cell line. Journal of bone and mineral research: the official journal of the American Society for Bone and Mineral Research. 1995;10:178-86
35. Fleming MD, Romano MA, Su MA, Garrick LM, Garrick MD, Andrews NC. Nramp2 is mutated in the anemic Belgrade (b) rat: evidence of a role for Nramp2 in endosomal iron transport. Proceedings of the National Academy of Sciences of the United States of America. 1998;95:1148-53
36. Salazar J, Mena N, Hunot S, Prigent A, Alvarez-Fischer D, Arredondo M. et al. Divalent metal transporter 1 (DMT1) contributes to neurodegeneration in animal models of Parkinson's disease. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:18578-83
37. Tan TC, Crawford DH, Jaskowski LA, Subramaniam VN, Clouston AD, Crane DI. et al. Excess iron modulates endoplasmic reticulum stress-associated pathways in a mouse model of alcohol and high-fat diet-induced liver injury. Laboratory investigation; a journal of technical methods and pathology. 2013;93:1295-312
38. Yang L, Meng H, Yang M. Autophagy protects osteoblasts from advanced glycation end products-induced apoptosis through intracellular reactive oxygen species. Journal of molecular endocrinology. 2016;56:291-300
39. Liu W, Dai N, Wang Y, Xu C, Zhao H, Xia P. et al. Role of autophagy in cadmium-induced apoptosis of primary rat osteoblasts. Scientific reports. 2016;6:20404
40. Wei M, Duan D, Liu Y, Wang Z, Li Z. Autophagy may protect MC3T3-E1 cells from fluoride-induced apoptosis. Molecular medicine reports. 2014;9:2309-15
41. Wang X, Feng Z, Li J, Chen L, Tang W. High glucose induces autophagy of MC3T3-E1 cells via ROS-AKT-mTOR axis. Molecular and cellular endocrinology. 2016;429:62-72
42. Guo Y, Huang C, Li G, Chen T, Li J, Huang Z. Paxilitaxel induces apoptosis accompanied by protective autophagy in osteosarcoma cells through hypoxia-inducible factor-1alpha pathway. Molecular medicine reports. 2015;12:3681-7
43. Pei J, Deng J, Ye Z, Wang J, Gou H, Liu W. et al. Absence of autophagy promotes apoptosis by modulating the ROS-dependent RLR signaling pathway in classical swine fever virus-infected cells. Autophagy. 2016:1-21
Author contact
![]() Corresponding author: Mao-Wei Yang, Department of Orthopedics, The First Hospital of China Medical University, 155 North Nanjing Street, Shenyang, Liaoning, 110001, China. E-mail: ymw69com; FAX: +86 24 83283360; Phone: +86 24 83283360.
Corresponding author: Mao-Wei Yang, Department of Orthopedics, The First Hospital of China Medical University, 155 North Nanjing Street, Shenyang, Liaoning, 110001, China. E-mail: ymw69com; FAX: +86 24 83283360; Phone: +86 24 83283360.
Received 2016-10-10
Accepted 2016-12-21
Published 2017-2-24