ISSN: 1449-1907International Journal of Medical Sciences

Int J Med Sci 2014; 11(12):1262-1269. doi:10.7150/ijms.10038 This issue Cite
Research Paper
Silencing SATB1 Inhibits the Malignant Phenotype and Increases Sensitivity of Human Osteosarcoma U2OS Cells to Arsenic Trioxide
Haiying Zhang1, Xuejin Su1, Li Guo2, Lingzhi Zhong1, Wenxue Li1, Zhen Yue1, Xiaotong Wang1, Yan Mu1, Xinna Li1, Ronggui Li1 ![]() , Zonggui Wang3
, Zonggui Wang3 ![]()
1. Key Laboratory of Pathobiology, Ministry of Education, Norman Bethune College of Medicine, Jilin University, Changchun, China
2. School of Public Health, Jilin University, Changchun, China
3. The Second Hospital of Jilin University, Changchun, China
Abstract
In a previous study, we found that the global genome organizer Special AT-rich binding protein 1 (SATB1) is highly expressed in mesenchymal-derived human osteosarcoma U2OS cells and that the knock-down of SATB1 results in the inhibition of cell proliferation. The present study was aimed at investigating the effect of silencing SATB1 on cell migration, invasion, apoptosis and resistance to the chemotherapeutic drug arsenic trioxide. Cell migration and invasion were detected by wound-healing assays and trans-well invasion assays, respectively. Cell apoptosis was analyzed by an in situ Cell Death Detection POD Kit, based on terminal deoxynucleotydyl transferase mediated dUTP nick-end labeling (TUNEL) staining and mRNAs were analyzed by real time qRT-PCR. We found that cell migration and invasion were inhibited and that the proportion of apoptotic cells and sensitivities to the chemotherapeutic drug arsenic trioxide were enhanced by knockdown of SATB1 in U2OS cells. Furthermore, mRNA of ABCC1 and ABCG2 were decreased strikingly after SATB1 silencing. It was concluded that the elevated expression of SATB1 in U2OS cells contributes to maintenance of the malignant phenotype and resistance to chemotherapeutic drugs ATO, suggesting that silencing SATB1 in the cells might improve the effects of arsenic trioxides in the treatment of osteosarcoma in which SATB1 is over-expressed and that ABCC1 and ABCG2 were involved in SATB1 mediated resistance of U2OS cells to ATO.
Keywords: SATB1, migration, invasion, apoptosis, arsenic trioxide
Introduction
Osteosarcoma is the most common malignant bone tumor in children and adolescents [1]. Many patients experience tumor recurrence following treatment with current osteosarcoma therapy, which consists of chemotherapy and surgery. Considering that the majority of high-grade osteosarcoma patients are children and adolescents whose remaining life expectancy is considerably longer than other cancer patients, the long-term effects of chemotherapy must be taken into account. Therefore, the development of novel approaches that can overcome osteosarcoma resistance to current drugs and have better toxicity profiles are urgently needed.
Methotrexate (MTX), Doxorubicin, cis-dichlorodiamineplatinum (CDDP), and ifosfamide (IFO) are the main chemotherapeutic agents used to treat osteosarcoma [2, 3]. However, cells within osteosarcoma tumors rapidly become less sensitive and/or develop resistance to these drugs [2, 3]. Furthermore, the application of these drugs increases the probability of collateral toxic effects [2-4]. Arsenic trioxide (ATO) has been successfully used to treat leukemia [5-8] and some solid malignant tumors [9, 10]. ATO has been shown to induce G2/M phase arrest in MGC803 gastric cancer cells and induces apoptosis in APL-derived NB4 cells [11]. ATO has also been shown to induce apoptosis in acute promyelocytic leukemia cells by inducing the phosphorylation and activation of Chk2 and p38 MAPK [12]. ATO has been shown to induce growth arrest in human hepatocellular carcinoma cells by increasing FOXO3a expression and its translocation from the cytoplasm to the nucleus [13]. Other research has shown that ATO inhibits the metastasis of Ewing's sarcoma cells [14].
Special AT-rich binding protein 1 (SATB1) is a global genome organizer that binds selectively to the nuclear matrix DNAs through a unique nuclear architecture [15, 16] and recruits chromatin remodeling factors to regulate chromatin structure and gene expression [17, 18]. It has been shown that SATB1 expression plays an important role in breast cancer progression and predicts a poor prognosis [19, 20]. SATB1 expression is also upregulated in multidrug resistant breast cancer cells that exhibit higher invasive potential than the parental cells [21]. In addition to breast cancer, it has been reported that SATB1 expression may have value in determining the invasive and metastatic potential of gastric cancer [22], and that it might serve as an independent prognostic marker for predicting outcome of cutaneous malignant melanoma [22]. Our previous study showed that SATB1 was highly expressed in mesenchymal derived human osteosarcoma U2OS cells and that the knock-down of SATB1 suppressed the proliferation of the cells [23].
The present study was carried out to seek evidence for a role of SATB1 in maintaining the malignancy of human osteosarcoma U2OS cells and their resistance to chemotherapeutic drugs. We investigated the effect of silencing SATB1 on cell migration, invasion, apoptosis and the resistance to ATO. It was found that cell migration and invasion were inhibited and that the apoptosis and sensitivities to ATO were enhanced by silencing of SATB1 in the cells. These results indicate that the increased expression of SATB1 in U2OS cells contributes to their maintenance of the malignant phenotype and resistance to ATO, suggesting that silencing SATB1 in the cells might improve the therapeutic effects of ATO on those osteosarcomas in which SATB1 is over-expressed.
Materials and methods
Materials
Arsenic trioxide was purchased from Pharmaceuticals Limited Company of Harbin Medical University (Harbin, China). High Glucose Dulbecco's Modified Eagle Medium (H-DMEM) was from Gibco BRL (Rockville, USA). Fetal bovine serum (FBS) was from HyClone Inc. (Logan, USA). Cell Counting Kit-8 (CCK-8) was from Dojindo (Tokyo, Japan), in situ Cell Death Detection Kit, POD from Roche (Penzberg, Germany) and G418 from Invitrogen Life Technologies (Carlsbad, USA).
Cell culture
Human osteosarcoma U2OS cells were purchased from the American Type Culture Collection (Manassas, USA). The cells in which SATB1 had been knocked down (U2OS SATB1-shRNA) and the control cells (U2OS control-shRNA) have been described and used in our previous study [23]. They were made by the stable transfection with a shRNA expressing vector against SATB1 and a scrambled shRNA, respectively. All the cells were maintained in H-DMEM supplemented with 10% FBS at 37°C in a 5% CO2 and humidified atmosphere.
Cell migration assay
Cell migration was determined by a wound-healing assay. The cells were grown to confluence on 6-cm dishes. A scratch was then made through the cell monolayer using a sterile 200-µl pipette tip. After washing twice with PBS, fresh culture medium and mitomycin C (25 μg/ml) were added and cells were incubated at 37°C for another 24 hours. Photographs of the wounded areas were taken at 0 h and 24 h after making the wound. The widths of the wounded areas were measured at 0 h (M0) and 24 h (M24). The relative migration distance (%)= (M0-M24)/M0×100.
Cell invasion assay
Cell suspensions (each with 5×104 cells in 0.1 ml of serum-free H-DMEM medium) were added to the top chamber of 24-well cell culture inserts with 8μm pores (Corning, Medfield, Massachusetts, USA) coated with Matrigel (Invitrogen, Carlsbad, USA). 200μl of conditioned medium from cultured NIH/3T3 cells were added to the bottom chamber to create a chemotactic gradient. Cells were incubated at 37°C for 24 hours, fixed with neutral formaldehyde, and then stained with hematoxylin and eosin. The number of migrated cells was counted in five high-power fields in each of 6 samples. For the preparation of conditioned medium (CM), NIH/3T3 cells were grown to 80% confluence in H-DMEM medium containing 10% fetal bovine serum in a 5% CO2 and 37°C incubator. The culture medium was changed to serum-free H-DMEM medium. The cells were incubated for another 24 hours, the culture media was collected and centrifuged, and the supernatants were stored at -20°C and used as CM.
Cell apoptosis assay
The cells were grown on glass microscope slides and then fixed with 4% paraformaldehyde solution after incubation for 24 hours. The slides were terminal deoxynucleotydyl transferase mediated dUTP nick-end labeled (TUNEL-stained) using an in situ cell death detection POD kit (Roche, Penzberg, Germany) in accordance with the manufacturer's instructions. All slides were counterstained with hematoxylin. As a negative control, the terminal transferase was omitted. The TUNEL-positive cells were found after diaminobenzidine (DAB) coupling. The total number of cells and number of TUNEL-positive cells were counted in five high-power fields in each of 6 samples. All slides were read by an experienced scientist who was blinded to the evaluation and scoring and the average ratio of the number of TUNEL-positive cells to the total number of cells was calculated.
Sensitivity of the tumor cells to arsenic trioxide
Cells were seeded into 96-well plates at 1000 cells/well and incubated overnight to allow for cell attachment. On the following day, ATO was added to the wells at the final concentrations as indicated in Figure 4, and the cultures were incubated for an additional 24 hours. Cell viability was evaluated using the Cell Counting Kit-8 (CCK-8) (Dojindo, Japan) according to the manufacturer's instructions. Briefly, the cells were incubated with CCK8 solution and then the absorbance was determined at 450 nm wavelength in a micro plate reader. The absorbance values were converted to cell numbers from a standard curve of cell numbers against absorbance value which resulted from a series of analyses of samples in which the cell numbers were known.
RNA purification and qRT-PCR
For RNA purification, the cells from different clones in exponential growth phase were plated in 6 cm diameter dishes (1 × 106 cells / dish) and allowed to grow for 3 days. Total RNA purification and qRT-PCR were performed with a detailed procedure as described previously [24]. The primer sets used for PCR amplification are shown in Table 1. After amplification, a melting curve was generated and data analysis was performed by using Dissociation Curves 1.0 software (Applied Biosystems). The normalized value was given by the ratio of mRNA of the target gene to mRNA of the reference gene (RPL13) in each sample and the data were expressed relative to the value of the control cells (Control-shRNA).
Statistical Analysis
Statistical analyses were performed using SPSS software (SPSS, Version 17.0, Chicago, IL, USA). Differences among treatment groups were analyzed by one-way analysis of variance (ANOVA) with post hoc analysis using Dunnett's test. Differences were considered statistically significant when P<0.05.
Primers for qRT-PCR
| Gene | Primers Sequences | GenBank | |
|---|---|---|---|
| RPL13 | Forward Reverse | 5'-CGAGGTTGGCTGGAAGTACC-3' 5'-CTTCTCGGCCTGTTTCCGTAG-3' | NM_012423 |
| SATB1 | Forward Reverse | 5'-TCGACCTTCCCAAGTACACC-3' 5'-CGACCTCTAAACCGGAATTG-3 | NM_002971 |
| Suvivin | Forward Reverse | 5'-GGACCACCGCATCTCTACAT-3' 5'-TCTGGCTCGTTCTCAGTGG -3 | NM_001987 |
| Bcl-2 | Forward Reverse | 5'-GAACTGGGGGAGGATTGTGG-3' 5'-CCGGTTCAGGTACTCAGTCA-3' | NM_000633 |
| MMP2 | Forward Reverse | 5'-CTTCCAAGTCTGGAGCGATGT -3' 5'-TACCGTCAAAGGGGTATCCAT -3 | NM_004530 |
| MMP9 | Forward Reverse | 5'-GGGACGCAGACATCGTCATC -3' 5'-TCGTCATCGTCGAAATGGGC -3' | NM_004994 |
| TIMP3 | Forward Reverse | 5'-CATGTGCAGTACATCCATACGG-3' 5'-CATCATAGACGCGACCTGTCA-3 | NM_000362 |
| ABCC1 | Forward Reverse | 5'-CCAGTGGGGATCGGACAGA-3' 5'-AGGGGATCATCGAAGAGGTAAAT-3' | NM_004996 |
| ABCG2 | Forward Reverse | 5'-AACCTGGTCTCAACGCCATC-3' 5'-GTCGCGGTGCTCCATTTATC-3' | NM_004827 |
Results
SATB1 silencing suppressed the migration and invasiveness of U2OS cells
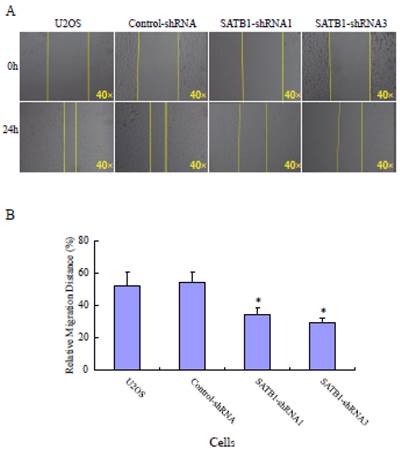
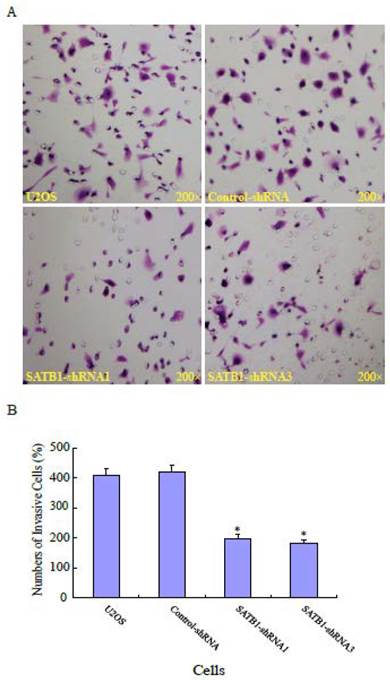
To explore the role of highly expressed SATB1 in maintaining the migratory and invasive traits of the cells, we examined whether knockdown of SATB1 in U2OS cells reduced their migratory and invasive traits using a monolayer wound-healing assay and a Matrigel based Boyden chamber invasion assay. Fig. 1A shows a representative photograph of cell migration and Fig. 1B shows a statistical analysis of the effects of silencing SATB1 on the migration. The percentage of relative migration distance in the two cell clones (SATB1-shRNA1 and SATB1-shRNA3) whose SATB1 had been silenced [23] were 34±4.7 and 29±3.3 respectively, when compared with the percentage of relative migration distance in parent (U2OS: 52±8.5 and transfection control (control-shRNA: 54±6.6). The data indicate that silencing SATB1 decreased the cell migration, suggesting increased expression of SATB1 in U2OS contributes to the elevated migration of these cells. Fig. 2A shows a representative photograph of the cell invasion and Fig. 2B shows the statistical analysis of the effects of silencing SATB1 on cell invasion. Numbers of invasive cells in the two cell clones (SATB1-shRNA1 and SATB1-shRNA3) whose SATB1 had been silenced were 197±15.0 and 182±12.4 respectively, when compared with numbers of invasive cells in parent (U2OS: 410±21.1) and transfection control (control-shRNA: 422±21.1). The data indicate that silencing SATB1 decreased the cell invasion, suggesting that high expression of SATB1 in U2OS contributes to the elevated cell invasion.
SATB1 Silencing decreased the cell migration. The cell migration assay and statistical analysis were carried out as described in the Materials and Methods. (A): A representative microscopic photograph of the cell migration. (B): Statistical results of cell migration. The data are expressed as the mean ± SD, N=9, *P < 0.05, versus non-silenced (Control-shRNA cells).
SATB1 silencing enhanced the apoptosis of U2OS cells
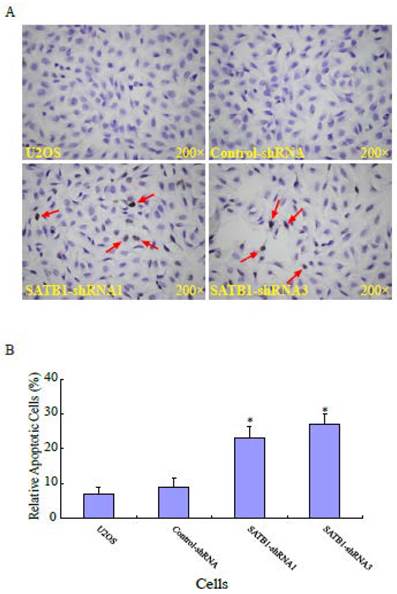
Apoptosis is the most common mechanism for cell death triggered by chemotherapeutic drugs [25, 26]. To study the effects of silencing SATB1 on apoptosis in human osteosarcoma U2OS cells, the apoptosis of the cells with different background of SATB1 was analyzed using a method based on terminal deoxynucleotydyl transferase mediated dUTP nick-end labeling (TUNEL) staining. Fig. 3A shows a representative photograph of the U2OS cells and the arrow indicates the apoptotic cells. As shown in Fig. 3B, the percentage of apoptotic cells in the two cell clones (SATB1-shRNA1 and SATB1-shRNA3) whose SATB1 had been silenced were 23± 3.5 and 27±2.9 respectively, when compared with the parent (U2OS: 7±2.1) and transfection control (control-shRNA: 9±2.7) cells. The data indicate that silencing SATB1 significantly increased the rates of apoptosis of the cells, suggesting the potential ability of highly expressed SATB1 in U2OS cells to prevent apoptosis of the cells.
SATB1 Silencing decreased cell invasion. The cell invasion assay and statistical analysis were carried out as described in the Materials and Methods. (A): A representative microscopic photograph of the cells that have passed through the membranes. (B): Statistical analysis showing that the number of cells passing through the membranes. The data are expressed as the mean ± SD, N=6, *P < 0.05, versus non-silenced (Control-shRNA) cells.
SATB1 silencing increased the sensitivities of U2OS cells to arsenic trioxide
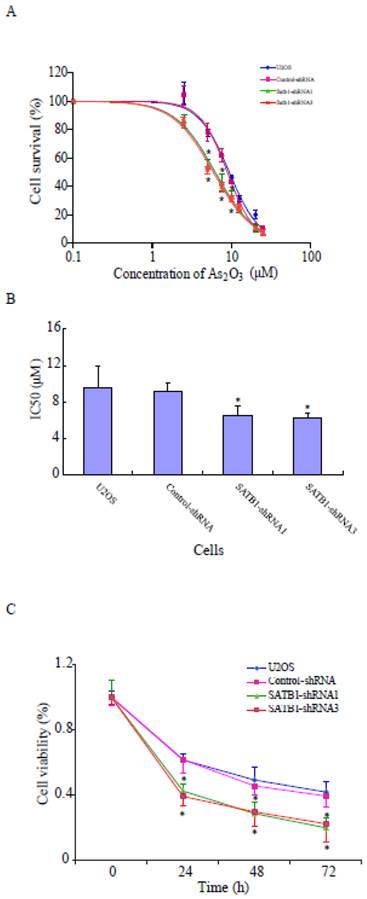
To study whether the increased expression of SATB1 in U2OS is responsible for the resistance of the cells to the chemotherapeutic drug ATO, its effects on the cells with different SATB1 background expression were evaluated using the CCK8 Cell viability assay. As shown in Fig. 4A, ATO caused dose-dependent cell death in both control cells (U2OS and Control-shRNA) and SATB1-silenced cells (SATB1-shRNA1 and SATB1-shRNA3). However, silencing SATB1 expression significantly increased the sensitivity of U2OS cells to arsenic treatment as reflected by a shift to the left in the dose-response curve in SATB1-silenced cells when compared with the control cells (Fig. 4A). As shown in Fig. 4B, the concentration of ATO (6.47 μM and 6.20 μM respectively) that caused 50% cell death in SATB1-shRNA cells was significantly less than that of the control cells (9.15 μM) and As shown in Fig. 4C, ATO induced cell death in a time-dependent manner; however, SATB1-silenced cells (SATB1-shRNA1 and SATB1-shRNA3) demonstrated more rapid and quantitatively greater cell death rates by ATO. All these data indicate that silencing SATB1 might help overcome the resistance of human osteosarcoma U2OS cells to arsenic-based chemotherapy.
SATB1 silencing enhanced apoptosis of U2OS cells. The cell apoptosis assay and statistical analysis were carried out as described in the Materials and Methods. (A): A representative microscopic photograph of the cell apoptosis assay. (B): Statistical results on the percentage of apoptotic cells. The data are expressed as the mean ± SD, N=6, *P < 0.05, versus non-silenced (Control-shRNA) cells.
SATB1 silencing enhanced ATO-mediated cell death. SATB1-silenced (SATB1-shRNA1 and SATB1-shRNA3) and non-silenced (Control-shRNA) cells and un-transfected U2OS cells were exposed to various concentrations of ATO (as indicated) for 24 hours, and cell viability was evaluated using the Cell Counting Kit-8 assay kit. Cell survival (%) resulting from exposure to ATO was calculated relative to the respective cell clone without exposure to ATO. Dose-effects curves (A) were plotted by using a non-linear regression model and IC50s shown in (B) were determined based on the fitted curves. Time responses curves are shown (C). The data are expressed as the mean ± SD, N=9, *P < 0.05, versus non-silenced (Control-shRNA) cells at the same concentration of ATO.
Effects of SATB1 silencing on the expression of related genes
| SATB1 | Survivin | Bcl-2 | MMP-2 | MMP-9 | TIMP-3 | ABCC1 | ABCG2 | |
|---|---|---|---|---|---|---|---|---|
| U2OS | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 |
| Control-shRNA | 0.92 ± 0.15 | 1.10 ± 0.17 | 0.77 ± 0.11 | 1.19 ± 0.17 | 0.91 ± 0.15 | 0.88 ± 0.17 | 1.06 ± 0.13 | 1.32 ± 0.19 |
| SATB1-shRNA1 | 0.12 ± 0.01* | 0.14 ± 0.03* | 0.25 ± 0.02* | 0.04 ± 0.02* | 0.04 ± 0.02* | 2.07 ± 0.16* | 0.02 ± 0.01* | 0.34 ± 0.02* |
| SATB1-shRNA3 | 0.14 ± 0.03* | 0.15 ± 0.01* | 0.27 ± 0.03* | 0.03 ± 0.01* | 0.05 ± 0.03* | 1.64 ± 0.09* | 0.03 ± 0.02* | 0.41 ± 0.06* |
The mRNAs of target genes were normalized by the internal standard, RPL13A mRNA and the data were expressed relative to the value of the control cells (Control-shRNA). Each value represents the Mean ± SD of triplicate samples. *p<0.05, versus the control cells.
SATB1 silencing changed the expression of related genes
Based on the above findings and the fact that SATB1 regulates the expression of more than 1,000 genes in tumor cells, we used qRT-PCR to quantify the expression of mRNAs of key genes related with cell migration, invasion, apoptosis and resistance to chemotherapeutic drugs in order to examine the possibility that SATB1 might exert its role through the regulation of gene expression. The results of these experiments are summarized in Table 2. Silencing of SATB1 (SATB1-shRNA1 and SATB1-shRNA3) broadly changed the expression of these genes significantly, as compared to their expression in the shRNA control (U2OS control-shRNA) cells. The mRNAs for two anti-apoptotic genes Survivin and B-cell lymphoma 2 (Bcl-2) decreased around 85% and 75% respectively in both two SATB1 silenced cell clones. The mRNAs for MMP-2 and MMP-9 genes encoding degradative enzymes which destroy local tissue architecture and basement membranes to allow tumor invasion and metastasis decreased more than 90% in SATB1 silenced cell clones. In contrast, mRNA for TIMP-3 which functions as a tissue inhibitor of MMP increased 2 and 1.6 fold respectively in two SATB1 silenced cell clones. The mRNAs for multidrug resistance protein 1 (MRP1/ABCC1) decreased more than 90% in SATB1 silenced cell clones. The mRNAs for breast cancer resistance protein (BCRP/ABCG2) decreased around 60% in both two SATB1 silenced cell clones. The data indicate that silencing SATB1 broadly changed gene expression, suggesting that highly expressed SATB1 in U2OS play its role through its control on the expression of targeted genes.
Discussion
U2OS is a comprehensively characterized human osteosarcoma cell line, which robustly represents clinical osteosarcoma providing researchers with the ability to use in vitro models to study the genetics and functional characteristics of this highly malignant neoplasm [27]. In the previous study, we found that SATB1 is highly expressed in human osteosarcoma U2OS cells and that silencing SATB1 by the shRNA expression vector inhibited the proliferation of the cells. In this study we found that cell migration and invasion were also suppressed and that rates of apoptosis and sensitivities to ATO were enhanced by silencing SATB1 in U2OS cells.
In this study, an ATO concentration of 9.15 μM was found to cause 50% cell death in human osteosarcoma U2OS cells after 24 hours of exposure. In two SATB1 silenced cell clones the IC50 was decreased to 6.47 μM and 6.20 μM respectively. ATO has been under investigation as treatment for a variety of solid tumors including bladder cancer, glioma, breast cancer, hepatocellular carcinoma, cervical cancer, colorectal cancer, esophageal cancer, germ cell tumors, liver cancer, lung cancer, and melanoma (www.clinicaltrials.gov) [5]. ATO in combination with chemotherapy has shown promising activity in osteosarcoma and Ewing sarcoma [28]. These results indicate that ATO plus SATB1 silencing may be a promising therapeutic approach for treating osteosarcomas in which SATB1 is over-expressed, even though more studies are needed to increase its efficiency and decrease adverse reactions. It has also been reported that SATB1 could be down-regulated by the 3-hydroxy-3-methylglutaryl coenzymeA (HMG-CoA) reductase inhibitors (statin drugs) [29] which are widely used to treat hypercholesterolemia. These results provide a practical way to down regulate SATB1, without invoking significant cytotoxicity.
Migration and invasion are a prerequisite for metastasis and are usually positively correlated with the metastatic potential of malignant tumor cells [30]. SATB1 has been reported to be responsible for the migration and invasion of some epithelial derived tumor cells. For example, SATB1 overexpression correlates with the progression of human rectal cancer [31], and upregulation of SATB1 promotes tumor metastasis in liver cancer [32]. Knockdown of SATB1 in human breast cancer MDA-MB-231 cells inhibited tumor growth and metastasis [19]. However little is known about the role of SATB1 in the migration and invasion of mesenchymal derived-osteosarcoma cells. The present study shows that silencing SATB1 significantly suppresses the migratory and invasive ability of human osteosarcoma U2OS cells, indicating that abnormal expression of SATB1 is responsible for the elevated migration and invasion of the cells, suggesting its potential role in the metastasis of human osteosarcoma.
Malignant cells can survive through the upregulation of cell proliferation and the escape from apoptosis. Our previous study showed that the knock-down of SATB1 suppressed the proliferation of human osteosarcoma U2OS cells via cell cycle arrest [23] and in the present study we show that the knock-down of SATB1 increases the rates of apoptosis of the cells, indicating that the abnormal expression of SATB1 in U2OS cells contributes to promote cell proliferation and to protect the cells from apoptosis. These results are also supported by earlier studies which have shown that elevated expression of SATB1 in human U251 glioma cells is associated with the suppressed apoptosis of the cells [33]. We also found that silenced SATB1 changed the expression of Bcl-2 and Survivin which generally known as two key anti-apoptotic proteins. It has been reported that the expression both SATB1 and Bcl-2 is significantly higher in human glioblastoma multiforme (GBM) tissues than in normal brain and their levels are associated with patient survival and that there exists a positive correlation between SATB1 and Bcl-2 expression [34]. It has also been demonstrated that the major breakpoint region (mbr) within the 3'-untranslated region (3'-UTR) of Bcl-2 gene could physically interact with Bcl-2 promoter through SATB1-mediated chromatin looping, which was required for epigenetic modifications of the promoter [35]. It has been reported that Epstein-Barr virus latent membrane protein 1 (LMP1) upregulates expression of SATB1 and Survivin in human nasopharyngeal cell lines and that silencing SATB1 decreases cell resistance to apoptosis induced by growth factor withdrawal and the LMP1-mediated Survivin expression [36]. All of these indicate that changed expression of Bcl-2 and Survivin by Knock down of SATB1 might be responsible for the increased apoptosis of human osteosarcoma U2OS cells.
The resistance of the malignant tumor cells to the drug frequently resulted in the failure of chemotherapy. In the present study we shows that the knock-down of SATB1 in U2OS cells increased cell sensitivities to ATO which is currently the most promising chemotherapeutic agent for the treatment of osteosarcoma in which SATB1 is over expressed and decreased mRNAs for multidrug resistance protein 1 (MRP1/ABCC1) and for breast cancer resistance protein (BCRP/ABCG2), indicating that elevated expression of SATB1 of U2OS cells was responsible for the resistance of the cells to ATO and may occur via the up-regulation of ABCC1 and ABCG2. The conclusion is supported by the studies that the higher expression of SATB1 contributes to multidrug resistance of breast cancer [21]. We also plan to conduct experiments to examine the effects of silencing SATB1 on the resistance of U2OS cells to additional chemotherapeutic agents used to treat osteosarcoma, including Methotrexate (MTX), Doxorubicin, cis-dichlorodiamineplatinum (CDDP), and ifosfamide (IFO) in the coming studies.
Taken together, the results in this study indicate that silencing SATB1 significantly inhibits the malignant phenotype of human osteosarcoma U2OS cells; and increases sensitivity of the cells to the chemotherapeutic drug, arsenic trioxide via the effects on the expression of related genes. These data suggest that silencing SATB1 may improve the therapeutic effects of ATO on those osteosarcomas in which SATB1 is over-expressed.
Conclusion
In this study we found that cell migration and invasion were suppressed and the apoptosis and sensitivities to ATO were enhanced by silencing SATB1 in U2OS cells and that ABCC1 and ABCG2 were involved in SATB1 mediated resistance of U2OS cells to ATO. These results indicate that the over-expression of SATB1 in U2OS cells contributes to maintenance of the malignant phenotype and resistance to chemotherapeutic drug. They suggest that silencing SATB1 in these cells might improve the therapeutic effects of arsenic trioxides on osteosarcomas in which SATB1 is over-expressed.
Acknowledgements
This study was supported by the National Natural Science Foundation of China (Grants: NSFC No. 21277057). We would like to express our great appreciation to Professor F. William Orr from the University of Manitoba in Canada for his great help in revising the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Siclari VA, Qin L. Targeting the osteosarcoma cancer stem cell. J Orthop Surg Res. 2010;5:78-87
2. Marina N, Gebhardt M, Teot L, Gorlick R. Biology and therapeutic advances for pediatric osteosarcoma. Oncologist. 2004;9:422-41
3. Ferrari S, Palmerini E. Adjuvant and neoadjuvant combination chemotherapy for osteogenic sarcoma. Curr Opin Oncol. 2007;19:341-6
4. Pakos EE, Nearchou AD, Grimer RJ, Koumoullis HD, Abudu A, Bramer JA. et al. Prognostic factors and outcomes for osteosarcoma: an international collaboration. Eur J Cancer. 2009;45:2367-75
5. Emadi A, Gore SD. Arsenic trioxide - An old drug rediscovered. Blood Rev. 2010;24:191-9
6. Lo-Coco F, Avvisati G, Vignetti M, Thiede C, Orlando SM, Iacobelli S. et al. Retinoic acid and arsenic trioxide for acute promyelocytic leukemia. N Engl J Med. 2013;369:111-21
7. Mayor S. Arsenic trioxide combination improves survival in APL. Lancet Oncol. 2013;14:e346
8. Mi JQ, Li JM, Shen ZX, Chen SJ, Chen Z. How to manage acute promyelocytic leukemia. Leukemia. 2012;26:1743-51
9. Chow SK, Chan JY, Fung KP. Inhibition of cell proliferation and the action mechanisms of arsenic trioxide (As2O3) on human breast cancer cells. J Cell Biochem. 2004;93:173-87
10. Mann KK, Wallner B, Lossos IS, Miller WH Jr. Darinaparsin: a novel organic arsenical with promising anticancer activity. Expert Opin Investig Drugs. 2009;18:1727-34
11. Li Y, Qu X, Qu J, Zhang Y, Liu J, Teng Y. et al. Arsenic trioxide induces apoptosis and G2/M phase arrest by inducing Cbl to inhibit PI3K/Akt signaling and thereby regulate p53 activation. Cancer Lett. 2009;284:208-15
12. Yoda A, Toyoshima K, Watanabe Y, Onishi N, Hazaka Y, Tsukuda Y. et al. Arsenic trioxide augments Chk2/p53-mediated apoptosis by inhibiting oncogenic Wip1 phosphatase. J Biol Chem. 2008;283:18969-79
13. Fei M, Lu M, Wang Y, Zhao Y, He S, Gao S. et al. Arsenic trioxide-induced growth arrest of human hepatocellular carcinoma cells involving FOXO3a expression and localization. Med Oncol. 2009;26:178-85
14. Zhang S, Guo W, Ren TT, Lu XC, Tang GQ, Zhao FL. Arsenic trioxide inhibits Ewing's sarcoma cell invasiveness by targeting p38(MAPK) and c-Jun N-terminal kinase. Anticancer Drugs. 2012;23:108-18
15. Dickinson LA, Joh T, Kohwi Y, Kohwi-Shigematsu T. A tissue-specific MAR/SAR DNA-binding protein with unusual binding site recognition. Cell. 1992;70:631-45
16. Tattermusch A, Brockdorff N. A scaffold for X chromosome inactivation. Hum Genet. 2011;130:247-53
17. Shannon MF. A nuclear address with influence. Nat Genet. 2003;34:4-6
18. Cai S, Han HJ, Kohwi-Shigematsu T. Tissue-specific nuclear architecture and gene expression regulated by SATB1. Nat Genet. 2003;34:42-51
19. Han HJ, Russo J, Kohwi Y, Kohwi-Shigematsu T. SATB1 reprogrammes gene expression to promote breast tumour growth and metastasis. Nature. 2008;452:187-93
20. Zheng J. Is SATB1 a master regulator in breast cancer growth and metastasis?. Womens Health (Lond Engl). 2008;4:329-32
21. Li QQ, Chen ZQ, Xu JD, Cao XX, Chen Q, Liu XP. et al. Overexpression and involvement of special AT-rich sequence binding protein 1 in multidrug resistance in human breast carcinoma cells. Cancer Sci. 2010;101:80-6
22. Cheng C, Lu X, Wang G, Zheng L, Shu X, Zhu S. et al. Expression of SATB1 and heparanase in gastric cancer and its relationship to clinicopathologic features. APMIS. 2010;118:855-63
23. Zhang H, Qu S, Li S, Wang Y, Li Y, Wang Y. et al. Silencing SATB1 inhibits proliferation of human osteosarcoma U2OS cells. Mol Cell Biochem. 2013;378:39-45
24. Wang Y, Wei Y, Zhang H, Shi Y, Li Y, Li R. Arsenic trioxide induces apoptosis of p53 null osteosarcoma MG63 cells through the inhibition of catalase. Med Oncol. 2012;29:1328-34
25. Yedjou CG, Moore P, Tchounwou PB. Dose- and time-dependent response of human leukemia (HL-60) cells to arsenic trioxide treatment. Int J Environ Res Public Health. 2006;3:136-40
26. Huan SY, Yang CH, Chen YC. Arsenic trioxide therapy for relapsed acute promyelocytic leukemia: an useful salvage therapy. Leuk Lymphoma. 2000;38:283-93
27. Mohseny AB, Machado I, Cai Y, Schaefer KL, Serra M, Hogendoorn PC. et al. Functional characterization of osteosarcoma cell lines provides representative models to study the human disease. Lab Invest. 2011;91:1195-205
28. Guo W, Tang XD, Tang S, Yang Y. [Preliminary report of combination chemotherapy including Arsenic trioxide for stage III osteosarcoma and Ewing sarcoma]. Zhonghua Wai Ke Za Zhi. 2006;44:805-8
29. Lakshminarayana Reddy CN, Vyjayanti VN, Notani D, Galande S, Kotamraju S. Down-regulation of the global regulator SATB1 by statins in COLO205 colon cancer cells. Mol Med Rep. 2010;3:857-61 doi:10.3892/mmr.2010.338
30. Entschladen F, Drell TLt, Lang K, Joseph J, Zaenker KS. Tumour-cell migration, invasion, and metastasis: navigation by neurotransmitters. Lancet Oncol. 2004;5:254-8
31. Meng WJ, Yan H, Zhou B, Zhang W, Kong XH, Wang R. et al. Correlation of SATB1 overexpression with the progression of human rectal cancer. Int J Colorectal Dis. 2011;27:143-50
32. Tu W, Luo M, Wang Z, Yan W, Xia Y, Deng H. et al. Upregulation of SATB1 promotes tumor growth and metastasis in liver cancer. Liver Int. 2012;32:1064-78
33. Chu SH, Ma YB, Feng DF, Zhang H, Zhu ZA, Li ZQ. et al. Upregulation of SATB1 is associated with the development and progression of glioma. J Transl Med. 2012;10:149-52
34. Chu SH, Ma YB, Feng DF, Li ZQ, Jiang PC. Correlation between SATB1 and Bcl-2 expression in human glioblastoma multiforme. Mol Med Rep. 2013;7:139-43
35. Gong F, Sun L, Wang Z, Shi J, Li W, Wang S. et al. The BCL2 gene is regulated by a special AT-rich sequence binding protein 1-mediated long range chromosomal interaction between the promoter and the distal element located within the 3'-UTR. Nucleic Acids Res. 2011;39:4640-52 doi:10.1093/nar/gkr023
36. Endo K, Shackelford J, Aga M, Yoshizaki T, Pagano JS. Upregulation of special AT-rich-binding protein 1 by Epstein-Barr virus latent membrane protein 1 in human nasopharyngeal cells and nasopharyngeal cancer. J Gen Virol. 2013;94:507-13 doi:10.1099/vir.0.046243-0
Author contact
![]() Corresponding author: Dr. Ronggui Li, The Key Laboratory of Pathobiology, Ministry of Education, Norman Bethune College of Medicine, Jilin University, Changchun, 130021, P.R. China. Tel.: 86-431 85619481; E-mail: lirgedu.cn and Dr. Zonggui Wang, The Second Hospital of Jilin University, Changchun, P.R. China. Tel.: 86-431 88796114; E-mail: zgw1965com
Corresponding author: Dr. Ronggui Li, The Key Laboratory of Pathobiology, Ministry of Education, Norman Bethune College of Medicine, Jilin University, Changchun, 130021, P.R. China. Tel.: 86-431 85619481; E-mail: lirgedu.cn and Dr. Zonggui Wang, The Second Hospital of Jilin University, Changchun, P.R. China. Tel.: 86-431 88796114; E-mail: zgw1965com
Received 2014-7-4
Accepted 2014-8-27
Published 2014-10-2